

Abstract
Neocortical long-term potentiation (LTP) consists of both presynaptic and postsynaptic components that rely on nitric oxide (NO) and the GluR1 subunit of the AMPA receptor, respectively. In this study, we found that hippocampal LTP, induced by theta-burst stimulation in mature (>8-week-old) GluR1 knock-out mice was almost entirely NO dependent and involved both the α splice variant of NO synthase-1 and the NO synthase-3 isoforms of NO synthase. Theta-burst induced LTP was also partly NO-dependent in wild-type mice and made up ∼50% of the potentiation 2 h after tetanus. Theta-burst stimulation reliably produced postsynaptic spikes, including a high probability of complex spikes. Inhibition of postsynaptic somatic spikes with intracellular QX314 or local TTX application prevented LTP in the GluR1 knock-out mice and also blocked the NO component of LTP in wild types. We conclude that theta-burst stimulation is particularly well suited to producing the postsynaptic somatic spikes required for NO-dependent LTP.
Keywords: plasticity, potentiation, nNOS, eNOS, working memory, long-term memory
Introduction
The mechanisms underlying hippocampal long-term potentiation (LTP) have been studied extensively since its original discovery (Bliss and Lomo, 1973). Recently, efforts have concentrated on the postsynaptic mechanisms of LTP, which involve insertion of AMPA receptors into the postsynaptic membrane (Malinow and Malenka, 2002). However, presynaptic components of hippocampal LTP have also been documented (Malinow and Tsien, 1990), most recently by direct imaging (Stanton et al., 2005; Bayazitov et al., 2007) and less is known of the incipient mechanisms involved in this form of LTP. Because LTP can be induced postsynaptically (Malenka et al., 1989) and yet is partly expressed presynaptically (Stanton et al., 2005; Bayazitov et al., 2007), some retrograde factor must be involved in coordinating presynaptic and postsynaptic components of transmission strength (Lisman and Raghavachari, 2006). Nitric oxide (NO) became an early candidate for this retrograde factor (Haley et al., 1992; Kantor et al., 1996; Son et al., 1996), but research in this area has slowed partly due to the difficulty of reproducing findings in different laboratories on the role of NO in LTP and memory [for review, see Hölscher (1997)].
Recent evidence from the neocortex has shown that layer II/III cells exhibit LTP that can be separated into presynaptic and postsynaptic components by manipulating GluR1 and NOS (Hardingham and Fox, 2006). While GluR1 is responsible for the postsynaptic component of LTP in the neocortex, the presynaptic component is dependent on postsynaptic NO synthase (NOS) activation (Hardingham and Fox, 2006). In the neocortex, LTP cannot be abolished entirely by blocking either NOS or GluR1, but blocking both simultaneously eliminates LTP.
The first reports of GluR1-dependent LTP in the hippocampus indicated that LTP was completely absent in GluR1 knock-outs (Zamanillo et al., 1999), but it was later discovered that GluR1 dependent LTP was present in younger animals and required a spike-timing protocol to induce it (Hoffman et al., 2002; Jensen et al., 2003). This raises the possibility that this residual component of LTP is also NO dependent in the hippocampus, as it is in the neocortex.
Therefore, we looked at LTP in the hippocampus of GluR1 null mutants (Zamanillo et al., 1999) to test whether LTP is NO-dependent. We also studied the isoforms of NOS involved using the endothelial NOS (NOS-3) knock-out and the α neuronal NOS (αNOS-1) knock-out mice. The αNOS-1 knock-out shows a 94.5% reduction in catalytic activity (Huang et al., 1993) and lacks the major α splice variant but not the β and gamma splice variants of NOS-1 (Eliasson et al., 1997). The α splice variant contains a PDZ domain which links NOS-1 to PSD-95 and hence to the postsynaptic density, whereas the β and gamma isoforms do not and so are cytoplasmic (Eliasson et al., 1997). Deletion of the synaptically located αNOS-1 isoform therefore makes the αNOS-1 knock-out particularly well suited to studying synaptic deficits. Our studies reveal that both major NOS isoforms play a role in hippocampal LTP and that postsynaptic spikes are necessary for the induction of the NO component of LTP in both GluR1 knock-outs and wild-type mouse hippocampus.
Materials and Methods
Animals.
Subjects were mice aged P45–64 (6–9 weeks) for the intracellular experiments and P57–64 (8–9 weeks) for the extracellular experiments. AMPA receptor subunit 1 (GluR1) knock-out mice, αNO synthase isoform 1 (αNOS-1) knock-out mice, NO synthase isoform 3 (NOS-3) knock-out mice and wild-type littermates were bred into a C57BL/6 background and maintained in the colony as heterozygotes. Experimental null mutants and wild-type littermates were bred from heterozygote crosses (cousin mating). Double knock-out animals were created by breeding heterozygous single knock-outs until double heterozygous males and females were produced. Double knock-outs were produced by mating double heterozygous animals, or on a few occasions by mating GluR1−/− NOS−/+ mice with double heterozygotes. The GluR1 knock-out mice were kindly supplied by Rolf Sprengel (Max Planck Institute for Medical Research, Heidelberg, Germany) via the Rawlins laboratory at Oxford University. The αNOS-1 and NOS-3 knock-outs were obtained from The Jackson Laboratory. We genotyped the animals used in this study by PCR using primers ordered from MWG. The following primer sequences were used, for αNOS-1: (oIMR13) 5′ CTT GGG TGG AGA GGC TAT TC 3′; oIMR14 5′AGG TGA GAT GAC AGG AGA TC 3′; (oIMR406) 5′ TCA GAT CTG ATC CGA GGA GG 3′; (oIMR407) 5′ TTC CAG AGC GCT GTC ATA GC 3′. For NOS-3: (oIMR94) 5′ TGG CTA CCC GTG ATA TTG CT 3′; (oIMR1823) 5′ ATT TCC TGT CCC CTG CCT TC 3′; (0IMR1824) 5′GGC CAG TCT CAG AGC CAT AC 3′. Jackson Laboratories supplied both αNOS-1 and NOS-3 primer sequences. For the GluR1 knock-outs we used (1005) 5′ AAT GCC TAG TAC TAT AGT GCA CG 3′; (MH60) 5′ CAC TCA CAG CAA TGA AGC AGG AC 3′; (3Int3) 5′ CTG CCT GGG TAA AGT GAC TTG G 3′. Rolf Sprengel supplied primer sequences for the GluR1 knock-outs.
Slice preparation.
Mice were killed via cervical dislocation and decapitated. Brains were quickly removed and immersed into ice-cold artificial CSF (aCSF) [composition (in mm): 124 NaCl, 2.3 KCl, 2 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26 NaHCO3, and 11 d-glucose] constantly bubbled with 95%O2/5%CO2 to maintain the pH at 7.4. Coronal sections (400 μm) were cut with a vibratome and incubated for at least 1 h in a submersion chamber kept at 32°C.
Extracellular field potentials.
Slices were transferred to a submerged recording chamber perfused with aCSF at 32°C. Extracellular field potentials were recorded in the stratum radiatum of the CA1 region of hippocampus using carbon fiber electrodes. Responses were evoked in control and test pathways using a 20 μs square voltage step applied at 0.05 Hz through two monopolar electrodes located in stratum radiatum test (S1) and control (S2) pathways. The S1 electrode was placed approximately equidistant from the molecular and pyramidal layers on the CA3 side of the recorded cell. To ensure pathway independence, the stimulating electrodes were placed at slightly different depths in the stratum radiatum. The S2 electrode was placed either higher or lower than S1 (in alternate experiments) and was always located on the subiculum side of the recorded cell. If any effect on the S2 control pathway was observed after tetanus given to the S1 pathway, the recording was discarded. Input/output (I/O) curves were produced by gradual increases in stimulus strength at the beginning of each experiment, until a stable baseline of evoked response was reached. The test stimulus pulse was then adjusted to produce a field EPSP (fEPSP) whose slope and amplitude was 40% that of the maximum possible fEPSP and was kept constant throughout the experiment. The negative going slope of each fEPSP was measured over the 20–80% range of the peak amplitude. Responses were amplified (Axoclamp 2B), digitized [Cambridge Electronic Design (CED) 1401], and recorded using Signal (CED).
Dual extracellular and whole-cell patch-clamp recordings.
Recordings were made in a submerged chamber perfused with aCSF at 32°C. To enable the results of the intracellular recordings to be compared directly to the extracellular recordings it was important to keep the stimulus strength constant between studies. This was achieved by recording an extracellular I/O curve before the intracellular recording was made. Intracellular recordings were then obtained from CA1 cells that were directly above the fEPSP recording electrode, perpendicular to the stratum radiatum. CA1 pyramidal cells were visualized using an Olympus Optical BH2 video microscope, using DIC optics. Patch electrodes with a resistance of 10–15 MΩ were pulled from borosilicate filamented glass capillaries using a horizontal puller (Sutter). Pipettes were filled with intracellular solution containing in mm: 110 K-gluconate, 10 KCl, 2 MgCl2, 0.3 Na2ATP, 0.03 Na2GTP, and 10 HEPES corrected to pH 7.3 (290 mOsm). QX314 (20 μm) was included in the electrode filling solution for one set of experiments as described in the results. Whole-cell recordings were made at the post break-in potential (average Em of –64 ± 3 mV for wild types, –68 ± 4 mV for GluR1 knock-outs) in the current-clamp mode but discarded if the input resistance changed by >20%. Responses were amplified (Axoclamp 2B), low-pass filtered below 4–5 kHz, digitized (CED 1401) and recorded using Signal (CED). When indicated in the text, 10 μm TTX dissolved in aCSF was pressure-applied through a patch pipette positioned near the soma under visual guidance. The flow of the solution was monitored by using 0.1% Fast Green in the TTX pipette and optimized to avoid TTX diffusion into the SR. Pressure application prevented postsynaptic action potential initiation and backpropagation while not affecting the EPSP amplitude.
Induction of synaptic plasticity.
Extracellular fEPSPs were recorded from two independent pathways for a baseline period of 20 min. Intracellular recording only permitted a 4–5 min baseline period to prevent LTP washout. LTP was induced by either 100 Hz or theta-burst stimulation (indicated in figure legends). Stimulation (100 Hz) consisted of 100 pulses at a frequency of 100 Hz; this was then repeated three times at 0.05 Hz. In theta-burst stimulation, the bursts consisted of four pulses at 100 Hz, repeated 10 times at 5 Hz (theta frequency); these were also repeated three times at 0.05 Hz. These protocols were used either at a low stimulus intensity (defined as a control voltage of 40% of maximum and a pulse-width of 20 μs) or at a high stimulus intensity (defined as a control voltage of 40% maximum and a double pulse-width of 40 μs).
We measured the number of spikes produced by theta-burst and 100 Hz stimulation in the intracellular recording experiments. For the purposes of the analysis the number of “spikes per train” was considered as the number of spikes produced in a train of 10 theta-bursts (40 stimuli) and a train of 100 stimuli for 100 Hz stimulation. The number of “spikes per stimulus” was considered to be the total number of spikes produced divided by the number of stimuli during the stimulus train, which was 120 stimuli for theta-burst stimulation and 300 stimuli for 100 Hz stimulation.
Drugs.
All drugs were applied in the aCSF unless otherwise stated. N-nitro-l-arginine (l-NNA), N-ω-nitro-l-arginine methyl ester (l-NAME), (d)-2-amiono-5-phosphopentanoate (d-AP5), N-(2,6-dimethylphenylcarbamoylmethyl) triethylammonium chloride (QX314), and octahydro-12-(hydroxymethyl)-2-imino-5,9:7,10a-dimethan-o-10aH-[1,3]dioxocino[6,5-d]pyrimidine-4,7,10,11,12-pentol (TTX) were all obtained from Tocris Bioscience. Autocamtide-2-inhibitory peptide (AIP) (KKALRRQEAVDAL) was obtained from Calbiochem [described by Ishida et al. (1998)].
Data analysis.
Responses were normalized with respect to the baseline and data groups expressed as mean ± SEM. For extracellular recordings, data comparisons were made between test and control pathways at the time points given in the text. For intracellular recordings, data were compared within pathway between baseline and 40–45 min after LTP induction. ANOVAs were then run to test for effects of genotype and drug application, together with any interaction terms using Graphpad software. Post hoc Bonferroni corrected t tests were then used to investigate the origin of effects further. For tests of statistical significance, p values are as reported in the text.
The mean amplitude and variance were measured for the whole control period plus stable periods of at least 50 (and usually 100) stimuli after the induction of LTP. At least two separate noise measurements were taken for each EPSP, from nonoverlapping parts of the baseline, to calculate the mean noise SD. This noise SD was then subtracted from the EPSP SD using the following equation: (EPSP SD)2 = (SD of combined EPSP + noise)2 − (noise SD)2.
Two periods were studied, the control period and the last 10 min of potentiation. We calculated the squared coefficient of variation (CV2) from the variance and mean amplitude (CV−2 = mean2/variance), normalized the mean amplitude and CV−2 values to the control period, and plotted values for the two time periods (Malinow and Tsien, 1990).
Paired-pulse facilitation (PPF) was measured (interstimulus interval 75 ms) during the control period and 40 min after the induction of LTP. PPF was expressed as a ratio, i.e., the amplitude of the second EPSP was divided by the amplitude of the first. The average PPF during the control period was then compared with the PPF ratio 40 min after the tetanus. The change in the PPF ratio (ΔPPF) was then calculated by subtracting the post tetanus PPF ratio from the control PPF ratio in each individual experiment and then averaged (Hardingham and Fox, 2006).
Results
LTP can be induced by orthodromic theta-burst stimulation in adult mice
A tetanic stimulus applied at 100 Hz to the schaeffer collateral-CA1 pathway produced robust LTP in wild-type mice (mean ± SEM = 153 ± 9%) but not in GluR1 knock-out mice (96 ± 4%) (Fig. 1A). The stimulus intensity was routinely set at 40% of the maximal value measured from the I/O curves plotted at the start of the experiment. Theta-burst stimulation at the same pulse width (20 μs) or 100 Hz stimulation at an increased stimulus strength, (produced by doubling the width of the stimulus pulse from 20 to 40 μs) were also ineffective at inducing LTP in the GluR1 knock-outs (Fig. 1B,C). However, theta-burst stimulation in combination with the double pulse-width stimulus produced LTP of similar magnitude both in wild types and GluR1 knock-outs (173 ± 7% in wild types vs 171 ± 16 in GluR1 knock-outs measured at 60 min after tetanus) (Fig. 1D). LTP was significant in both cases here (p < 0.001, Bonferroni corrected post hoc t test).
Figure 1.
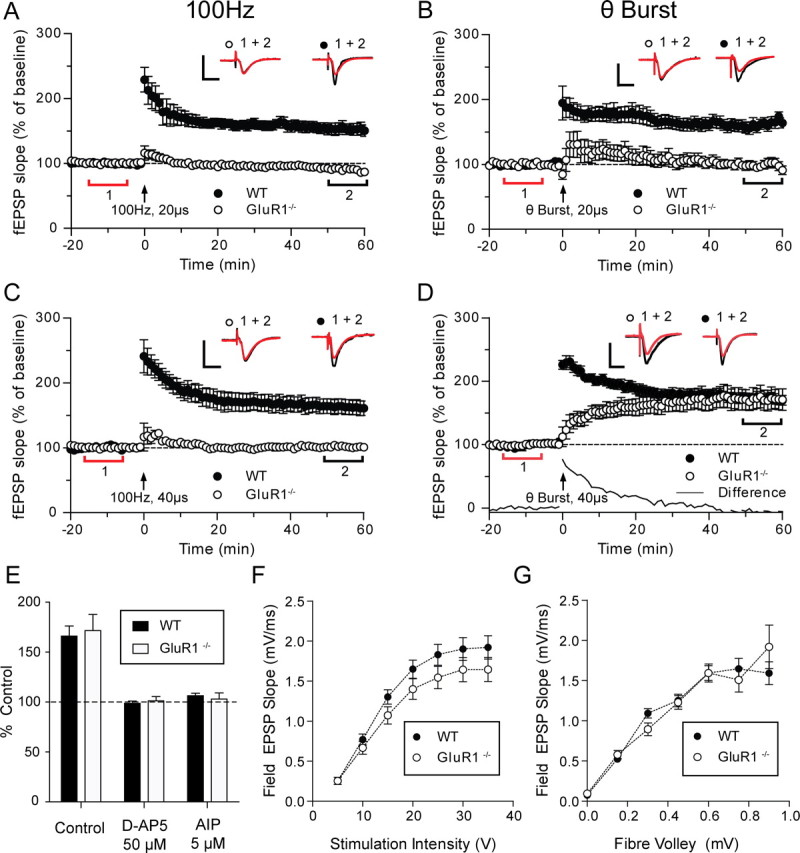
High-intensity theta-burst stimulation produces GluR1- independent LTP that depends on NMDAR and CaMKII. A, C, In GluR1−/− mice (○), 100 Hz stimulation (100 pulses at 100 Hz, repeated three times at 0.05 Hz, delivered at arrow) at either low intensity (A) (test pulse-width 20 μs) or high intensity (C) (double test pulse-width 40 μs), produces no significant potentiation of the fEPSP, compared with a highly significant potentiation in wild-type mice (●). B, Low-intensity theta-burst stimulation [four pulses at 100 Hz repeated 10 times at 5 Hz (theta) repeated three times at 0.05 Hz] also produces no fEPSP potentiation in the GluR1−/− mice (○), whereas significant potentiation is seen in wild-type mice (●). D, High-intensity theta-burst stimulation produces a slowly rising form of potentiation in GluR1−/− mice (○) that is indistinguishable from the potentiation in wild-type mice (●) at 60 min. Each point plots the average amplitude of four successive fEPSPs normalized with respect to the baseline and expressed as mean ± SEM. Insets are representative traces taken at time points indicated by the bars (red, control period; black, 50–60 min) with the symbols identifying individual experimental conditions. Calibration: 1 mV, 10 ms. E, 50 μm d-AP5 or 5 μm AIP completely block LTP in wild-type (filled bars) and GluR1−/− mice (gray bars). F, I/O curves for wild-type (●) and GluR1−/− (○) mice show no difference in baseline transmission [p > 0.05, not significant (NS)]. G, Fiber volley, I/O curves for wild-type (●) and GluR1−/− (○) mice also show no difference in baseline transmission (p > 0.05, NS). WT, Wild type.
Over the first 20 min, LTP in the GluR1 knock-outs increased more slowly than LTP in the wild types. Figure 1D (bottom, solid line) illustrates the difference in the time course of the potentiation by subtracting the potentiation seen in the GluR1 knock-outs from the potentiation seen in wild types; it is very similar to the LTP described by Hoffman et al. (2002) and Jensen et al. (2003). However, two factors were different in the present study; first LTP was produced purely by orthodromic stimuli and did not require postsynaptic current injection to ensure spike pairing. Second, the animals were at least 8 weeks of age and therefore the LTP was not restricted to immature synases.
We found that the induction of LTP in the GluR1 knock-outs depended not only on the intensity of stimulation but also on the parameters of the tetanus protocol. Neither theta-burst stimulation with a 20 μs stimulus pulse-width (Fig. 1B) nor 100 Hz stimulation with a 40 μs stimulus pulse-width (Fig. 1C) reliably induced LTP in our hands. The dependence of LTP on stimulus intensity in the GluR1 knock-outs could not be accounted for by lower levels of synaptic transmission (when compared with wild types), as the I/O curves were not significantly different between the two genotypes (Fig. 1F) (Bonferroni corrected t test, p > 0.05). To analyze this result further, the I/O function was also assessed relative to the size of the fiber volley. The fiber volley amplitude is proportional to the number of axons activated, allowing for an independent measurement of input strength and compensating for any small differences in stimulating and recording electrode placement between experiments. However, when the I/O response was plotted against the fiber volley we still found no difference between wild types and GluR1 knock-outs (Bonferroni corrected t test, p > 0.05) (Fig. 1G). As mentioned above, the stimulus intensity was routinely set at 40% of maximum response saturation corresponding to a mean value of ∼10V (Fig. 1F). As can be seen from the I/O curve the response averages for the two genotypes at the 40% setting are very similar (Fig. 1F,G).
We investigated whether the LTP seen in the GluR1 knock-outs depended on the same receptors and signaling cascades as LTP in the wild types (Fig. 1E). We conclude that induction of LTP in the GluR1 knock-outs by theta-burst stimulation applied using a 40 μs stimulus pulse was dependent on NMDA receptors because it was blocked by 50 μm d-AP5 applied extracellularly (101 ± 4%; significantly different from control, p < 0.001; t(27) = 4.2) and dependent on CaMKII because it was blocked by 5 μm autocamtide inhibitory peptide (AIP) applied intracellularly (103 ± 6, p < 0.001, t(27) = 4.1).
Efficacy of LTP protocols is strongly correlated with spike production
To understand more about the differences between the theta-burst and 100 Hz stimulation protocols, we recorded intracellularly from postsynaptic cells during LTP induction. We found that theta-burst stimulation only produced a significant number of postsynaptic action potentials at the higher stimulation intensity (40 μs pulse-width) as shown in Figure 2B,D. To our surprise, we found that in our hands, 100 Hz stimulation was not at all effective in producing postsynaptic spikes. Action potentials did not follow the high rate of stimulation and rapidly failed over time, either due to depolarization block or perhaps due to spike accommodation (Fig. 2A,C).
Figure 2.

Number of postsynaptic spikes differs significantly between induction protocols. Example traces of intracellular recording during (A), 100 Hz stimulation at low intensity; B, theta-burst stimulation at low intensity; C, 100 Hz stimulation at high intensity; and D, theta-burst stimulation at high intensity. Left (panels) are the first 100 ms of each burst. E, Bars indicate the number of spikes produced per stimuli in the train (total number of spikes in train/total number of stimuli given in the train). The probability of generating a spike is significantly greater during high-intensity (gray bars) theta-burst stimulation compared with low-intensity (white bars) 100 Hz stimulation, high-intensity 100 Hz stimulation and low-intensity theta-burst stimulation. F, The total number of spikes per train is significantly greater during high-intensity (gray bars) theta-burst stimulation compared with low-intensity (white bars) 100 Hz stimulation, high-intensity 100 Hz stimulation or low-intensity theta-burst stimulation.
We quantified these effects and found that the spike probability (per stimulus) was ∼40-fold greater during theta-burst stimulation (44 ± 8% in wild-type, 53 ± 6% in GluR1 knock-outs) than for 100 Hz stimulation (1 ± 6% in wild-type, 2 ± 7% in GluR1 knock-outs) (Fig. 2E,F), using the same intensity of stimulation in each case (40% of maximum, 40 μs pulse-width). Consequently, applying more presynaptic stimuli during a 100 Hz protocol produced many fewer spikes than with a theta-burst protocol. Although it is possible that other experimenters produced action potentials using 100 Hz stimulation, we were not able to do so, and, as described below, this made 100 Hz stimulation a useful tool for some of the experiments in these studies.
Blocking somatic spikes prevents LTP in GluR1 knock-outs
To determine the importance of somatic spikes in LTP induction we recorded from CA1 pyramidal neurones using electrodes containing the sodium channel blocker QX314 and used the theta-burst LTP protocol (40 μs duration pulses). We found that after breaking into the cell QX314 rapidly abolished action potentials (Fig. 3A). Although spikes were eliminated during the theta-burst tetanus, the degree of EPSP summation with QX314 was almost identical to control levels (Control = 22.0 ± 0.7 mV; QX314 = 23.0 ± 1.1 mV). (Fig. 3B,C). We found that QX314 prevented induction of LTP in the GluR1 knock-out mice (Fig. 3D) but had no effect on wild-type LTP.
Figure 3.
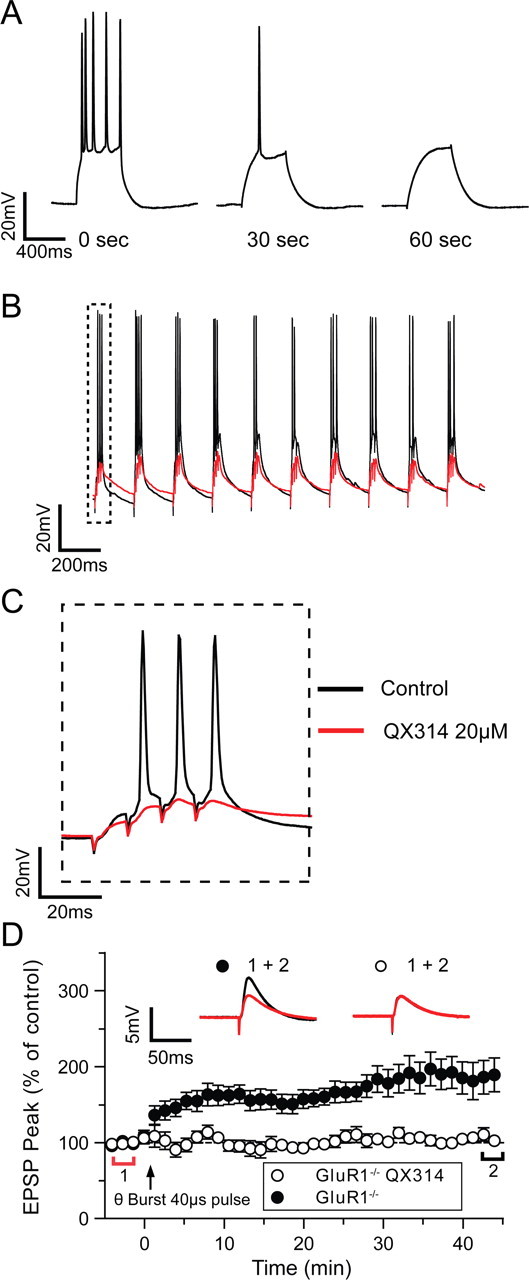
Intracellular QX314 blocks plasticity in the GluR1−/− mice. A, Spikes generated by depolarizing current injection (2.5 nA, 500 ms) are quickly blocked (<30 s) by QX314 (20 μm) as it dialyses into the cell (legends show time after gaining access to the cell). B, Spiking during high-intensity theta-burst stimulation (black line) is blocked when QX314 is included in the patch electrode (red line). C, EPSP summation is unaffected by the inclusion of QX314 in the electrode (control black line, QX314 red line) D, LTP was induced after a 5 min control period by a high-intensity theta-burst stimulation at t = 0 (arrow). Potentiation in GluR1−/− mice (●) was blocked by QX314 in the patch pipette (○). Each point plots the average amplitude of 9 successive EPSPs normalized with respect to the baseline and is expressed as the mean ± SEM. Insets are representative traces taken at time points indicated by the bars (red, control period; black, 42–45 min), with the symbols identifying the experimental conditions.
It was possible that QX314 acted by eliminating somatic or dendritic orthodromic spikes or by affecting targets other than sodium channels. Therefore, as a more specific test of the need for somatic action potentials in LTP induction in the GluR1 knock-outs, we applied the specific sodium channel blocker TTX extracellularly via a micropipette carefully positioned close to the soma of the cell being recorded from, under visual control (Fig. 4A). The bath aCSF flowed from dendrites to soma to further localize TTX to the soma. Using this technique it was possible to pressure eject TTX onto the soma and reversibly block action potentials (Fig. 4B). We found that blockade of somatic action potentials had little effect on the degree of EPSP summation caused by the stimulus (Control = 22.0 ± 0.7 mV, TTX = 19.0 ± 2.1 mV) (Fig. 4C,D).
Figure 4.

Somatic spikes are required for plasticity in GluR1−/− mice. A, Schematic diagram of the experimental setup. Action potential generation and propagation can be blocked by local pressure application of TTX (10 μm) to the soma. The slice is positioned so that TTX does not perfuse on the stratum (s.) radiatum. B, Example trace illustrating how spikes generated by a depolarizing current injection (2.5 nA, 500 ms) can be reversibly blocked by the local somatic application of TTX (10 μm). C, Spiking during high-intensity theta-burst stimulation (black line) is blocked if TTX is perfused on the soma (red line). D, EPSP summation during the theta-burst stimulation is unaffected by somatic TTX application. E, LTP was induced after a 5 min control period by a high-intensity theta-burst stimulation at t = 0 (arrow). Somatic TTX application (○) has a small effect on wild-type (WT) LTP at 45 min (●). F, The LTP observed in GluR1−/− mice (●) is completely abolished when somatic spikes are blocked with local TTX application (○). Each point plots the average amplitude of eight successive EPSPs normalized with respect to the baseline and expressed as mean ± SEM. Insets are representative traces taken at time points indicated by the bars (red, control period; black, 42–45 min) with the symbols identifying the experimental conditions.
Blocking somatic action potentials with TTX did not prevent LTP in wild types (mean = 155 ± 17%) (Fig. 4E), but it did prevent LTP in GluR1 knock-out mice (109 ± 5%) (Fig. 4F). Experiments with TTX were interleaved with control experiments. A two-way ANOVA showed an interaction between TTX treatment and genotype (F(1,64) = 5.08, p < 0.03) and post hoc tests revealed that this was because the GluR1 knock-out only showed significant LTP without TTX (t(35) = 3.1, p < 0.01), while wild types showed LTP with or without TTX (t(23) = 1.68, p > 0.05). On several occasions we were able to hold the postsynaptic cell long enough to reverse the effects of TTX and recover normal action potentials (Fig. 4B) (n = 3). LTP was not induced when action potentials were blocked, but subsequently could be induced when action potential firing was restored (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
We further analyzed the data from the experiments described above to see if the depolarization level produced by the induction protocol affected whether LTP was induced successfully. We found that the level of depolarization produced by the different protocols had little influence on the level of LTP in the GluR1 knock-out animals (Fig. 5B,C) while the total number of spikes the protocol produced had a much bigger effect on the level of LTP (Fig. 5A). Of course, the action potentials themselves produced a substantial depolarization, but this was not an important factor in controlling LTP induction in wild types, which showed LTP both with and without spikes. On average, theta-burst stimulation using a double pulse-width produced more than one spike per train (mean = 1.75 spikes/train) and produced a number of complex spikes in the postsynaptic cells (Fig. 2D). From the 44 cells recorded, there was on average four complex spikes per theta-burst train and all cases showed at least one complex spike. Single pulse-width theta-burst stimulation produced far fewer spikes per train (mean = 0.22 spikes/train) and rarely produced complex spikes (2 from 16 cells), which might explain why it was less effective in producing LTP in the GluR1 knock-out mice.
Figure 5.
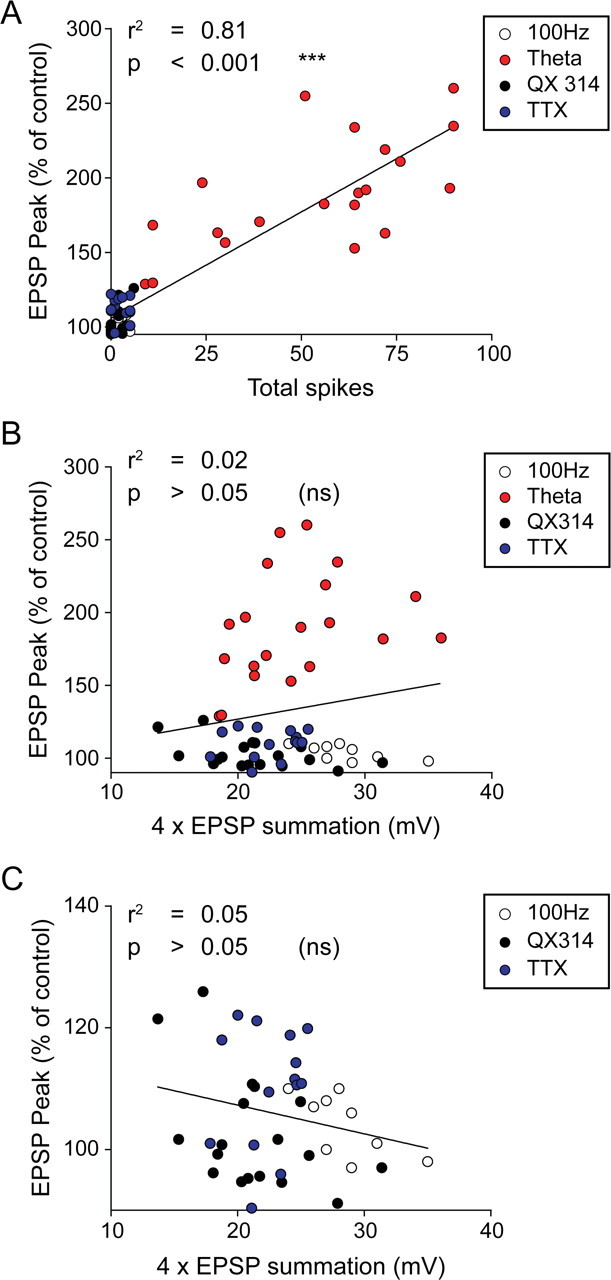
The level of potentiation in the GluR1−/− mice is correlated to the number of spikes observed in the burst and not to the level of depolarization in the burst. A, Magnitude of LTP in the GluR1 −/− depends on the total number of spikes during the burst. Individual experiments show a correlation between the number of spikes in a burst and the increase in EPSP observed with LTP. B, Magnitude of LTP in the GluR1−/− is not correlated to the average amplitude of summated EPSPs during the theta-burst stimulation. C, Same data as shown in B with control theta-burst stimulation data excluded as spiking will be related to the level of depolarization. Data are pooled from LTP experiments generated by high-intensity 100 Hz stimulation (white circle), theta-burst stimulation (red circle), theta-burst stimulation with 0.2 mm QX 314 (black circle), and theta-burst stimulation with 10 μm TTX (blue circle).
Spike-dependent LTP in GluR1 knock-outs is largely NO dependent
In barrel cortex, a large part of the LTP expressed in GluR1 knock-outs is dependent on NO (Hardingham and Fox, 2006). To determine whether a similar dependency exists in the CA1 region of the hippocampus, we perfused alternate GluR1 knock-out slices with the NOS inhibitor l-NNA (Fig. 6A). With extracellular l-NNA, LTP was reduced to 115 ± 11% at 60 min after a theta-burst tetanus compared with 172 ± 16% in untreated controls, which was a highly significantly reduction (Fig. 6D p < 0.001). The small amount of residual LTP present with l-NNA application was however still significantly different from the untetanised control pathway [using a paired t test (t(10) = 3.29, p < 0.05, Bonferroni corrected)].
Figure 6.

Comparison of LTP in GluR1−/− single-knock-out and GluR1−/−αNOS-1−/− and GluR1−/− NOS-3−/− double- mutant mice with or without NOS inhibitor. A, LTP in the GluR1−/− (○) mice is significantly reduced by a 5 min application of 100 μm
l-NNA (●). B, LTP in the GluR1−/− αNOS-1−/− double-mutant mice ( ) is reduced when compared with the GluR1−/− single-mutant mice (○). The remaining LTP in double-mutant mice is further reduced by 100 μm
l-NNA (●) and is similar to LTP observed in single GluR1−/− with 100 μm
l-NNA. C, LTP in the GluR1−/− NOS-3−/− double-mutant mice () is also slightly reduced compared with the GluR1−/− single-mutant mice (○) and is comparable with the LTP in GluR1−/−αNOS-1−/− double-mutant mice. The remaining LTP in the double-mutant mice can be further reduced by 100 μm
l-NNA (●). Each point plots the average amplitude of three successive fEPSPs normalized with respect to the baseline and expressed as mean ± SEM. Insets are representative traces taken at time points indicated by the bars (red, control period; black, 50–60 min) with the symbols identifying the experimental conditions. Calibration: 1 mV, 10 ms. D, The average levels of LTP at 50–60 min are plotted for the S1 and S2 pathways in the three genotypes. Note the similarity of the LTP in the presence of l-NNA for all three genotypes. E, F, I/O curves for GluR1−/− single-mutant (●) and GluR1−/− NOS-1−/− double-mutant (E) and GluR1−/− NOS-3−/− double-mutant (F) (○) mice show no differences in baseline transmission (p > 0.05, NS).
) is reduced when compared with the GluR1−/− single-mutant mice (○). The remaining LTP in double-mutant mice is further reduced by 100 μm
l-NNA (●) and is similar to LTP observed in single GluR1−/− with 100 μm
l-NNA. C, LTP in the GluR1−/− NOS-3−/− double-mutant mice () is also slightly reduced compared with the GluR1−/− single-mutant mice (○) and is comparable with the LTP in GluR1−/−αNOS-1−/− double-mutant mice. The remaining LTP in the double-mutant mice can be further reduced by 100 μm
l-NNA (●). Each point plots the average amplitude of three successive fEPSPs normalized with respect to the baseline and expressed as mean ± SEM. Insets are representative traces taken at time points indicated by the bars (red, control period; black, 50–60 min) with the symbols identifying the experimental conditions. Calibration: 1 mV, 10 ms. D, The average levels of LTP at 50–60 min are plotted for the S1 and S2 pathways in the three genotypes. Note the similarity of the LTP in the presence of l-NNA for all three genotypes. E, F, I/O curves for GluR1−/− single-mutant (●) and GluR1−/− NOS-1−/− double-mutant (E) and GluR1−/− NOS-3−/− double-mutant (F) (○) mice show no differences in baseline transmission (p > 0.05, NS).
Application of l-NNA did not decrease the probability of spike induction. Spike probability was 0.64 ± 0.09 in untreated GluR1 knock-outs and 0.56 ± 0.06 in l-NNA treated GluR1 knock-outs (F(1,29) = 1.16, p = 0.56). This data therefore implies that NOS is significantly involved in hippocampal LTP. This conclusion was corroborated by evidence from double knock-out mice in which LTP was reduced in both GluR1/αNOS-1 and GluR1/NOS-3 double knock-out animals (Fig. 6B,C). In both cases, application of l-NNA further reduced LTP in the double knock-outs, indicating that both isoforms of NOS (endothelial (NOS-3) and neuronal (αNOS-1) are involved in LTP in the schaeffer collateral CA1 pathway. The residual component of LTP present in the GluR1/αNOS-1 knock-outs (142 ± 8%) was significantly different from the untetanised control pathway (t(28) = 3.92, p < 0.001). Treatment of the double knock-outs with l-NNA (an unspecific NOS inhibitor) reduced but did not totally block LTP (mean = 117 ± 11%, t(9) = 1.7, p < 0.05) (Fig. 6D, summary bars).
As was the case with l-NNA application to wild-type slices, reduction of LTP in the double knock-outs was not due to an inability to produce action potentials in the theta-burst tetanus, while the I/O curves were again indistinguishable from those of the single GluR1 knock-outs (Fig. 6E,F). These results therefore imply that NOS is involved in a substantial component of LTP in the GluR1 knock-outs.
Spike-dependent LTP in wild types is partly NO dependent
As the LTP in GluR1 knock-outs requires action potentials and is also largely NO dependent, we hypothesized that the same is true of a component of wild type LTP. We therefore again used two LTP induction protocols, one that caused consistent spike production (theta-burst) and one that in our hands only sparingly produced spikes (100 Hz). Both protocols produced LTP in the wild-type mice (Fig. 7). However, application of l-NNA reduced only the LTP produced by theta-burst stimulation and not that produced by 100 Hz stimulation. The level of LTP induced by theta-burst stimulation was almost halved by application of l-NNA (from 60 + 9% to 28 + 5%) (Fig. 7B), and this was statistically significantly different (t(25) = 4.74, p < 0.001). In contrast, the level of LTP induced by 100 Hz stimulation was not significantly affected by l-NNA treatment (t(20) = 0.49, p > 0.05) (Fig. 7A). We also tested whether LTP might have been accidentally underestimated in the control 100 Hz condition by including cases of spuriously unstable LTP (supplemental Table 1, available at www.jneurosci.org as supplemental material). However, we found that even if we discounted cases of control LTP that had not reached an asymptotic value at the end of 2 h, the comparison with the l-NNA cases showed no significant difference (t(17) = 0.81, p > 0.05).
Figure 7.

An NO sensitive component to LTP is only observed in wild types if LTP induction involves action potential firing. A, Stimulation (100 Hz) at high-intensity stimulation produces LTP that is stable for >2 h (●). This LTP is unaffected by the application of 100 μm l-NNA (○). B, High-intensity theta-burst stimulation produces a significantly larger potentiation (●) than the potentiation induced by 100 Hz stimulation and is more sensitive to 100 μm l-NNA (○). l-NNA is applied for 10 min starting at t = −5 min. Each point plots the average amplitude of six successive fEPSPs normalized with respect to the baseline and expressed as mean ± SEM. Right insets are representative traces taken at time points indicated by the bars (red, control period; black, 110–120 min) with the symbols identifying the experimental conditions. Left insets are example intracellular traces recorded to illustrate the type of spiking that occurs during induction. Calibration (unless otherwise stated): 1 mV, 10 ms.
Finally, we tested to see which isoforms of NOS might be involved in wild-type LTP by looking at expression of LTP in αNOS-1 and NOS-3 knock-out mice. The level of LTP was significantly reduced in both knock-outs to 136 ± 4% (NOS-1) and 137 ± 11% (NOS-3) (p < 0.001), although as can be seen from supplemental Figure 2 (available at www.jneurosci.org as supplemental material), levels of LTP were still quite substantial in both cases. These studies show that ∼50% of LTP is NOS dependent when induced by theta-burst stimulation (which evokes action potentials during induction) and that both αNOS-1 and NOS-3 isoforms are involved in the NOS-dependent component of LTP.
Evidence regarding the presynaptic origin of NO-dependent LTP
NO signaling has been implicated in the presynaptic modulation of transmitter release in LTP (O'Dell, 1991). Since LTP in the GluR1 knock-outs is almost fully blocked by NOS inhibition, while wild-type LTP is only partly blocked, one might predict that the locus of expression of the LTP would also be almost entirely presynaptic in the GluR1 knock-outs and a mixture of presynaptic and postsynaptic in the wild types.
To test this, we monitored PPF before and 40 min after LTP induction. In the wild types, there was no overall change in PPF after LTP induction (ΔPPF = 0.02 ± 0.07, t(19) = 0.68, p = 0.5) (Fig. 8C). There was a large variability in ΔPPF between individual recordings; six cells showed an increase, six cells show no change, and eight show a decrease in ΔPPF after LTP (Fig. 8A). However, in the GluR1 knock-outs there was far lower variability in the ΔPPF, with only one cell showing a substantial increase, six remaining unchanged while 14 cells showed a decrease in the PPF ratio after LTP (Fig. 8B). Using a paired t test and comparing each cell before and after LTP we found a significant decrease in ΔPPF for the GluR1 knock-outs following LTP (ΔPPF = 0.28 ± 0.04, t (20) = 2.9, p < 0.01). Consequently, ΔPPF was significantly different in wild types and GluR1 knock-outs (t(32) = 2.7, p < 0.05) (Fig. 8C).
Figure 8.

The locus of plasticity in wild types and GluR1 knock-outs. A, Changes in PPF (75 ms interpulse interval) 40 min after LTP induction are highly variable in wild types (6 substantially increased, 6 do not change, and 8 decreased). B, Less variability in ΔPPF is observed in the GluR1 knock-outs (1 substantially increased, 6 do not change, and 14 decreased), and there is a general decrease in the PPF 40 min after LTP induction. Insets are representative traces taken during the control period (red line) and 40 min after LTP induction (black line). C, Bars represent the average ΔPPF for the wild types (open bars) and GluR1 knock-outs (black bars) (see Materials and Methods for definition of ΔPPF). PPR, Paired-pulse ratio. D, The normalized mean response versus CV−2 trajectory is more vertical for GluR1 knock-outs than wild types. The origin (1,1) represents the baseline condition, whereas the points to the right are taken at 40 min after LTP induction. Both CV−2 and mean amplitude are normalized. WT, Wild type.
The initial PPF ratio has been shown to be inversely related to the magnitude and sign of ΔPPF following LTP in the hippocampus, for instance by Schulz et al. (1994). In agreement with these data, we also saw a negative correlation between the initial PPF ratio and the ΔPPF after LTP both in wild types (r = 0.47, n = 19, p < 0.05) and GluR1 knock-outs (r = 0.55, n = 21, p < 0.01) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). In the GluR1 knock-outs there was also a negative correlation between the ΔPPF ratio and the magnitude of LTP at 45 min (r = 0.52, n = 21, p = 0.03) and a positive correlation between the control PPF ratio and the magnitude of LTP (r = 0.47, n = 21, p = 0.01) (supplemental Fig. 3B,C, available at www.jneurosci.org as supplemental material); however, neither of these correlations were apparent in the wild types. The initial PPF and ΔPPF failed to predict the magnitude of LTP in wild types, presumably due to additional postsynaptic mechanisms involving GluR1, while in the GluR1 knock-outs these postsynaptic mechanisms are not available so presynaptic mechanisms dominate. Increased dependence of LTP on presynaptic mechanisms in GluR1 knock-outs has also been reported in the barrel cortex (Hardingham and Fox, 2006)
The presynaptic locus of LTP in GluR1 knock-outs was further corroborated by normalized mean CV−2 analysis (Malinow and Tsien, 1990). Purely postsynaptic changes would produce a plot with a horizontal trajectory (Fig. 8D), whereas changes in N or Pr would cause more vertical trajectories. This is because CV−2 is proportional to NPr (1 − Pr) −1 and is therefore not dependent on Q, whereas the mean amplitude is proportional to NPrQ and is therefore proportional to Q (in which N is the number of release site, Pr is the probability of release, and Q is the quantal size). In wild types, the trajectory of the CV−2 plot was approximately diagonal, indicative of a mixed locus of potentiation [consistent with the work of Hardingham and Fox (2006)] (Fig. 8D). In GluR1 knock-outs, the trajectory of the CV–2 plot was significantly steeper than in wild types (linear fits were made to individual experiments and the average slope and error was calculated for wild types and GluR1 knock-outs, wild-type slope = 1.07 ± 0.05, GluR1 knock-out slope = 1.30 ± 0.07, t test t(34) = 2.5, p < 0.05), indicating that in GluR1 knock-outs the locus of LTP expression is more presynaptic than wild types.
The conclusion of the CV−2 analysis seems consistent with the paired-pulse analysis in that they both suggested a predominantly presynaptic component of LTP in the GluR1 knock-outs and a mixed locus of LTP expression in the wild types.
In wild types, theta-burst stimulation produces an additional component of LTP to 100 Hz stimulation
These experiments suggest that two mechanistically distinct components of LTP are generated in wild types, dependent on the induction protocol used and on whether somatic spikes are produced during the tetanus. If this is true one might predict that theta-burst stimulation, which induces both GluR1- and NO-dependent forms of LTP should occlude subsequent LTP induced by 100Hz stimulation, whereas the converse would not be true. One would predict that since 100 Hz stimulation does not in our hands, induce NO-dependent LTP, it would be possible to produce additional NO-dependent LTP with theta-burst stimulation following the 100 Hz stimulation.
We tested this hypothesis in studies where we induced LTP with a strong stimulation protocol (3× theta-burst or 3× 100 Hz) using the 40 μs stimulus pulse width. Thirty minutes after LTP the stimulus intensity was turned down to return the field EPSP to its control value and we then tried to induce LTP a second time. We found that theta-burst stimulation produced LTP that occluded further LTP induced by 100 Hz stimulation (Fig. 9A). Transient potentiation (STP) was produced by the second tetanus but it fell back to baseline within 30 min (101 ± 2%) (Fig. 9C). However, if we swapped the order of the stimulus protocols so that the 100 Hz tetanus occurred before the theta-burst tetanus, a small LTP was observed (Fig. 9B). There was no clear post-tetanic potentiation episode and the potentiation rose then remained at a steady state level of (117 ± 7%) for the 50 min we followed it (Fig. 9C). Statistical analysis showed that the theta-burst LTP was significantly different from baseline (p < 0.05). This experiment supports the hypothesis that postsynaptic action potentials (recruited using theta-burst stimulation) activate a mechanistically different and additional component of LTP to that induced by depolarization without postsynaptic action potentials (produced, in our hands, using 100 Hz stimulation). It also suggests that the LTP component produced only by theta-burst is smaller than the LTP component common to both protocols.
Figure 9.
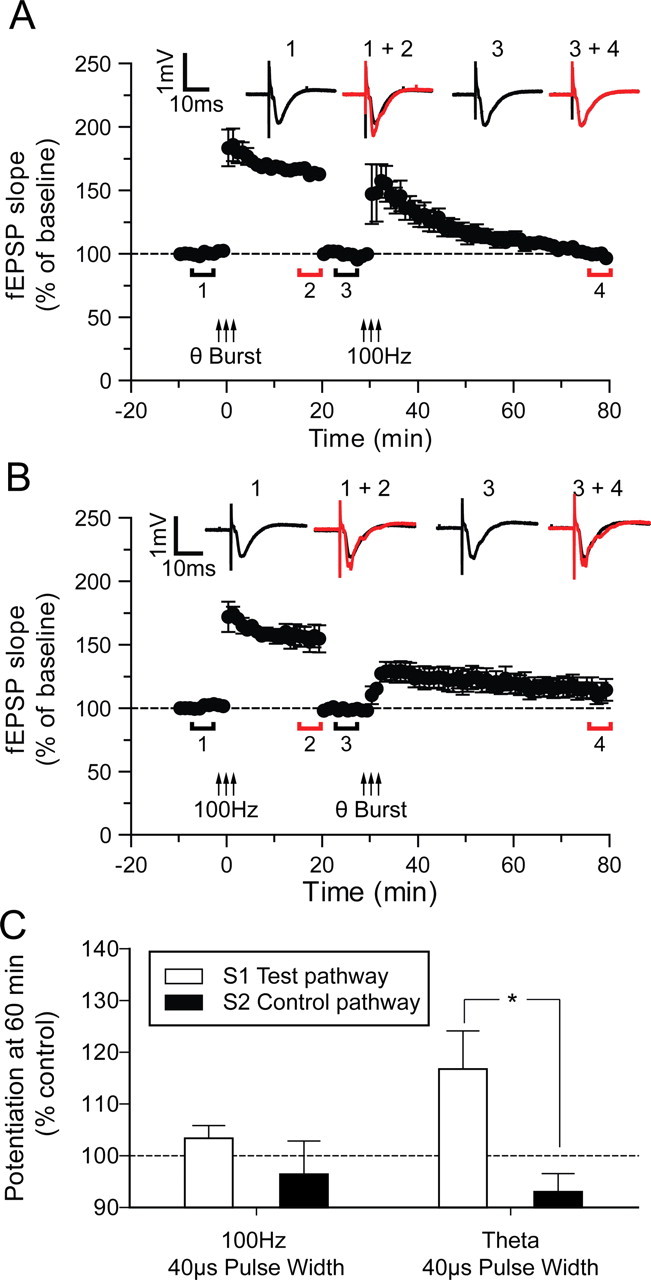
LTP induced by theta-burst stimulation occludes subsequent 100 Hz stimulation induced LTP, whereas LTP induced by 100 Hz stimulation does not completely occlude subsequent LTP induced by theta-burst stimulation. A, LTP was initially induced in the S1 pathway by high- intensity theta-burst stimulation; after 20 min of further recording, the baseline was reset to the initial control level by decreasing the stimulus intensity. A further 10 min baseline period was then recorded before a second high-intensity 100 Hz stimulation was applied to the same pathway. The potentiation induced by the 100 Hz stimulation was transient (STP) and returned back to baseline within 45 min. B, The same dual LTP protocol treatment as in A but performed in reverse: first, high-intensity 100 Hz stimulation was given to S1 followed by high-intensity theta-burst stimulation after resetting the baseline again. The potentiation induced by the high-intensity theta-burst stimulation did not return to baseline and was significantly different from the S2 pathway at 60 min after tetanus (p < 0.05). Each point plots the average amplitude of three successive fEPSPs normalized with respect to the initial baseline and is expressed as mean ± SEM. Insets are representative traces taken at time points indicated by the bars. C, Bars represent mean levels of potentiation as indicated taken 60 min after the second tetanus.
Discussion
The main findings of this study are that postsynaptic action potentials are necessary for the NO-dependent component of hippocampal LTP in both GluR1 knock-out and wild-type mice. In GluR1 knock-out mice, almost all the LTP is NO sensitive, while in wild types the later stages of LTP are NO sensitive. In wild types, NO-dependent LTP accounts for ∼50% of the potentiation 2 h post-tetanus. Presumably, the remaining component of LTP in wild types is GluR1 dependent, which would account for the large difference in the size of the NO-dependent component between the two genotypes.
Comparison with previous studies on the role of NO in LTP
Previous studies on the role of NO in LTP have investigated the source of discrepant results in different labs. One of the primary factors appears to be the differing levels of NOS present in different rat (Hölscher, 2002) and mouse strains (Blackshaw et al., 2003). The wild-type and mutant mice used in these studies are from a C57/Black 6 background in which NOS-1 is expressed in CA1 cells at higher levels than 129sv mice or rats, but at a level not dissimilar to humans (Blackshaw et al., 2003). The present study exposes two further sources of possible confusion when investigating the NO-dependent component of LTP; first, LTP can occur in the hippocampus despite inhibition of NOS and second, activation of the NO mechanism depends on postsynaptic spike production during the tetanus, which is rarely monitored in extracellular field studies (which comprise practically all studies on the role of NOS in LTP). Both factors could lead to under-estimating the role that NO plays in hippocampal LTP. To take the first of these factors; the GluR1 component of LTP would still be present even in cases where NOS activity was completely pharmacologically or genetically inactivated. This explains why many studies have found only a partial block of LTP with NOS inhibition (O'Dell et al., 1994; Son et al., 1996; Hölscher, 2002). The second factor concerns the production of postsynaptic action potentials; it is certainly our experience that increasing stimulus strength tends to inactivate sodium channels and reduce spike production during a 100 Hz tetanus. Absence of postsynaptic spikes eliminates the NO component of LTP. However, the GluR1 component of LTP does not rely on action-potentials and therefore an increased stimulus strength does not affect it in the same way. The combined effect of increasing the stimulus strength is therefore to decrease the NO-dependent component of LTP relative to the GluR1-dependent component. This explains several reports in the literature that increasing stimulus intensity reduces the NO-dependent component of LTP (Gribkoff and Lum-Ragan, 1992; Chetkovich et al., 1993; Haley et al., 1993; O'Dell et al., 1994).
In our hands we found that theta-burst stimulation produced spikes more readily than 100 Hz stimulation. However, this is not to say that it is impossible to produce NO-dependent LTP with 100 Hz stimulation. In fact, some of the pioneering studies on the role of NO in LTP found that 100 Hz stimulation produced NO-dependent LTP, provided that the stimulus used was of a weak intensity (O'Dell et al., 1991,1994). We assume that in these cases the stimulus was weaker than we used in our studies and that it more successfully produced postsynaptic spikes during the tetanus.
Comparison with previous studies on GluR1 knock-outs
We found no differences in baseline levels of synaptic efficacy between GluR1 knock-out animals and wild types, consistent with previous studies (Zamanillo et al., 1999). It has also been shown that GluR1 knock-outs have normal levels of whisker evoked responses in layers II/III, IV and V of the barrel cortex and normal levels of synaptic response in the layer IV to II/III and II/III to V pathway (Wright et al., 2008). However, synaptic scaling is known to require GluR1 containing AMPA receptors in the hippocampus, which might predict a reduction in distal synaptic currents in the GluR1 knock-outs (Andrásfalvy et al., 2003). It may be that presynaptic plasticity mechanisms are able to compensate for the lack of postsynaptic scaling.
Earlier studies on GluR1 knock-outs concluded that LTP relied on spike pairing protocols that were effective in younger but not older animals (Jensen et al., 2003) or that it required pairing a burst of postsynaptic spikes with presynaptic stimulation (Hoffman et al., 2002). Here we show that spikes are essential for induction of LTP in the GluR1 knock-outs but that it is sufficient that they are produced naturally from orthodromic stimulation. Previous studies have shown that somatic action potentials are usually generated by prior dendritic spikes during theta-burst stimulation (Golding et al., 2002). Here we found that theta-burst stimulation reliably evoked postsynaptic action potentials for approximately half the presynaptic stimuli and induced a significant number of complex spikes when a higher presynaptic stimulus strength was used. Our studies were all performed on animals older than 6 weeks of age (P45–P64) and the extracellular field experiments were on animals older than 8 weeks of age (P57–64); animals were therefore certainly not immature. The inability to see LTP in adult GluR1 knock-out animals in previous studies could be due to the stimulus protocols used, for example 100 Hz stimulation (Jensen et al., 2003), which in our hands did not produce many postsynaptic action potentials. We therefore conclude that LTP in GluR1 knock-outs can be produced by orthodox stimuli and is not restricted to immature animals.
It has been suggested that because GluR1 knock-outs show normal water maze learning, hippocampal LTP might not be necessary for spatial memory in this structure (Zamanillo et al., 1999). While the present studies do not provide any evidence for hippocampus LTP being involved in spatial memory, they do argue against rejection of this theory due to the lack of hippocampal LTP in GluR1 knock-out mice. This study shows that LTP can be induced by both conventional and physiologically relevant stimuli in GluR1 knock-outs while previous studies also show that spatial memory still occurs in GluR1 knock-outs (Zamanillo et al., 1999).
Recently, a more specific memory deficit has been identified in GluR1 knock-outs. While GluR1 knock-out mice are able to perform reference memory tasks such as the Morris water maze, where information needs to be recalled from previous trials (Zamanillo et al., 1999), they are impaired in working memory tasks that require within trial recall of recently acquired information (Sanderson et al., 2008). For example, GluR1 knock-out mice trained to retrieve food rewards from a radial arm maze made many more working memory errors (reentering arms which had previously been visited) when the food rewards were not replaced during the task, thereby requiring the animal to remember where it had just been (Schmitt et al., 2003). The working memory deficit in the GluR1 knock-outs is restored by forebrain expression of transgenic GluR1 (Schmitt et al., 2005) as is LTP in the hippocampus (Mack et al., 2001). It is conceivable that the GluR1 dependence of working memory is related to the early GluR1-dependent phase of LTP shown in this and previous studies (Hoffman et al., 2002; Hardingham and Fox, 2006). It is intriguing to think that the reference memory component that remains in the GluR1 knock-out animals might be NO-dependent, given that LTP in GluR1 knock-outs is largely NO-dependent. If so, it may rely on the slowly developing presynaptic mechanisms of plasticity recently described in the neocortex and hippocampus (Hardingham and Fox, 2006; Bayazitov et al., 2007).
A compound LTP with an early postsynaptic component and later presynaptic component has recently been described in the hippocampus (Bayazitov et al., 2007). Presynaptic function was monitored directly using a transgenic mouse strain expressing a pH sensitive fluorescent VAMP2 marker in neurons. The presynaptic component of LTP was only induced by theta-burst stimulation and not by 100 Hz stimulation (Bayazitov et al., 2007). Our study suggests that only theta-burst activity evokes presynaptic LTP in the Bayazitov study because it produces postsynaptic spikes more effectively than 100 Hz stimulation and hence enables the NO-dependent component of LTP. There is evidence that NO can play a postsynaptic role in LTP by producing direct nitrosylation of NSF and hence affecting insertion of AMPA receptors into the membrane (Huang et al., 2005). While we cannot rule out some postsynaptic action of NO in these studies, the paired pulse and CV2 analysis in the present data suggest that NO mainly acts presynaptically, in common with the conclusions of several other studies (O'Dell et al., 1991; Hawkins et al., 1998). Finally, one further study has also reported that postsynaptic spikes are necessary during theta-burst LTP induction for the persistence of LTP (Raymond, 2008) again suggesting that the slower developing, NO-dependent component of LTP induced by theta-burst stimulation requires postsynaptic spikes.
In conclusion, the recent discovery of different temporal components of memory formation is paralleled by the discovery of different temporal components of LTP. This study and previous studies suggest that the early and late components have different presynaptic and postsynaptic loci. In this study, we further show that the later component relies strongly on NO, which in turn relies on postsynaptic spike production and may provide a means for dissecting different components of hippocampus-dependent memory in the future.
Footnotes
This work was supported by the Medical Research Council (UK) and the National Institute of Mental Health (Conte Center). We thank them for supporting this work and Rob Malenka for critically reading a previous version of this manuscript.
References
- Andrásfalvy BK, Smith MA, Borchardt T, Sprengel R, Magee JC. Impaired regulation of synaptic strength in hippocampal neurons from GluR1-deficient mice. J Physiol. 2003;552:35–45. doi: 10.1113/jphysiol.2003.045575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayazitov IT, Richardson RJ, Fricke RG, Zakharenko SS. Slow presynaptic and fast postsynaptic components of compound long-term potentiation. J Neurosci. 2007;27:11510–11521. doi: 10.1523/JNEUROSCI.3077-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackshaw S, Eliasson MJ, Sawa A, Watkins CC, Krug D, Gupta A, Arai T, Ferrante RJ, Snyder SH. Species, strain and developmental variations in hippocampal neuronal and endothelial nitric oxide synthase clarify discrepancies in nitric oxide-dependent synaptic plasticity. Neuroscience. 2003;119:979–990. doi: 10.1016/s0306-4522(03)00217-3. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetkovich DM, Klann E, Sweatt JD. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. Neuroreport. 1993;4:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- Eliasson MJ, Blackshaw S, Schell MJ, Snyder SH. Neuronal nitric oxide synthase alternatively spliced forms: prominent functional localizations in the brain. Proc Natl Acad Sci U S A. 1997;94:3396–3401. doi: 10.1073/pnas.94.7.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Staff NP, Spruston N. Dendritic spikes as a mechanism for cooperative long-term potentiation. Nature. 2002;418:326–331. doi: 10.1038/nature00854. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Lum-Ragan JT. Evidence for nitric oxide synthase inhibitor-sensitive and insensitive hippocampal synaptic potentiation. J Neurophysiol. 1992;68:639–642. doi: 10.1152/jn.1992.68.2.639. [DOI] [PubMed] [Google Scholar]
- Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- Haley JE, Malen PL, Chapman PF. Nitric oxide synthase inhibitors block long-term potentiation induced by weak but not strong tetanic stimulation at physiological brain temperatures in rat hippocampal slices. Neurosci Lett. 1993;160:85–88. doi: 10.1016/0304-3940(93)90919-c. [DOI] [PubMed] [Google Scholar]
- Hardingham N, Fox K. The role of nitric oxide and GluR1 in presynaptic and postsynaptic components of neocortical potentiation. J Neurosci. 2006;26:7395–7404. doi: 10.1523/JNEUROSCI.0652-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Son H, Arancio O. Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Prog Brain Res. 1998;118:155–172. doi: 10.1016/s0079-6123(08)63206-9. [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Sprengel R, Sakmann B. Molecular dissection of hippocampal theta-burst pairing potentiation. Proc Natl Acad Sci U S A. 2002;99:7740–7745. doi: 10.1073/pnas.092157999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hölscher C. Nitric oxide, the enigmatic neuronal messenger: its role in synaptic plasticity. Trends Neurosci. 1997;20:298–303. doi: 10.1016/s0166-2236(97)01065-5. [DOI] [PubMed] [Google Scholar]
- Hölscher C. Different strains of rats show different sensitivity to block of long-term potentiation by nitric oxide synthase inhibitors. Eur J Pharmacol. 2002;457:99–106. doi: 10.1016/s0014-2999(02)02641-9. [DOI] [PubMed] [Google Scholar]
- Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- Huang Y, Man HY, Sekine-Aizawa Y, Han Y, Juluri K, Luo H, Cheah J, Lowenstein C, Huganir RL, Snyder SH. S-nitrosylation of N-ethylmaleimide sensitive factor mediates surface expression of AMPA receptors. Neuron. 2005;46:533–540. doi: 10.1016/j.neuron.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Ishida A, Shigeri Y, Tatsu Y, Uegaki K, Kameshita I, Okuno S, Kitani T, Yumoto N, Fujisawa H. Critical amino acid residues of AIP, a highly specific inhibitory peptide of calmodulin-dependent protein kinase II. FEBS Lett. 1998;427:115–118. doi: 10.1016/s0014-5793(98)00405-0. [DOI] [PubMed] [Google Scholar]
- Jensen V, Kaiser KM, Borchardt T, Adelmann G, Rozov A, Burnashev N, Brix C, Frotscher M, Andersen P, Hvalby O, Sakmann B, Seeburg PH, Sprengel R. A juvenile form of postsynaptic hippocampal long-term potentiation in mice deficient for the AMPA receptor subunit GluR-A. J Physiol. 2003;553:843–856. doi: 10.1113/jphysiol.2003.053637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor DB, Lanzrein M, Stary SJ, Sandoval GM, Smith WB, Sullivan BM, Davidson N, Schuman EM. A role for endothelial NO synthase in LTP revealed by adenovirus-mediated inhibition and rescue. Science. 1996;274:1744–1748. doi: 10.1126/science.274.5293.1744. [DOI] [PubMed] [Google Scholar]
- Lisman J, Raghavachari S. A unified model of the presynaptic and postsynaptic changes during LTP at CA1 synapses. Sci STKE. 2006;2006:re11. doi: 10.1126/stke.3562006re11. [DOI] [PubMed] [Google Scholar]
- Mack V, Burnashev N, Kaiser KM, Rozov A, Jensen V, Hvalby O, Seeburg PH, Sakmann B, Sprengel R. Conditional restoration of hippocampal synaptic potentiation in Glur-A-deficient mice. Science. 2001;292:2501–2504. doi: 10.1126/science.1059365. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Kauer JA, Perkel DJ, Mauk MD, Kelly PT, Nicoll RA, Waxham MN. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nature. 1989;340:554–557. doi: 10.1038/340554a0. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ, Hawkins RD, Kandel ER, Arancio O. Tests of the roles of two diffusible substances in long-term potentiation: evidence for nitric oxide as a possible early retrograde messenger. Proc Natl Acad Sci U S A. 1991;88:11285–11289. doi: 10.1073/pnas.88.24.11285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Huang PL, Dawson TM, Dinerman JL, Snyder SH, Kandel ER, Fishman MC. Endothelial NOS and the blockade of LTP by NOS inhibitors in mice lacking neuronal NOS. Science. 1994;265:542–546. doi: 10.1126/science.7518615. [DOI] [PubMed] [Google Scholar]
- Raymond CR. Different requirements for action potentials in the induction of different forms of long-term potentiation. J Physiol. 2008;586:1859–1865. doi: 10.1113/jphysiol.2008.151035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson DJ, Good MA, Seeburg PH, Sprengel R, Rawlins JN, Bannerman DM. The role of the GluR-A (GluR1) AMPA receptor subunit in learning and memory. Prog Brain Res. 2008;169:159–178. doi: 10.1016/S0079-6123(07)00009-X. [DOI] [PubMed] [Google Scholar]
- Schmitt WB, Deacon RM, Seeburg PH, Rawlins JN, Bannerman DM. A within-subjects, within-task demonstration of intact spatial reference memory and impaired spatial working memory in glutamate receptor-A-deficient mice. J Neurosci. 2003;23:3953–3959. doi: 10.1523/JNEUROSCI.23-09-03953.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt WB, Sprengel R, Mack V, Draft RW, Seeburg PH, Deacon RM, Rawlins JN, Bannerman DM. Restoration of spatial working memory by genetic rescue of GluR-A-deficient mice. Nat Neurosci. 2005;8:270–272. doi: 10.1038/nn1412. [DOI] [PubMed] [Google Scholar]
- Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci. 1994;14:5325–5337. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Winterer J, Zhang XL, Müller W. Imaging LTP of presynaptic release of FM1–43 from the rapidly recycling vesicle pool of Schaffer collateral-CA1 synapses in rat hippocampal slices. Eur J Neurosci. 2005;22:2451–2461. doi: 10.1111/j.1460-9568.2005.04437.x. [DOI] [PubMed] [Google Scholar]
- Wright N, Glazewski S, Hardingham N, Phillips K, Pervolaraki E, Fox K. Laminar analysis of the role played by GluR1 in experience-dependent and synaptic depression of sensory responses in barrel cortex. Nat Neurosci. 2008;11:1140–1142. doi: 10.1038/nn.2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, Kaiser KM, Köster HJ, Borchardt T, Worley P, Lübke J, Frotscher M, Kelly PH, Sommer B, Andersen P, Seeburg PH, Sakmann B. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284:1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]