

Conspectus
The aggregation of proteins is tightly controlled in living systems, and misfolded proteins are normally removed before aggregation of the misfolded protein can occur. But for reasons not clearly understood, in some individuals this degradation process breaks down, and misfolded proteins accumulate in insoluble protein aggregates (amyloid deposits) over time. Of the many proteins expressed in humans, a small but growing number have been found to form the long, highly ordered β-sheet protein fibers that comprise amyloid deposits. Despite a lack of obvious sequence similarity, the amyloid forms of diverse proteins are strikingly similar, consisting of long, highly ordered insoluble fibers with a characteristic crossed β-sheet pattern. Amyloidogenesis has been the focus of intense basic and clinical research, as a high proportion of amyloidogenic proteins has been linked to common degenerative diseases, including Alzheimer’s, type II diabetes, and Parkinson’s.
The apparent link between amyloidogenic proteins and disease was initially attributed to the amyloid form of the protein; however, increasing evidence suggests the toxicity is due to intermediates generated during the assembly of amyloid fibers. These intermediates have been proposed to attack cells in a variety of ways, such as by generating inflammation, creating reactive oxygen species, and overloading the misfolded protein response pathway. One common, well-studied mechanism is the disruption of the plasma and organelle membranes.
In this Account, we examine the early molecular-level events in the aggregation of the Islet amyloid polypeptide (IAPP, also called amylin) and its ensuing disruption of membranes. IAPP is a 37-residue peptide secreted in conjunction with insulin; it is highly amyloidogenic and often found in amyloid deposits in type II diabetics. IAPP aggregates are highly toxic to the β-cells that produce insulin, and thus IAPP is believed to be one of the factors involved in the transition from early to later stages of type II diabetes. Using variants of IAPP that are combinations of toxic or non-toxic and amyloidogenic or nonamyloidogenic, we have shown that formation of amyloid fibers is a sufficient but not necessary condition for the disruption of β-cells. Instead, the ability to induce membrane disruption in model membranes appears to be related to the peptide’s ability to stabilize curvature in the membrane, which in turn is related to the depth of penetration in the membrane.
Although many similarities exist between IAPP and other amyloidogenic proteins, one important difference appears to be the role of small oligomers in the assembly process of amyloid fibers. In many amyloidogenic proteins, small oligomers form a distinct metastable intermediate that is frequently the most toxic species; however, in IAPP, small oligomers appear to be transient and are rapidly converted to amyloid fibers. Moreover, the aggregation and toxicity of IAPP is controlled by other cofactors present in the secretory granule from which it is released, such as zinc and insulin, in a control mechanism that is somehow unbalanced in type II diabetics. Investigations into this process are likely to give clues to the mysterious origins of type II diabetes on the molecular level.
Introduction
Islet Amyloid Polypeptide (IAPP)1 is a 37 residue amino acid hormone secreted in conjunction with insulin to regulate glucose metabolism (sequence shown in Fig. 1).1 Since its discovery in 1986, IAPP has been intensely investigated for its possible role in the loss of β-cell mass in type II diabetes, an important step in the progression of the disease.1,2 The reason for this interest is the strong correlation between the formation of IAPP aggregates in β-cells and type II diabetes; IAPP aggregates are found in over 90% of diabetics but only in a minority of non-diabetic subjects.1 In addition, the loss of β-cell mass appears to be spatially correlated with IAPP protein deposition, as β-cell mass is reduced strongly in islets containing IAPP deposits while neighboring islets lacking the deposits were relatively unaffected. These results suggest IAPP aggregation is either a possible causative agent or an epiphenomenon associated with type II diabetes. The cytotoxicity of human IAPP (hIAPP) aggregates argues for a causative effect, human IAPP has a strong cytotoxic effect on β-cells while the non-aggregating rat version of IAPP (rIAPP) has little effect on β-cell survival rates even when massively over expressed. These findings have led to speculation that a cycle started by the formation of IAPP aggregates may play a central role in the transition from an earlier stage of insulin resistance to the later stages of diabetes mellitus.2 In the first stage of this cycle, insulin resistance causes a greater demand for insulin. Since the expression of insulin and IAPP are intrinsically linked, a higher production of insulin translates into higher production of IAPP. Due to IAPP’s cytotoxicity, higher production of IAPP leads to a greater rate of β-cell apoptosis. The result is a greater rate of production of IAPP in the surviving islets to meet the demand for insulin. IAPP is concentrated in these islets, translating to even greater β-cell death rates. The cycle continues until the pancreas is significantly depleted of β-cells.
Figure 1.

Amino acid sequences of human and rat islet amyloid polypeptides with nonconserved residues shown in red color. Both peptides are physiologically expressed with an amidated C-terminus and a disulfide bridge from C2 to C7.
IAPP aggregates have a characteristic structure shared by other aggregates that commonly occur in Alzheimer’s, Huntington’s, Parkinson’s and other degenerative diseases and are therefore grouped together as a class called amyloid.3 Amyloids are defined by their primarily β-sheet structure and their supramolecular organization into long fibers with the protein backbone orthogonal to the fiber axis. Besides these structural characteristics, amyloids share some common ligand binding partners, some common physical properties, and a common general mechanism of assembly despite the lack of sequence similarity among the individual proteins.1 Amyloids form through a nucleation dependent process leading to characteristic sigmoidal type kinetics; in which amyloid formation is minimal (the lag-phase) until a critical concentration of nuclei is reached, at which point amyloid growth proceeds explosively. The specific kinetics of assembly are typically complicated and involve a variety of on and off-pathway intermediates, and differ among amyloidogenic proteins. In addition, almost all amyloids bind a specific set of cellular components and most (but not all) are toxic in some stage of their formation.
The molecular mechanism behind cytotoxicity is not clear; however, several mechanisms have been proposed. The possibilities include the formation of reactive oxygen species caused by metal complexation, endoplasmic reticulum stress caused by the accumulation of misfolded proteins, an inflammatory response induced by amyloid formation, and membrane disruption among others.1 In the absence of convincing cellular data, it is difficult to ascertain which of the mechanisms is most prevalent; it is possible that all of the above mechanisms and others play a role in β-cell death. The membrane disruption hypothesis remains the most studied for IAPP and will be the main focus of this article, with a special focus on the structural changes that occur in IAPP and the membrane upon membrane binding and aggregation as researched in our laboratory.
Mechanism of membrane disruption
There are essentially two general theories of membrane disruption by hIAPP and other amyloid proteins, although the mechanisms are not exclusive and may be both operative at once.4 In the pore theory, amyloid proteins form discrete pores within the membrane, whether of a traditional ion channel type (“barrel stave”) or in localized ruptures of the membrane that resemble discrete pores in some respects. In the non-specific model, the collective action of large oligomeric species reduces the structural integrity of the membrane resulting in either membrane fragmentation or in membrane thinning and greatly increased membrane conductance. Controversy over the mechanism is not merely of theoretical importance but has great practical implications for the prevention of membrane damage by amyloidosis. Membrane damage primarily mediated by discrete ion channels can be alleviated by channel blockers designed to plug the channels formed by toxic amyloid species, while a non-specific mechanism requires a different approach.
The strongest evidence for a pore-like mechanism of membrane disruption by hIAPP comes from single channel recordings of hIAPP on planar membranes. The addition of hIAPP at low (10 mM KCl) salt concentrations causes step-like fluctuations in membrane conductance that are reflective of the opening and closing of individual channels.5–7 The channels can be reversibly blockaded by zinc,6 indicating the conductance jumps are likely related to a specific pore structure. Additionally, permeabilization by hIAPP is strongly selective for small molecules, arguing against the large-scale disruptions predicted by detergent-like effects.8
On the other hand, a detergent-like mechanism with mosaic-like opening and closing of transient defects within the membrane is supported by AFM studies which show macroscale defects in the lipid bilayer upon prolonged exposure to hIAPP.9 The time-course of membrane disruption is strongly correlated with amyloid fibril formation and can be altered by seeding amyloid formation, implying a direct link between fibril formation and membrane disruption.10 It is difficult to reconcile this observation with a pore-dominated mechanism, as the dimensions of a fiber or protofiber are too large to form pores of a traditional type.
Our first steps into investigating membrane disruption by hIAPP was to examine the oriented 31P spectra of DMPC membranes after addition of hIAPP20–29, using an aluminum oxide matrix for alignment of the membrane.11 The 20–29 segment of hIAPP has been shown to be critical for the aggregation of the peptide into amyloid fibers; the majority of the sequence differences between non-amyloidogenic variants of IAPP (such as rIAPP) and amyloidogenic versions of IAPP are found within this sequence. Like full-length hIAPP, hIAPP20–29 inserts into lipid bilayers, albeit with a lower binding affinity and without the preference for anionic lipids shown by full-length hIAPP.12
If hIAPP20–29 fragments the bilayer into vesicle-like structures, the rapidly tumbling the vesicles should give rise to a motionally averaged isotropic 31P NMR signal near 0 ppm. This isotropic 31P signal was indeed detected for a narrow concentration range of hIAPP20–29, suggesting a detergent-like mechanism of membrane-disruption is involved.11 The absence of membrane fragmentation below a threshold concentration of peptide is consistent with the requirement for a threshold concentration of membrane defects to be achieved before fragmentation and is predicted by the standard detergent-like model for membrane disruption.13 However, the disappearance of the isotropic peak at higher concentrations is unusual and suggests highly cooperative aggregation on the membrane surface can interfere with membrane disruption by hIAPP20–29.
Linkage between amyloidogenicity and membrane disruption
The isotropic 31P NMR signals were not detected for the non-amyloidogenic rIAPP20–29 sequence and disappeared when an inhibitor of amyloidogenesis, Congo Red, was added.11 This finding suggests an intimate connection between amyloidogenesis and membrane damage. This connection was noted based on the observation of amyloid fibrils located at the surface of islet cells in vivo and are correlated with membrane morphology irregularities.14 The morphological irregularities are absent when cells are incubated with non-amyloidogenic rIAPP, apparently corresponding to the non-toxic nature of this variant. In addition, inhibition of hIAPP fiber formation generally (but not always) inhibits hIAPP induced membrane damage, leading to the hypothesis that amyloidogenesis and membrane disruption are linked processes. The strongest evidence for this hypothesis is the kinetics of membrane disruption by hIAPP in mixed DOPC/DOPG membranes largely matches that of amyloid fiber formation, suggesting a link between these two processes.10
If membrane disruption is linked to amyloidogenicity, it is reasonable to assume that non-amyloidogenic rIAPP will have little affinity for membranes. However, rIAPP binds to membranes at a similar surface pressure as hIAPP12 and can disrupt model membranes at high concentrations.15 Interestingly, disruption by rIAPP is linked to the formation of non-amyloid helical aggregates, suggesting aggregation on the membrane, but not amyloid formation per se, is a prerequisite for membrane disruption.15 Furthermore, the membrane binding site of hIAPP is largely localized to the N-terminal 19 residues of the peptide, which are similar in the human and nontoxic rat versions of the peptide.12 Given the involvement of the N-terminus in membrane binding and its apparent low amyloidogenicity, hIAPP1–19 provides a reasonable test for the influence of amyloidogenicity on membrane disruption. hIAPP1–19 causes severe membrane disruption of anionic model membranes despite not forming fibers when bound to the membrane (hIAPP1–19 slowly forms fibers in the absence of the membrane).16 This finding suggests that amyloid formation may be a sufficient but not necessary condition for membrane disruption.
Since hIAPP differs from rIAPP at only one residue in the 1–19 region, it is useful to compare the abilities of the 1–19 fragments from both peptides to disrupt membranes. While hIAPP1–19 caused leakage at low concentrations, rIAPP1–19 was only able to disrupt membranes at higher peptide to lipid ratios.17 This finding was mirrored in tests measuring the calcium influx caused by IAPP into islet cells (see Fig. 2), suggesting it is not an artifact of the model vesicle system used and IAPP1–19 has a similar effect on β-cell membranes.17 Interestingly, while hIAPP1–19 efficiently disrupts anionic liposomes and cellular membranes, it does not have that effect on primarily zwitterionic membranes.
Figure 2. Membrane disruption caused by IAPP variants in islet cells.

Disruption of the cellular membrane of intact islet cells causes an influx of extracellular calcium detected by the dye Fura-2AM localized in the cell. Human IAPP1–37, hIAPP1–19, and rIAPP1–19 cause a significant amount of influx immediately after addition (red line) while rIAPP1–37 does not, largely correlating with their ability to disrupt model membranes. The decrease in the signal after 250 seconds is most likely related to the escape of Fura-2 from the cell. Adapted from reference 17.
IAPP may disrupt membranes by imposing excessive curvature on the membrane
What force drives the disruption of the membrane by IAPP? The fragmentation of the bilayer by hIAPP20–29 suggests binding of the amphipathic peptide to the membrane creates an unfavorable stress in the membrane, which is relieved by the release of the peptide into peptide-lipid micelles. Several groups have reported distortions of lipid vesicle shape when bound to hIAPP amyloid fibers.10,18 This finding has led to the theory that the growth of the fiber on the membrane generates an excess of curvature strain on the membrane, possibly due to the inability of a flat membrane surface to accommodate the twist found in β-sheet fibers.18
The ability of proteins to induce curvature can be measured by monitoring the transition temperature of DiPOPE from the liquid crystalline (Lα) to the inverted hexagonal state (HII). The inverted hexagonal state is highly curved (see Fig. 3A) and it is expected that ligands that stabilize highly curved lipid structures will lower the temperature of this phase transition. Human IAPP1–37, hIAPP1–19, and rIAPP1–19, all of which caused calcium influx in beta-cells (see Fig. 2), caused a significant decrease in the transition temperature (see Fig. 3B), while rIAPP1–37, which failed to cause such an increase, had much less of an effect on the transition temperature.19
Figure 3. Toxic variants of IAPP stabilize negative curvature.

(A) Cartoon of the inverted hexagonal phase, showing the highly curved surface around the lipid headgroup. (B) DSC chromatograms showing a significant lowering of the phase transition of DiPOPE to the inverted hexagonal phase for the toxic variants of IAPP (hIAPP1–37, hIAPP1–19, and rIAPP1–19) but not for the non-toxic rIAPP1–37, suggesting the ability to stabilize curvature of the membrane may be related to IAPP’s toxicity. Adapted from reference 19.
While this correspondence is indirect evidence that curvature strain is directly related to membrane disruption by IAPP, it is complicated somewhat in that the inverted hexagonal phase transition temperature is dependent not only on the spontaneous curvature of the membrane but also on the bilayer elasticity, as a more flexible membrane favors curvature in both directions.20 To more directly probe the role of curvature in IAPP membrane binding, we examined the binding of IAPP to bicelles at the atomic-level. Bicelles are a membrane mimetic system composed of mixtures of short (DHPC) and long (DMPC and DMPG) chain lipids. At certain ratios of short to long chain lipids they form perforated lamellar bilayers in which the curved perforations are lined with the short chain DHPC lipid molecules (see Fig. 4C). Because the short and long chain lipids localize into the regions of high and low curvature respectively, bicelles can be used to test the binding preferences of peptides for curved vs. flat regions.
Figure 4. Toxic variants of IAPP.

(A) 31P NMR spectra showing binding of toxic rIAPP1–19 to DHPC in bicelles. (B) 13C NMR spectra showing binding of non-toxic rIAPP1–37 to DMPC and DMPG. (C) Cartoons of bicelles showing rIAPP1–19 (dark blue) localized to DHPC in the curved perforation (light blue) and rIAPP1–37 (red) localized to DMPC and DMPG in the flat lamellar region (yellow). Adapted from reference 19.
For the two IAPP variants that could be successfully incorporated into the bicelle, solid-state NMR shows that the toxic rIAPP1–19 fragment strongly associates with DHPC in the highly curved perforation while non-toxic rIAPP1–37 is mostly associated with DMPC and DMPG in the flat lamellar region (fig. 4), in agreement with the DSC results.19 Preliminary results also indicate that membranes incorporating POPE, a lipid favoring negative curvature, also favor a detergent-like membrane disruption by hIAPP. Taken together these results indicate that the excess curvature caused my membrane binding of toxic IAPP variants destabilizes the membrane.
IAPP does not form a substantial population of the small oligomers typical of other amyloidogenic proteins
Substantial controversy exists about the nature of the membrane disruptive species. Amyloid proteins typically form a variety of amyloid-like and non-amyloid oligomeric intermediates along the aggregation pathway. Some of these species, such as the mature amyloid fiber, are known to have a low toxicity while others are believed to be strongly toxic. Attention has focused on a particular set of small oligomers of approximately 5 nm in size due to the apparently high toxicity displayed by these species (for most amyloid proteins) and their pore-like morphology. Small oligomers of this type are typically formed in solution after a lag period but before amyloid fibers are formed. Although oligomers of this type have been heavily studied for Aβ1–40 and α-synuclein (among other amyloidogenic proteins),21 data for oligomers of this specific type has been relatively scant for IAPP.22
Since the presence of membranes can strongly affect the pathway of aggregation, it is appropriate to ask if the membrane disruptive species form on the membrane or in solution before inserting. To address this question, we tracked the size particles generated during IAPP amyloidogenesis by PFG-NMR spectroscopy. In the PFG 1H proton spectra most of the resonances decay rapidly with increasing field gradient consistent with rapid diffusion, as would be expected for monomeric hIAPP.23 However, a broad peak near 0 ppm corresponds to a large, slowly tumbling oligomeric species with a radius around 100 nm corresponding to approximately 1 million monomers.23 Similar peaks have also been detected for other amyloidogenic proteins including Aβ1–40 and calcitonin,24 and the peak has been used as a diagnostic for oligomeric species in coexistence with the monomer. Interestingly the peak is absent for PAP248–286, an amyloidogenic protein with a long lag-time for amyloid formation.25 The experiment was repeated over 10 hours without changes in the spectra that would be reflective of aggregation.23 This result is somewhat surprising in the context of nucleation dependent aggregation theory, which predicts that once a nucleus is formed aggregation should occur rapidly. The absence of a substantial degree of aggregation in these samples argues that the large aggregates detected by PFG are not likely to be nuclei for further aggregation.
Although the PFG experiment is not sensitive enough in that size range to determine the exact size of the large oligomers, a lower limit of approximately 50 nm can be definitively established.23 Surprisingly, oligomers of less than 50 nm in radius could not be detected, suggesting they do not accumulate to a significant amount during aggregation.23 These findings are consistent with a study that showed by sedimentation velocity experiments under a different set of conditions that oligomers composed of < 100 monomers form < 1% of the total population of hIAPP before fibrillation,26 in marked contrast to Aβ, α-synuclein, and others in which small oligomers play a crucial role in aggregation.21 The aggregation of IAPP in solution appears to be largely an all or nothing process, once small oligomers are formed they appear to be quickly converted into large protofibrillar aggregates and therefore a significant population of small oligomers is never accumulated (see Fig. 5). It is a reminder that, in spite of their many similarities, substantial differences exist among amyloidogenic proteins.
Figure 5. Hypothetical mechanism for membrane disruption by IAPP.
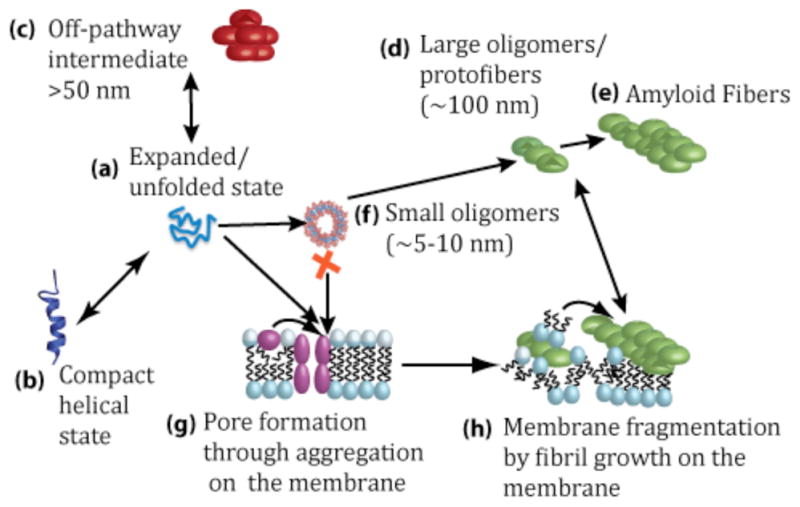
In solution, IAPP initially exists in equilibrium between expanded (a) and compact (b) monomer states as well as disordered large aggregates (c). In solution, IAPP aggregates rapidly to form amyloid fibers (d) and (e), likely through a helical intermediate (b). Small oligomers (f) are rapidly converted to large oligomers (d). Alternatively, the monomeric IAPP can bind to the membrane and aggregate to form pores (g). Continued aggregation results in amyloid formation and membrane fragmentation (h). Drawing is not to scale. Adapted from reference 23.
Structures of membrane-bound IAPP
Aggregation of IAPP on the membrane proceeds through a different pathway than in solution. Both hIAPP and rIAPP initially bind to the membrane in a largely helical state before hIAPP aggregates on the surface of the membrane to form amyloid. To investigate the initial steps of this process, we along with others have solved the structures of IAPP variants of varying degrees of toxicity and amyloidogenicity in detergent micelles, where the low number of peptide molecules per micelle keeps the peptide in a monomeric conformation. The structures of hIAPP1–19 and rIAPP1–19 are very similar in DPC micelles (see Fig. 6),27 suggesting a structural difference is not responsible for the difference in the behavior of the two peptides. However, while hIAPP1–19 and rIAPP1–19 adopt similar α-helical folds, they differ in their orientation of binding to the membrane. Rat IAPP1–19 binds near the surface of the membrane while human IAPP1–19 penetrates deeper into the hydrophobic core.17,27 This change is entirely dependent on the charge of H18 as hIAPP1–19 binds near the surface similar to rIAPP1–19 at acidic pH.17,27 This change in orientation is correlated with a change in the potential for membrane disruption, as hIAPP1–19 at pH 6 causes a low level of membrane disruption almost identical to rIAPP1–19.17
Figure 6.

NMR structures of hIAPP1–19 (A), hIAPP (B), rIAPP (C) and rIAPP1–19 (D). The hydrophobic residues that have been implicated in coiled-coil interactions stabilize the IAPP oligomer are colored in blue. Adapted from reference 29.
The similarities between the membrane-bound structures of hIAPP1–19 and rIAPP1–19 are preserved in full-length hIAPP and rIAPP, as the N-terminus adopts a similar α-helical fold in both structures in detergent micelles (see Fig. 6).28–30 Instead, the structural differences between hIAPP and rIAPP largely lie at the C-terminal end of the peptide. In rIAPP, the C-terminus is almost completely disordered.29 Human IAPP, on the other hand, adopts a helix-kink-helix structure in micelles in which the C-terminus is largely helical. The degree of helicity in the C-terminus is higher in the amidated form of hIAPP compared to the free acid form of hIAPP, correlating with the greater amyloidogenicity of the former.28 Another correspondence between structure and membrane activity is the degree of exposure of the hydrophobic face of the N-terminal helix, which is largely occluded by the N-terminal disulfide ring (Cys2-Cys7) in non-toxic variants of IAPP but is exposed in toxic ones.29
Structures of membrane-bound IAPP share many similarities with the putative helical intermediates of hIAPP in solution
It is useful to compare the membrane bound structures of IAPP to the structural propensities of the peptide in solution. In solution, both hIAPP and rIAPP are predominantly unstructured. However, the absence of a significant degree of secondary structure does not mean that the peptide is completely unfolded, as a variety of techniques have shown that IAPP adopts a more compact structure than would be predicted for a random polymer, with hIAPP being substantially more compact than rIAPP.23,31,32
The compaction of the peptide and possible secondary structure formation is important as a partially folded conformation is intrinsically more amyloidogenic than a completely folded or unfolded conformation. In a partially folded conformation, aggregation prone sequences can be favorably positioned to interact with each other, which is difficult to achieve in completely unfolded and folded states.33 In particular, there is evidence for a mechanistic role for helical intermediates in amyloid aggregation.3 Helical intermediates have not been directly observed as major species along the aggregation pathway for hIAPP, as they have been for the Alzheimer’s amyloid Aβ1–40,34 possibly because the highly transitory nature of the species prevents a stable population from being formed.
Solution NMR suggests the monomeric states of both hIAPP and rIAPP transiently sample helical states.35,36 Unlike CD, solution NMR directly reports on the monomeric state as the long rotational correlation times of large oligomers broadens their signal beyond detection. The transient nature of the helical state is reflected in chemical shifts but an absence of the medium range (i,i+3) NOEs indicates a lack of stable secondary structure.35,36 The helical state of hIAPP is apparently a low-lying excited state conformer, with the helical state lower in energy in hIAPP than rIAPP. The importance of a low percentage of helical conformers of IAPP is reflected by the influence of the helix-inducing solvent HFIP on aggregation. A low percentage of HFIP (1%) has a strong catalytic effect on aggregation while higher percentages trap the protein in a helical monomeric state.
Some specific details of the membrane-associated IAPP structures appear to be preserved in the helical state as well. A comparison of chemical shifts indicates the helical propensity is greatest in the N-terminus (residues 8–18) and decreases near the C-terminus for both hIAPP and rIAPP.35,36 Similar to what is observed in membranes, this difference between the two termini is much more pronounced in rIAPP than in hIAPP. The relative orientations of the disulfide ring may be preserved in solution as well. In hIAPP, the disulfide ring of monomeric hIAPP is positioned towards the helical face in models of both the solution and membrane structures.36 It is interesting to note that corresponding NOEs were not found in the NOESY spectra of rIAPP, suggesting the disulfide ring in the rIAPP structure is not as constrained in both solution and in the membrane as it is in hIAPP.35,36
Influence of Secretory Granule Components on Human IAPP Aggregation and Toxicity Pathways
The strong amyloidogenic propensity and membrane disruptive characteristics of hIAPP suggest that there must be factors for attenuating these effects in vivo. IAPP is stored at millimolar concentrations and would be expected to aggregate in minutes under these conditions yet the lifetime of the secretory granule in which it is stored is on the order of hours.37 Several factors can explain this discrepancy including pH (IAPP amyloid formation is strongly disfavored at low pH) and other components of the secretory granule.38 In particular, IAPP is stored with high (millimolar) concentrations of zinc and insulin, both of which impact amyloidogenesis as explained below.
Insulin and IAPP are intimately linked biologically. IAPP works in tandem with insulin to regulate glucose levels at the molecular-level and for this reason its production is tightly coordinated with insulin. Insulin and IAPP are both initially produced as immature hormones in the secretory granule that are cleaved by the same enzyme to produce the active form and are released nearly simultaneously from the secretory granule. Insulin has a strong inhibitory effect on the fibrillization of hIAPP at substoichiometric levels by preventing fiber elongation by blocking the reactive ends of the fiber.39
The affect of insulin on hIAPP disruption of cellular membranes has yet to be determined; however, model membranes provide evidence that insulin has a complicated effect on membrane-mediated aggregation and membrane disruption by hIAPP. While insulin is a strong inhibitor of IAPP fiber formation in the absence of membranes,39 its effect on membrane mediated aggregation is greatly attenuated.40 Insulin’s effect on membrane disruption is similarly complex. Membrane disruption by hIAPP in the absence of insulin is two-phased, with a first phase occurring in the lag-time of fiber formation and a second phase strongly correlated with fibrillization.10,41 Insulin completely blocks the second fiber-dependent phase of membrane disruption but has essentially no effect on the first.41 In addition, by blocking formation of the inactive fiber form insulin maintains hIAPP in the membrane disruptive form for a longer period of time.41 Insulin’s ability to block the time-dependent decrease in hIAPP permeabilization activity and only the secondary membrane disruption phase indicates the interaction is relatively complex and makes it difficult to extrapolate the results from model systems to in vivo situations.
Zinc is another factor found in millimolar concentrations in the secretory granule and has been shown to decrease the aggregation propensity of human IAPP in vitro.38 Zinc binds unaggregated hIAPP at micromolar concentrations at a 1:6 ratio despite lacking a traditional zinc-binding motif and having only 1 residue, H18, that should bind zinc tightly.42 By contrast, the amyloid form of hIAPP has low affinity for zinc a difference that should disfavor amyloid formation in the presence of zinc.42 This difference is reflected in strongly decreased elongation rates that favor the formation of smaller oligomers in the presence of zinc. Physiologically, zinc may chaperone hIAPP in the dangerous phase following secretion where hIAPP local concentrations are high and pH values are neutral, as zinc is released slowly in comparison to IAPP from the secretory granule due to the slow dissolution of zinc binding insulin hexamers (see Fig. 7).38
Figure 7. A cartoon schematic of how zinc may influence IAPP aggregation and toxicity.
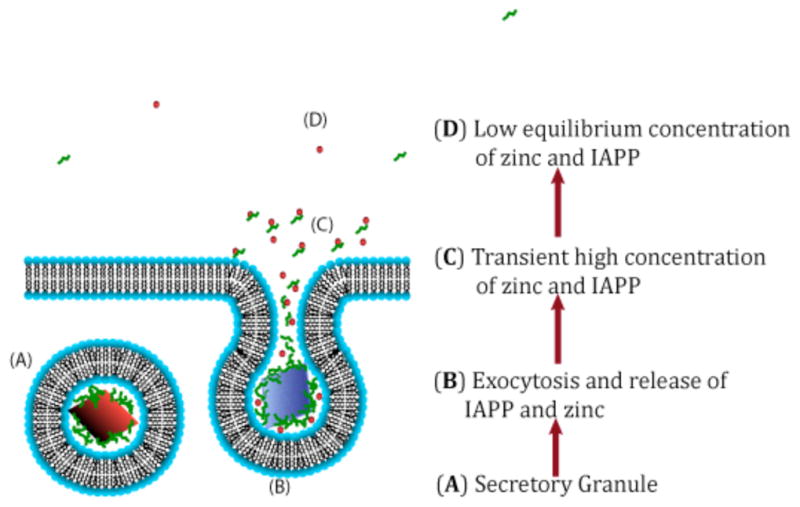
Adapted from reference 38.
Integrating biophysics and cellular biochemistry: the future of IAPP and amyloid research
We have shown that by isolating specific steps in the aggregation process a reductionist approach can yield useful information on the extremely complicated behavior of IAPP at the molecular level. However, the reductionist approach used here can run into complications when confronted with the complexity of the cell’s biochemistry. As an example of the complications that can arise, Fig. 2 shows the influx of calcium caused by the exogenous addition of IAPP1–19 to islet cells,17 suggesting IAPP1–19 is disrupting the plasma membrane in agreement with results obtained in anionic model membranes.16,17 However, hIAPP1–19 does not cause membrane disruption in mixed model membranes with a lower anionic content intended to more accurately mimic cell surfaces.43 This discrepancy suggests either: a.) IAPP1–19 does not directly induce membrane disruption in β-cells but instead activates an unidentified endogenous calcium channel b.) IAPP1–19 does induce membrane disruption in cells and anionic membranes but does not in simple mixed zwitterionic/anionic model membrane systems. Some evidence for the latter has emerged with studies showing the importance of lipid rafts and gangliosides for the membrane interaction of IAPP.44,45 Further work on IAPP in cells and more realistic model systems, in particular using advanced tools like in cell NMR and advanced imaging techniques,46,47 is likely to yield the answer to this and other questions. The integration of biophysical and in vivo techniques remains an outstanding frontier in IAPP research, and will likely be a fruitful area of research in the future.
Acknowledgments
This work was supported by funds from the National Institutes of Health. We thank the members of the Ramamoorthy group for their contributions to various research projects on amyloids.
Lipid abbreviations
- DMPC
1,2-dimyristoyl-sn-glycero-3-phosphocholine
- DHPC
1,2-dihexanoyl-sn-glycero-3-phosphocholine
- DMPG
1,2-dimyristoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- DPC
n-dodecylphosphocholine
- DiDOPE
1,2-dioleoyl-sn-glycero-3-phosphoethanolamine
- PFG
pulsed field gradient
- HFIP
hexafluoroisopropanol
- DSC
differential scanning calorimetry
Biographies
Jeffrey R. Brender is a postdoctoral fellow working under the guidance of Prof. Ramamoorthy on the interactions of proteins and drugs with membranes, with a special emphasis on the effect of amyloid proteins on biological membranes. He received his PhD from the University of Michigan, working on single molecule enzymology in the labs of Duncan Steel and Ari Gafni.
Samer Salamekh is an undergraduate student in the laboratory of Prof. Ramamoorthy whose research focuses on the interaction of IAPP with zinc and its implication type II diabetes.
Ayyulasamy Ramamoorthy has been on the faculty of Biophysics and Chemistry at the University of Michigan since 1996. His current research interests are on the development and application of NMR spectroscopy to study the structure, dynamics and function of membrane protein complexes, amyloid proteins, antimicrobial peptides, and bone. Details about his current research can be found at http://www.umich.edu/~ramslab.
References
- 1.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev. 2011;91:795–826. doi: 10.1152/physrev.00042.2009. [DOI] [PubMed] [Google Scholar]
- 2.Hoppener JWM, Ahren B, Lips CJM. Islet amyloid and type 2 diabetes mellitus. N Engl J Med. 2000;343:411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- 3.Harrison RS, Sharpe PC, Singh Y, Fairlie DP. Amyloid peptides and proteins in review. Rev Physiol Biochem P. 2007;159:1–77. doi: 10.1007/112_2007_0701. [DOI] [PubMed] [Google Scholar]
- 4.Capone R, Quiroz FG, Prangkio P, Saluja I, Sauer AM, Bautista MR, Turner RS, Mayer M. Amyloid-beta ion channels in artificial lipid bilayers and neuronal cells. Neurotox Res. 2008;15:608–650. doi: 10.1007/s12640-009-9033-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirzabekov TA, Lin MC, Kagan BL. Pore formation by the cytotoxic islet amyloid peptide amylin. J Biol Chem. 1996;271:1988–1992. doi: 10.1074/jbc.271.4.1988. [DOI] [PubMed] [Google Scholar]
- 6.Hirakura Y, Yiu WW, Yamamoto A, Kagan BL. Amyloid peptide channels: blockade by zinc and inhibition by Congo red (amyloid channel block) Amyloid. 2000;7:194–199. doi: 10.3109/13506120009146834. [DOI] [PubMed] [Google Scholar]
- 7.Quist A, Doudevski L, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc Natl Acad Sci US A. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anguiano M, Nowak RJ, Lansbury PT. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 2002;41:11338–11343. doi: 10.1021/bi020314u. [DOI] [PubMed] [Google Scholar]
- 9.Green JD, Kreplak L, Goldsbury C, Blatter XL, Stolz M, Cooper GS, Seelig A, Kist-Ler J, Aebi U. Atomic force microscopy reveals defects within mica supported lipid bilayers induced by the amyloidogenic human amylin peptide. J Mol Biol. 2004;342:877–887. doi: 10.1016/j.jmb.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 10.Engel MF, Khemtemourian L, Kleijer CC, Meeldijk HJ, Jacobs J, Verkleij AJ, de Kruijff B, Killian JA, Hoppener JW. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc Natl Acad Sci US A. 2008;105:6033–8. doi: 10.1073/pnas.0708354105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brender JR, Durr UHN, Heyl D, Budarapu MB, Ramamoorthy A. Membrane fragmentation by an amyloidogenic fragment of human Islet Amyloid Polypeptide detected by solid-state NMR spectroscopy of membrane nanotubes. Biochim Biophys Acta. 2007;1768:2026–2029. doi: 10.1016/j.bbamem.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engel MFM, Yigittop H, Elgersma RC, Rijkers DTS, Liskamp RMJ, de Kruijff B, Hoppener JWM, Killian JA. Islet amyloid polypelptide inserts into phospholipid monolayers as monomer. J Mol Biol. 2006;356:783–789. doi: 10.1016/j.jmb.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 13.Bechinger B, Lohner K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim Biophys Acta. 2006;1758:1529–1539. doi: 10.1016/j.bbamem.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 15.Knight JD, Hebda JA, Miranker AD. Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry. 2006;45:9496–9508. doi: 10.1021/bi060579z. [DOI] [PubMed] [Google Scholar]
- 16.Brender JR, Lee EL, Cavitt MA, Gafni A, Steel DG, Ramamoorthy A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J Am Chem Soc. 2008;130:6424–6429. doi: 10.1021/ja710484d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brender JR, Hartman K, Reid KR, Kennedy RT, Ramamoorthy A. A single mutation in the nonamyloidogenic region of islet amyloid polypeptide greatly reduces toxicity. Biochemistry. 2008;47:12680–12688. doi: 10.1021/bi801427c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knight JD, Miranker AD. Phospholipid catalysis of diabetic amyloid assembly. J Mol Biol. 2004;341:1175–1187. doi: 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- 19.Smith PES, Brender JR, Ramamoorthy A. Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: The case of islet amyloid polypeptide. J Am Chem Soc. 2009;131:4470–4478. doi: 10.1021/ja809002a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundbaek JA. Regulation of membrane protein function by lipid bilayer elasticity - a single molecule technology to measure the bilayer properties experienced by an embedded protein. J Phys -Condes Matter. 2006;18:S1305–S1344. doi: 10.1088/0953-8984/18/28/S13. [DOI] [PubMed] [Google Scholar]
- 21.Ferreira ST, Vieira MNN, De Felice FG. Soluble protein oligomers as emerging toxins in Alzheimer’s and other amyloid diseases. IUBMB Life. 2007;59:332–345. doi: 10.1080/15216540701283882. [DOI] [PubMed] [Google Scholar]
- 22.Zraika S, Hull RL, Verchere CB, Clark A, Potter KJ, Fraser PE, Raleigh DP, Kahn SE. Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence? Diabetologia. 2010;53:1046–1056. doi: 10.1007/s00125-010-1671-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soong R, Brender JR, Macdonald PM, Ramamoorthy A. Association of highly compact type II diabetes related islet amyloid polypeptide intermediate species at physiological temperature revealed by diffusion NMR spectroscopy. J Am Chem Soc. 2009;131:7079–7085. doi: 10.1021/ja900285z. [DOI] [PubMed] [Google Scholar]
- 24.Narayanan S, Reif B. Characterization of chemical exchange between soluble and aggregated states of beta-amyloid by solution-state NMR upon variation of salt conditions. Biochemistry. 2005;44:1444–52. doi: 10.1021/bi048264b. [DOI] [PubMed] [Google Scholar]
- 25.Brender JR, Nanga RPR, Popovych N, Soong R, Macdonald PM, Ramamoorthy A. The amyloidogenic SEVI precursor, PAP248-286, is highly unfolded in solution despite an underlying helical tendency. Biochim Biophys Acta. 2011;1808:1161–1169. doi: 10.1016/j.bbamem.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vaiana SM, Ghirlando R, Yau WM, Eaton WA, Hofrichter J. Sedimentation studies on human amylin fail to detect low-molecular-weight oligomers. Biophys J. 2008;94:L45–7. doi: 10.1529/biophysj.107.125146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nanga RPR, Brender JR, Xu JD, Veglia G, Ramamoorthy A. Structures of rat and human islet amyloid polypeptide IAPP(1–19) in micelles by NMR spectroscopy. Biochemistry. 2008;47:12689–12697. doi: 10.1021/bi8014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nanga RPR, Brender JR, Vivekanandan S, Ramamoorthy A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim Biophys Acta. 2011;1808:2337–2342. doi: 10.1016/j.bbamem.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nanga RPR, Brender JR, Xu JD, Hartman K, Subramanian V, Ramamoorthy A. Three-dimensional structure and orientation of rat islet amyloid polypeptide protein in a membrane environment by solution NMR spectroscopy. J Am Chem Soc. 2009;131:8252–8261. doi: 10.1021/ja9010095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patil SM, Xu SH, Sheftic SR, Alexandrescu AT. Dynamic alpha-helix structure of micelle-bound human amylin. J Biol Chem. 2009;284:11982–11991. doi: 10.1074/jbc.M809085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Padrick SB, Miranker AD. Islet amyloid polypeptide: Identification of long-range contacts and local order on the fibrillogenesis pathway. J Mol Biol. 2001;308:783–794. doi: 10.1006/jmbi.2001.4608. [DOI] [PubMed] [Google Scholar]
- 32.Vaiana SM, Best RB, Yau WM, Eaton WA, Hofrichter J. Evidence for a Partially Structured State of the Amylin Monomer. Biophys J. 2009;97:2948–2957. doi: 10.1016/j.bpj.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall D, Hirota N, Dobson CM. A toy model for predicting the rate of amyloid formation from unfolded protein. J Mol Biol. 2005;351:195–205. doi: 10.1016/j.jmb.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 34.Kirkitadze MD, Condron MM, Teplow DB. Identification and characterization of key kinetic intermediates in amyloid beta-protein fibrillogenesis. J Mol Biol. 2001;312:1103–1119. doi: 10.1006/jmbi.2001.4970. [DOI] [PubMed] [Google Scholar]
- 35.Williamson JA, Miranker AD. Direct detection of transient alpha-helical states in islet amyloid polypeptide. Protein Sci. 2007;16:110–117. doi: 10.1110/ps.062486907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yonemoto IT, Kroon GJ, Dyson HJ, Balch WE, Kelly JW. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry. 2008;47:9900–9910. doi: 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hutton JC. The insulin secretory granule. Diabetologia. 1989;32:271–281. doi: 10.1007/BF00265542. [DOI] [PubMed] [Google Scholar]
- 38.Brender JR, Hartman K, Nanga RP, Popovych N, de la Salud Bea R, Vivekanandan S, Marsh EN, Ramamoorthy A. Role of zinc in human islet amyloid polypeptide aggregation. J Am Chem Soc. 2010;132:8973–83. doi: 10.1021/ja1007867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Larson JL, Miranker AD. The mechanism of insulin action on islet amyloid polypeptide fiber formation. J Mol Biol. 2004;335:221–231. doi: 10.1016/j.jmb.2003.10.045. [DOI] [PubMed] [Google Scholar]
- 40.Knight JD, Williamson JA, Miranker AD. Interaction of membrane-bound islet amyloid polypeptide with soluble and crystalline insulin. Protein Sci. 2008;17:1850–1856. doi: 10.1110/ps.036350.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brender JR, Lee EL, Hartman K, Wong PT, Ramamoorthy A, Steel DG, Gafni A. Biphasic effects of insulin on islet amyloid polypeptide membrane disruption. Biophys J. 2011;100:685–692. doi: 10.1016/j.bpj.2010.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salamekh S, Brender JR, Hyung SJ, Nanga RPR, Vivekanandan S, Ruotolo BT, Ramamoorthy A. A two-site mechanism for the inhibition of IAPP amyloidogenesis by zinc. J Mol Biol. 2011;410:294–306. doi: 10.1016/j.jmb.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khemtemourian L, Engel MFM, Liskamp RMJ, Hoppener JWM, Killian JA. The N-terminal fragment of human islet amyloid polypeptide is non-fibrillogenic in the presence of membranes and does not cause leakage of bilayers of physiologically relevant lipid composition. Biochim Biophys Acta. 2010;1798:1805–1811. doi: 10.1016/j.bbamem.2010.05.022. [DOI] [PubMed] [Google Scholar]
- 44.Wakabayashi M, Matsuzaki K. Ganglioside-induced amyloid formation by human islet amyloid polypeptide in lipid rafts. FEBS Lett. 2009;583:2854–2858. doi: 10.1016/j.febslet.2009.07.044. [DOI] [PubMed] [Google Scholar]
- 45.Weise K, Radovan D, Gohlke A, Opitz N, Winter R. Interaction of hIAPP with Model Raft Membranes and Pancreatic beta-Cells: Cytotoxicity of hIAPP Oligomers. Chem Bio Chem. 2010;11:1280–1290. doi: 10.1002/cbic.201000039. [DOI] [PubMed] [Google Scholar]
- 46.Roberti MJ, Bertoncini CW, Klement R, Jares-Erijman EA, Jovin TM. Fluorescence imaging of amyloid formation in living cells by a functional, tetracysteine-tagged alpha-synuclein. Nat Methods. 2007;4:345–51. doi: 10.1038/nmeth1026. [DOI] [PubMed] [Google Scholar]
- 47.Pielak GJ, McNulty BC, Young GB. Macromolecular crowding in the Escherichia coli periplasm maintains alpha-synuclein disorder. J Mol Biol. 2006;355:893–897. doi: 10.1016/j.jmb.2005.11.033. [DOI] [PubMed] [Google Scholar]