

Abstract
Aims/hypothesis
The carcino-embryonic antigen-related cell adhesion molecule (CEACAM)2 is produced in many feeding control centres in the brain, but not in peripheral insulin-targeted tissues. Global Ceacam2 null mutation causes insulin resistance and obesity resulting from hyperphagia and hypometabolism in female Ceacam2 homozygous null mutant mice (Cc2 [also known as Ceacam2]−/−) mice. Because male mice are not obese, the current study examined their metabolic phenotype.
Methods
The phenotype of male Cc2−/− mice was characterised by body fat composition, indirect calorimetry, hyperinsulinaemic–euglycaemic clamp analysis and direct recording of sympathetic nerve activity.
Results
Despite hyperphagia, total fat mass was reduced, owing to the hypermetabolic state in male Cc2−/− mice. In contrast to females, male mice also exhibited insulin sensitivity with elevated β-oxidation in skeletal muscle, which is likely to offset the effects of increased food intake. Males and females had increased brown adipogenesis. However, only males had increased activation of sympathetic tone regulation of adipose tissue and increased spontaneous activity. The mechanisms underlying sexual dimorphism in energy balance with the loss of Ceacam2 remain unknown.
Conclusions/interpretation
These studies identified a novel role for CEACAM2 in the regulation of metabolic rate and insulin sensitivity via effects on brown adipogenesis, sympathetic nervous outflow to brown adipose tissue, spontaneous activity and energy expenditure in skeletal muscle.
Keywords: Brown adipogenesis, CEACAM2, Energy balance, Energy dissipation, Hypermetabolism, Hyperphagia, Insulin resistance, Insulin sensitivity, Sexual dimorphism, Sympathetic nervous activation
Introduction
The carcino-embryonic antigen-related cell adhesion molecule (CEACAM)2 is highly homologous to the well characterised CEACAM1, a plasma membrane glycoprotein with ubiquitous production and multiple functions. These include cell adhesion [1], hepatic insulin clearance [2-4], angiogenesis [5, 6], tumour suppression [7-10] and anti-inflammation [11].
Except for cell adhesion, which requires an intact extracellular domain [1], all other functions ascribed to CEACAM1 are regulated by phosphorylation events on its cytoplasmic tail [11, 12]. Because CEACAM1 and CEACAM2 share 70 of 73 amino acids in the cytoplasmic tail, it is likely that these two proteins exert overlapping functions. However, in light of their different tissue-specific distribution, it is also reasonable to speculate that each performs unique functions.
Unlike CEACAM1, CEACAM2 production is limited to brain, kidney, uterus, spleen and small intestine [13-17]. Moreover, its function has not been as well elucidated. In contrast to CEACAM1, CEACAM2 does not act as a cell adhesion molecule, a fact correlated to divergent amino acid sequences on the extracellular tails of these proteins [16]. Using Ceacam2 homozygous null mutant (Cc2 [also known as Ceacam2]−/−) mice, with global null deletion of Ceacam2, we recently identified a role for CEACAM2 in the regulation of energy balance, consistent with its production in brain centres involved in food intake behaviour [17]. Female, but not male Cc2−/− mice display obesity [17]. This results from hyperphagia and reduced energy expenditure. Hyperphagia causes peripheral insulin resistance and defective glucose disposal in skeletal muscle, as determined by pair-feeding analysis. Given that CEACAM2 is produced in the ventromedial nucleus of the hypothalamus (VMH) among other brain nuclei involved in the regulation of food intake, but is not produced in skeletal muscle, which is the main site of energy expenditure, the altered energy balance and glucose disposal in female Cc2−/− mice are attributed to deranged central control [18-22].
The present studies further investigated the mechanism underlining the lean phenotype of male Cc2−/− mice. They reveal that in contrast to that in females, hyperphagia in male mice does not cause insulin resistance or obesity, owing to increased energy expenditure that could be, at least in part, mediated by increased brown adipogenesis and activated sympathetic nervous outflow to adipose tissues.
Methods
Generation of Cc2−/− mice
The generation of the mice has been briefly described [17]. A 345 bp XbaI-SacI fragment containing exon 1 and flanking regions in the promoter and the proximal region of intron 1 was replaced with sequences derived from plasmid pGK Neor (electronic supplementary material [ESM] Fig. 1a). The ApaI-linearised targeting vector was electroporated into J-1 embryonic stem cells (ES-129 Sv; Genome Systems, St Louis, MO, USA). We screened 200 embryonic stem cell clones by Southern blot analysis. As expected, digestion of genomic DNA with XbaI/NsiI produced a 6.7- and 8.2-kb fragment from the wild-type and recombinant (knockout) allele, respectively (ESM Fig. 1b). Embryonic stem cells positive for homologous recombination events were injected into mouse blastocysts (C57BL/6J). Three resultant chimeric mice that gave rise to germ line transmission were bred with C57BL/6 mice, and Cc2+/− heterozygous mice were identified by PCR, using gDNA as template and (as indicated in ESM Fig. 1a) an antisense primer from exon 2 (α-123) with the (S-107) sense primer from exon 1 to detect the wild-type sequence, or the (S-Neor) sense primer from the Neor cassette to detect the knockout sequence (not shown). F1 heterozygous mice were bred to homozygosity by brother–sister mating to produce wild-type littermate (Cc2+/+) and Cc2−/− homozygous mice, identified by the 950 bp and the 1.9 kb PCR products, respectively, using the primer sets described above (ESM Fig. 1c). Interestingly, the Ceacam1 allele was intact in genomic isolates from Cc2+/+ and Cc2−/− mice (ESM Fig. 1c).
Fig. 1.
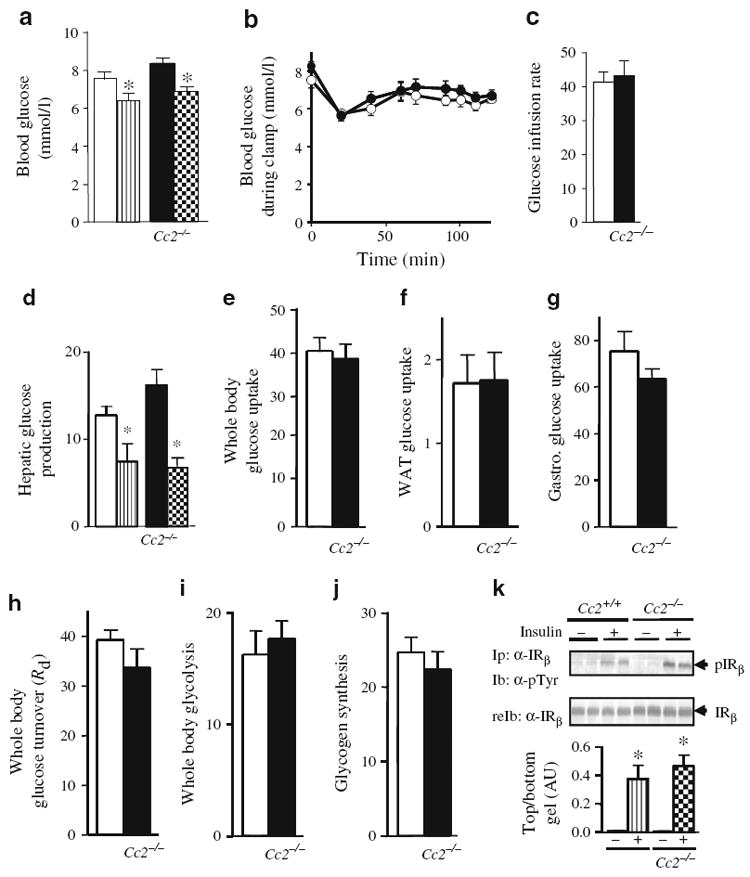
Peripheral insulin action. Overnight-fasted, awake wild-type Cc2+/+ and Cc2−/− mice (5 months old; n≥8 per group) were subjected to a 2 h hyperinsulinaemic–euglycaemic clamp. a Whole blood glucose at basal (white Cc2+/+, black Cc2−/− all panels) and clamp (striped Cc2+/+, checked Cc2−/− all panels) states, and b at time 0 and up to 120 min of clamp (white, Cc2+/+; black Cc2−/−). c Steady-state glucose infusion rate during clamp. d Hepatic glucose production as above (a). e–g Glucose uptake as indicated; WAT, white adipose tissue and Gastro., gastrocnemius muscle. h Whole-body glucose turnover (Rd). i Glycolysis during clamp. j Whole-body glycogen synthesis. Values are all expressed as mean±SEM in milligrams per kilogram per minute, except for blood glucose levels (mmol/l). *p<0.05 vs basal. k Ex vivo analysis of insulin signalling. Liver was excised from 5 months old male mice, lysed, and treated with insulin (+) or buffer alone (−) prior to immunoprecipitating (Ip) with an antibody against α-IRβ and immunoblotting (Ib) with anti-phosphotyrosine (α-pTyr) antibody to assess the amount of tyrosine phosphorylated insulin receptor. Gels were reprobed (reIb) with α-IRβ to normalise to the amount of insulin receptor loaded. The gel is representative of three experiments, performed on one pair of mice from each group in each experiment. The band density was scanned and analysed relative to loaded protein, and is quantified in arbitrary units (AU); *p<0.05 vs untreated
Ceacam2 gene deletion was confirmed by RT-PCR, followed by Southern blotting, using mRNA from the liver of wild-type, Cc2+/− and Cc2−/− mice (ESM Fig. 1d). A 50% and 100% loss of Ceacam2 cDNA content was detected in Cc2+/− and Cc2−/− mice, respectively (ESM Fig. 1d). The specificity of the probes was examined by DNA blotting and hybridising with a γ32P-labelled ATP Cc2-specific probe (ESM Fig. 1d).
All animals were housed in a 12 h dark–light cycle and had free access to standard chow (Harlan Teklad 2016; Harlan, Haslett, MI, USA). All procedures were approved by the Institutional Animal Care and Utilization Committee.
Metabolic analysis
Following an overnight fast, mice were anaesthetised with sodium pentobarbital at 11:00 hours. Whole venous blood was drawn from the retro-orbital sinuses to carry out radioimmunoassay analysis of serum levels of insulin and leptin (Linco Research, Billerica, MA, USA). Serum NEFA and serum triacylglycerol were determined using kits (NEFA C; Wako, Richmond, VA, USA; Triacylglycerol Kit; Pointe Scientific, Canton, MI, USA). Tissue triacylglycerol content was assayed, as described previously [23].
To determine oestrogen levels, daily vaginal smears in 50 μl normal saline were taken at 09:00 hours. The cytology was immediately examined in lavage samples under a light microscope at magnification ×10 to determine the dioestrus stage of the oestrus cycle (predominantly leucocytes). At this stage, mice were anaesthetised with isoflurane and retro-orbital blood was drawn to determine serum 17-beta oestradiol levels following the manufacturer’s instructions (mouse/rat oestradiol ELISA, catalogue number ES180S-100; Calbiotech, Spring Valley, CA, USA).
Body composition and hyperinsulinaemic–euglycaemic clamp
Gonadal visceral white adipose tissue (WAT) was excised and weighed, and visceral adiposity expressed as percentage of total body weight. Whole-body fat and lean mass were measured by 1H-magnetic resonance spectroscopy (MRS) (Echo Medical Systems, Houston, TX, USA), and a 2-h hyperinsulinaemic–euglycaemic clamp was performed in overnight-fasted awake mice (n=10–12) with primed and continuous infusion of human regular insulin (Humulin; Eli Lilly, Indianapolis, IN, USA) at a rate of 2.5 mU kg−1 min−1, as previously described [3, 23]. Glucose metabolism was estimated with a continuous infusion of [3-3H]glucose (PerkinElmer Life and Analytical Sciences, Waltham, MA, USA) for 2 h prior to (1,850 Bq/min) and throughout the clamps (3,700 Bq/min).
Daily food intake
Mice (n≥5 per age group) were housed individually in a 12 h dark–light cycle with free access to standard chow. Food was weighed daily at 12:00 hours for seven consecutive days. Daily food intake was calculated as food mass from previous day minus remaining food in the cage.
Indirect calorimetry
Indirect calorimetry was performed in awake mice (4 months old; n=6 per group), as previously described [24]. Mice were placed in metabolism cages (TSE Systems, Bad Homburg, Germany) for 3 days and had free access to food and water. Spontaneous locomotor activity was measured with an optical beam device used for quantifying horizontal and vertical movement (x-, y- and z-axes). Oxygen consumption (V̇O2) and CO2 production (V̇CO2) were sampled every 30 min for 5 s and data normalised per fat-free lean mass. Respiratory exchange ratio was calculated from the V̇CO2:V̇O2 ratio.
Recording of sympathetic nerve activity
Baseline sympathetic nerve activity (SNA) was measured directly from the nerves subserving brown adipose tissue (BAT) or WAT as described previously [25, 26]. Briefly, mice were anaesthetised by intraperitoneal injection of ketamine (91 mg/kg) and xylazine (9.1 mg/kg), and a nerve fibre innervating interscapular BAT or gonadal WAT was identified, placed on the bipolar platinum–iridium electrode and secured with silicone gel (Kwik-Cast; World Precision Instruments, Sarasota, FL, USA). Each electrode was attached to a high-impedance probe (HIP-511; Grass Instruments, West Warwick, RI, USA) and the nerve signal was amplified 105 times with an AC pre-amplifier (P5; Grass Instruments), and filtered at 100- and 1,000-Hz cut-offs using a nerve traffic analysis system (model 706C; University of Iowa Bioengineering, Iowa City, IA, USA). The signal was then routed (1) to an oscilloscope (model 54501A; Hewlett-Packard, Palo Alto, CA, USA) to monitor the quality of the sympathetic nerve recording and (2) to a resetting voltage integrator (model B600c; University of Iowa Bioengineering). SNA measurements were made for 30 min. To exclude electrical noise in the assessment of SNA, each recording was corrected for post mortem (death induced by lethal dose of ketamine/xylazine) background BAT and WAT sympathetic activities.
Immunohistochemistry
Gonadal WAT was formalin-fixed and paraffin-embedded. After deparaffinising and hydration, slides were placed in antigen retrieval solution, quenched in hydrogen peroxide solution for 30 min and blocked with normal goat serum (Vectastain Elite ABC peroxidase enzyme systems kit PK-6101; Vector Laboratories, Burlingame, CA, USA). Slides were then incubated overnight at 4°C with rabbit polyclonal α-uncoupling protein (UCP)1 antibody (1:100) (ab23841; Abcam, Cambridge, UK). After three washes with TBS (5 min each), slides were incubated at room temperature in biotinylated goat anti-rabbit IgG secondary antibody (ABC Vectastain) for 30 min, treated with ABC solution for 30 min at room temperature and stained with DAB Vectastain kit SK-4100 (Vector Laboratories).
Ex vivo palmitate oxidation
Ex vivo palmitate oxidation was analysed as previously described [17]. Briefly, mice (7 months old, n=5) were fasted overnight and anaesthetised before soleus and gastrocnemius muscles were removed, weighed, mixed, homogenised and injected into a sealed beaker at 30°C to initiate the reaction in incubation buffer containing 0.2 mmol/l [1-14C]palmitate at 18,500 Bq/ml in the presence of 2 mmol/l l-carnitine, 0.1 mmol/l malic acid, 2 mmol/l ATP and 0.05 mmol/l coenzyme A. After 45 min, the reaction was terminated with glacial acetic acid. Radioactive oxidised CO2 was trapped in a well, which was suspended above the medium and filled with benzothonium hydroxide, and counted by liquid scintillation (CytoCint; MP Biomedicals, Solon, OH, USA).
Western analysis and ex vivo phosphorylation
Tissues were lysed and the protein concentrations determined by BCA protein assay (Pierce Biotechnology, Rockford, IL, USA) prior to analysis by 7% or 4% to 12% gradient SDS-PAGE (Invitrogen, Carlsbad, CA, USA) and immunoprobing with specific antibodies. Antibodies included polyclonal antibodies against fatty acid transporter1 (FATP1) (I-20; Santa Cruz Biotechnology, Santa Cruz, CA, USA), AKT and phospho-AKT (Cell Signaling Technology, Danvers, MA, USA), in addition to monoclonal antibodies against actin (Sigma-Aldrich, St Louis, MO, USA) to normalise for the amount of proteins analysed. Blots were incubated with horseradish peroxidase-conjugated anti-goat IgG (Santa Cruz Biotechnology), anti-mouse IgG (Amersham Biosciences, Sunnyvale, CA, USA) or anti-rabbit IgG (Amersham) antibodies prior to detection by enhanced chemiluminescence (Amersham) and quantification by densitometry. For CEACAM2 quantification in hypothalami, 30 μg protein of tissue lysates from overnight-fasted mice were immunoprecipitated with custom-made anti-peptide mouse CEACAM2 antibody (α-mCC2; Bethyl Laboratories, Montgomery, TX, USA) prior to SDS-PAGE analysis and immunoblotting with the same antibody.
For ex vivo phosphorylation experiments, 50 μg tissue lysates were treated with 100 nmol/l insulin for 5 min prior to immunoprecipitation with antibodies against the β-subunit of the insulin receptor (α-IRβ) (Santa Cruz Biotechnology), SDS-PAGE analysis and immunoblotting with α-phosphotyrosine antibody (Upstate Biotechnology, Billerica, MA, USA), followed by reprobing with α-IRβ to normalise against the amount of insulin receptor in the immunopellet.
Semi-quantitative real-time PCR analysis
Total RNA was prepared using a kit (PerfectPure RNA Tissue kit; 5Prime, Gaithersburg, MD, USA) following the manufacturer’s instructions. cDNA was synthesised with oligo dT primers and Improm II Reverse Transcriptase (Promega, Madison, WI, USA), using 1 μg total RNA and primers (ESM Table 1). cDNA was evaluated with semi-quantitative real-time PCR (Fast SYBR Green PCR mix on a StepOne Plus Real-Time PCR system; Applied Biosystems, Foster City, CA, USA). Standard curves for each primer set were generated using serial 1:4 dilutions of pooled samples. The relative amounts of mRNA were calculated by comparison with the corresponding standards and normalised relative to Gapdh. Results are expressed as mean±SEM in fold change relative to controls.
Table 1.
Metabolic variables of male Cc2+/+ and Cc2−/− mice
| Variable | At 1 month, by mouse group
|
At 5–6 months by mouse group
|
||
|---|---|---|---|---|
| Cc2+/+ | Cc2−/− | Cc2+/+ | Cc2−/− | |
| Visceral adiposity (% BWT) | N/A | N/A | 3.13±0.26 | 2.38±0.46 |
| Lean mass (g) | N/A | N/A | 23.9±0.81 | 22.1±0.82 |
| Fat mass (g) | N/A | N/A | 5.00±0.30 | 4.01±0.21 * |
| Serum insulin (pmol/l) | 114±18.7 | 114±9.11 | 62.5±3.43 | 76.0±7.15 |
| Random blood glucose (mmol/l) | 7.33±0.29 | 7.66±0.33 | 6.49±0.27 | 6.33±0.27 |
| Serum triacylglycerol (mmol/l) | 0.54±0.04 | 0.59±0.04 | 0.90±0.09 | 0.52±0.10 * |
| Serum NEFA (mEq/l) | 0.83±0.03 | 0.67±0.05 | 0.85±0.06 | 0.76±0.02 * |
| Leptin (ng/ml) | 2.38±0.23 | 2.13±0.37 | 12.8±2.28 | 18.9±5.90 |
Mice were fasted overnight prior to blood drawing, except for leptin analysis (mice fasted for 6 h) and random blood glucose (blood removed at 23:00 hours)
p<0.05 for Cc2−/− vs Cc2+/+ mice; n≥7
BWT, body weight; N/A, not applicable
Statistical methods
Statistical significance was determined by two-tailed Student’s t test unless otherwise indicated. Welch’s correction was used if variances were significantly different. Statistical significance was set at 1% to 5%.
Results
Body composition of male Cc2−/− mice
Unlike female Cc2−/− mice, which exhibit increased body weight, male Cc2−/− mice maintain a body mass similar to that of Cc2+/+ counterparts throughout their life [17]. 1H-MRS imaging revealed that 5-month-old male Cc2−/− mice had similar lean mass, but reduced fat mass compared with Cc2+/+ mice (Table 1). The decrease in fat mass did not reflect a change in body weight, perhaps owing to compensation by minor changes in body water and/or lean mass that would fall below the detection limit. Male mice also exhibited no difference in their visceral adipose mass compared with wild-type controls (Table 1). Their leptin levels were consistently normal at all ages examined (Table 1).
Male Cc2−/− mice are insulin-sensitive
Male Cc2−/− mice displayed normal serum insulin and blood glucose levels (Table 1). To assess insulin sensitivity in vivo, a 2 h hyperinsulinaemic–euglycaemic clamp was performed on 5-month-old mice. Basal glucose at the beginning of the clamp was similar in both groups of mice (Fig. 1a). During the clamp, which maintains comparable glucose levels in awake overnight-fasted mice (Fig. 1b), the glucose infusion rate required to maintain euglycaemia was normal (Fig. 1c), indicating overall insulin sensitivity. Hepatic glucose production at the beginning and during the clamp was similar to that in controls (Fig. 1d), indicating normal suppression by insulin. Liver responsiveness to insulin was shown to be normal by the intact tyrosine phosphorylation (and activation) of insulin receptor β in response to insulin in liver lysates, as shown by western blot analysis of insulin receptor immunopellets with α-phosphotyrosine antibody (Fig. 1k).
Glucose uptake in the whole body (Fig. 1e), including WAT (Fig. 1f) and gastrocnemius muscle (Fig. 1g), was unaltered in male Cc2−/− mice. Consistently, whole-body glucose turnover was normal (Fig. 1h). Whole-body glycolysis (Fig. 1i) and glycogen synthesis (Fig. 1j) were also intact.
Increased fat metabolism in skeletal muscle of male Cc2−/− mice
Liver triacylglycerol content in Cc2−/− mice was similar to that in controls (16.5±1.6 μmol/g wet tissue [mean±SEM] vs 13.5±1.2 in Cc2+/+). However, fasting serum triacylglycerol and NEFA levels were reduced significantly, starting at 2 to 3 months of age (triacylglycerol 0.48±0.06 vs 0.86±0.07 mmol/l in Cc2+/+; NEFA 0.50±0.07 vs 0.72±0.04 mEq/l in Cc2+/+). Skeletal muscle Fatp1 (also known as Slc27a1) mRNA (Fig. 2a) and protein (Fig. 2b) levels were also increased. Together with reduced fed serum triacylglycerol (0.40±0.05 vs 1.20±0.08 mmol/l in Cc2+/+; p<0.05) and NEFA levels (0.40±0.03 vs 0.60±0.03 mEq/l in Cc2+/+; p<0.05), with no change in visceral obesity (Table 1), this points to increased fatty acid uptake in muscle relative to adipose tissue. Increased uptake in skeletal muscle was followed by increased β-oxidation, as determined by V̇CO2 from palmitate (Fig. 2c), and as supported by increased expression of Ppara mRNA and its target genes, including Cpt1 (also known as Cpt1a), which is involved in fatty acid transport to mitochondria (Fig. 2a). Increased β-oxidation lowered triacylglycerol content in the skeletal muscle of male Cc2−/− mice (Fig. 2d).
Fig. 2.
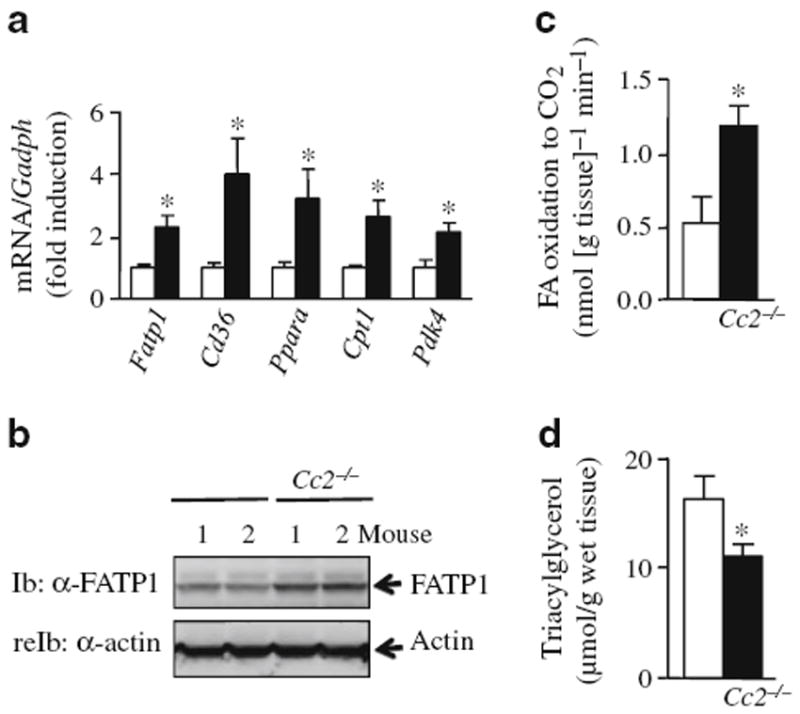
Biochemical analysis of lipid metabolism in skeletal muscle of male mice. Mixed skeletal muscle was isolated (gastrocnemius and soleus) from overnight-fasted 5 to 7 months old male mice (white bars, Cc2+/+; black bars, Cc2−/− all panels). a mRNA expression of genes involved in fatty acid uptake and oxidation was determined by semi-quantitative real-time PCR and normalised to Gapdh. b FATP1 abundance by western blot analysis, using α-FATP1 antibody to immunoblot (Ib) and α-actin antibody to reprobe (reIb) and normalise against the amount of protein loaded. c Ex vivo palmitate oxidation. d triacylglycerol content. Values are expressed as mean±SEM; n=4–8 per group; *p<0.05 vs Cc2+/+
Male Cc2−/− mice display a hypermetabolic state
Beginning at 2 months of age, Cc2−/− mice displayed an increase in daily food intake, tested over a period of 7 days (Fig. 3a).
Fig. 3.
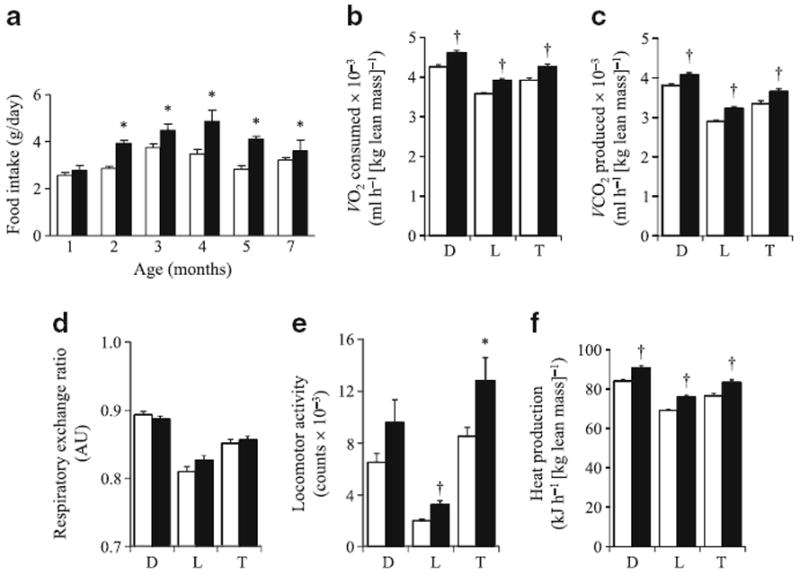
Characterisation of the metabolic state of male mice. Cc2+/+ (white bars) and Cc2−/− (black bars) mice were individually caged, given free access to food and subjected to: a analysis of daily food intake over seven consecutive days (n>6 per group) and b–f indirect calorimetry analysis (performed over a 24 h period at age 4 months, and values as indicated (b–f) were measured). T, total light–dark cycle; D, dark period; L, light period; n=5–6 mice per group. Values are expressed as mean±SEM; *p<0.05 and †p<0.005 vs Cc2+/+
Indirect calorimetry analysis over a 24 h period (Fig. 3b–f) revealed a 7% increase in total V̇O2 (Fig. 3b) and V̇CO2 (Fig. 3c) during a dark–light cycle in 4-month-old Cc2−/− mice compared with controls. Due to the combined increases of both variables, the respiratory exchange ratio remained unchanged (Fig. 3d). Moreover, total spontaneous locomotor activity was increased by 31% (Fig. 3e) and heat generation by 7% (Fig. 3f). Taken together, these data show that Cc2−/− mice are in a hypermetabolic state.
Male Cc2−/− mice display elevated brown adipogenesis
We next examined whether the hypermetabolic state and increased heat production in males is associated with increased brown adipogenesis. mRNA levels of the key BAT genes Ucp1 and Dio2 were elevated by three- and sixfold, respectively, in BAT derived from male Cc2−/− mice (Fig. 4a). In females, however, mRNA levels of BAT-specific genes tended to increase by around twofold without reaching statistical significance (Fig. 4a). Levels of Prdm16 mRNA were unaltered in BAT of male and female Cc2−/− mice (Fig. 4a), suggesting that deletion of Ceacam2 does not affect programming of brown adipogenesis [27].
Fig. 4.
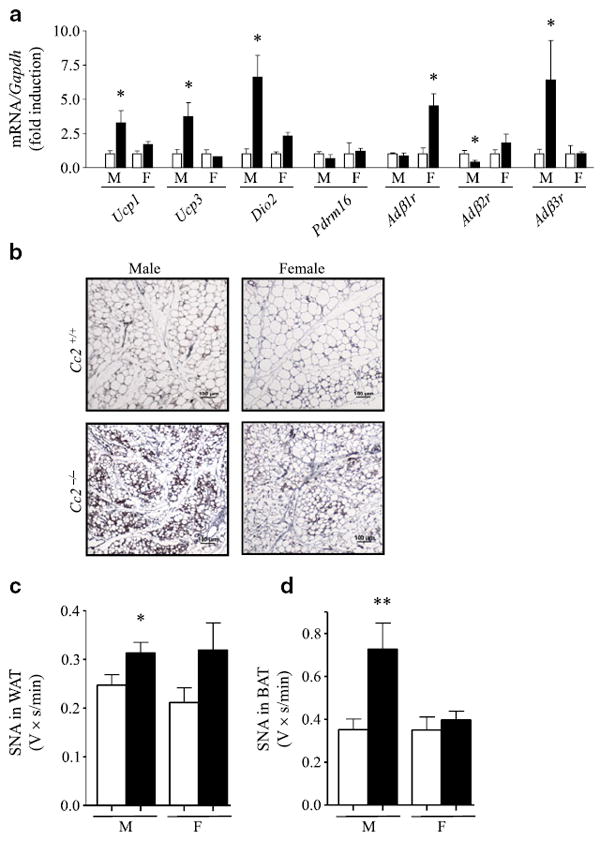
Elevated brown adipogenesis and SNA in male Cc2−/− mice. a mRNA content was assessed by semi-quantitative real-time PCR and normalised to Gapdh in BAT from 3-month-old Cc2+/+ (white bars) and Cc2−/− (black bars) mice; n≥6 per group. b Immunohistochemical analysis of WAT from four of each mouse group at age 3 months. Tissues were fixed and immunostained with α-UCP1 antibody (brown). Representative images from three sections per mouse are shown. Magnification ×20. Scale bar, 100 μm. c, d Baseline SNA, recorded directly from the nerve subserving (c) WAT or (d) BAT, was compared between male (M) and female (F) mouse groups as above (a), but at 5 months of age; n=9–10 per group. Values (a, c, d) are expressed as mean±SEM; *p<0.05 vs Cc2+/+ and **p<0.01 vs Cc2+/+
Immunohistochemical analysis revealed increased UCP1 levels (brown staining), relative to controls, and discrete multiloculated brown adipocytic foci dispersed between white adipocytes (Fig. 4b) in gonadal WAT derived from male Cc2−/− mice. Female Cc2−/− mice also had increased UCP1 immunostaining in WAT compared with controls (Fig. 4b). This suggests that null mutation of Ceacam2 causes a preferentially greater increase in brown adipogenesis in male than in female Cc2−/− mice [28]. Together with the fourfold increase in Ucp3 mRNA levels in BAT from male, but not female animals (Fig. 4a), this is consistent with the increased insulin sensitivity, overall lean phenotype and increased basal metabolic rate observed in male Cc2−/− mice.
Increased sympathetic tone to adipose tissue in male, but not in female Cc2−/− mice
Serum oestrogen levels in male (26.1±1.24 pg/ml in Cc2−/− vs 24.5±1.09 pg/ml in Cc2+/+ [mean±SEM]; p>0.05) and female null mice (24.7±1.32 pg/ml in Cc2−/− vs 24.1±0.94 pg/ml in Cc2+/+; p>0.05) were intact. However, mRNA levels of Adβ3r (also known as Adrb3), the predominant adrenergic receptor in brown adipogenesis with reduced expression in adipose tissue of obese animals [29], was elevated in BAT of male, but not of female null mice (Fig. 4a). With the different mRNA levels of the other adrenergic isoforms in BAT, i.e. Adβ1r (also known as Adrb1) and Adβ2r (also known as Adrb2) (Fig. 4a), this raised the possibility that the sympathetic tone in BAT is activated to different degrees in the two groups of mice. Thus we compared baseline SNA subserving different fat depots in Cc2−/− mice and controls. Consistent with the elevated energy expenditure, male Cc2−/− mice had a significant increase in SNA to WAT (Fig. 4c) and BAT (Fig. 4d). In contrast, BAT SNA was unaltered in female Cc2−/− mice (Fig. 4d), while WAT SNA tended to be elevated relative to littermate controls, but without reaching statistical significance (Fig. 4c). Given that an elevation of brown adipogenesis promotes energy dissipation and insulin sensitivity [27, 30, 31], it is likely that the dispersion of brown adipocytic foci in WAT also contributes to the enhanced energy dissipation in male mice. This suggests that the increased sympathetic tone in adipose depots (WAT and BAT) plays an important role in the different increase in energy dissipation in males, and that elevated β(1)-adrenergic receptor does not suffice to mediate an increase in sympathetic tone in BAT females in the absence of increased β(3)-adrenergic receptor [32]. Together, these data suggest that loss of CEACAM2 triggers an increase in sympathetic nerve outflow to adipose tissues in males, but not in females, leading to increased energy expenditure through changes in the expression of key genes.
Comparable level of hypothalamic CEACAM2 in male and female mice
We then tested whether the sexually dimorphic increase in sympathetic nerve outflow to adipose tissues in Cc2−/− mice is mediated, at least in part, by different levels of brain CEACAM2 in the two groups of mice. To this end, we analysed hypothalamic CEACAM2 levels in age-matched (5 months old) wild-type Cc2+/+ male and female mice that had undergone an overnight fast to induce CEACAM2 production [17]. mRNA and western blot analyses indicated a comparable level of hypothalamic CEACAM2 in these mice (Fig. 5a, b, respectively). This suggests that other mechanisms are involved in the sexual dimorphism of energy balance in Cc2−/− mice.
Fig. 5.
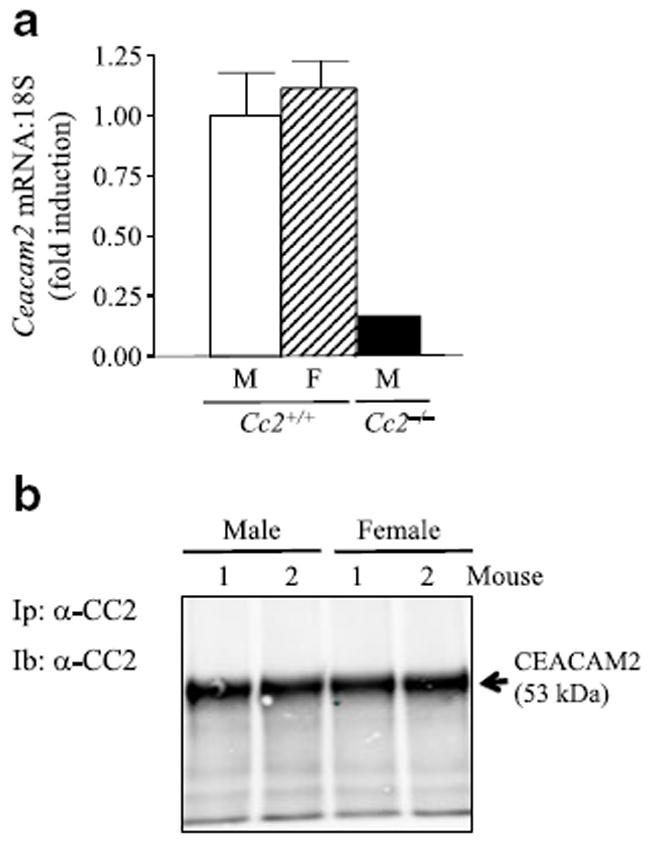
Comparable hypothalamic CEACAM2 levels in male and female Cc2+/+ mice. Male (M) and female (F) Cc2+/+ mice aged 5 months (n≥4 per group) were fasted overnight and their hypothalami removed to analyse (a) Ceacam2 mRNA levels by semiquantitative real-time PCR (Values are expressed as mean±SEM) and (b) CEACAM2 abundance by immunoblotting (Ib) of CEACAM2 immunopellet (Ip) with α-CEACAM2 (CC2) polyclonal antibody
Discussion
CEACAM2 is a protein related to CEACAM1, but has a relatively more limited tissue distribution [14]. The function of CEACAM1 in regulating insulin action by promoting insulin clearance in liver has been well documented [2-4]. The phenotype of female Cc2−/− mice demonstrates that CEACAM2 plays a role in the central regulation of food intake, energy balance and peripheral insulin action [17]. The present study uncovers a novel sexually dimorphic function of CEACAM2 in the sympathetic regulation of brown adipogenesis and in the oxidative state of skeletal muscle, which is a major site of energy expenditure.
Like their female counterparts [17], male Cc2−/− mice develop hyperphagia, suggesting a primary role for CEACAM2 in the regulation of feeding behaviour. The normal circulating levels of leptin at the onset of hyperphagia in both sexes suggest that hyperphagia in Cc2−/− mice is not secondary to changes in leptin sensitivity and/or signals [33]. Given that brain Ceacam2 mRNA is induced by fasting and is markedly reduced upon refeeding [17], it is likely that hypothalamic CEACAM2 is involved in food intake suppression and that hyperphagia in these mice is due primarily to CEACAM2 deletion from brain centres involved in feeding behaviour and rewards, including the VMH, the globus and ventral pallidus, the striatum, the olfactory bulb and the hippocampus [17, 34, 35].
Like female mice [17], male mice exhibit increased fatty acid uptake in skeletal muscle and in the mitochondria, as suggested by increased levels of FATP1 and of Ppara mRNA plus its target genes. However, while in females β-oxidation of fatty acids is incomplete, and lipotoxicity develops to cause insulin resistance and reduced glucose uptake in skeletal muscle [36, 37], in male mice fatty acid oxidation in skeletal muscle is increased, leading to insulin sensitivity and lower total fat mass. Thus, complete β-oxidation in skeletal muscle, together with increased brown adipogenesis in BAT and WAT [31], and increased sympathetic tone to adipose tissue, is likely to offset the effect of hyperphagia and render male Cc2−/− mice hypermetabolic and insulin-sensitive.
Given that CEACAM2 is not endogenously produced in adipocytes, it is likely that the changes in adipose depots of male Cc2−/− mice are driven by a cell non-autonomous effect brought about by Ceacam2 gene deletion. The enrichment of CEACAM2 content in metabolic sensing centres of the brain, including VMH, a nucleus associated with critical involvement in the sympathetic regulation of fatty acid mobilisation from adipose tissue [38, 39], points to loss of central CEACAM2 as a likely cause of the preferential increase in brown adipogenesis in male Cc2−/− mice. Consistent with such a possibility, direct assessment of sympathetic nervous system activity revealed elevated sympathetic traffic to adipose tissues in male Cc2−/− mice. This is further supported by a strong induction of key genes in BAT (Ucp1, Dio2 and Adb3r [also known as Adrb3]) and WAT (Ucp1) of male Cc2−/− mice, providing additional evidence that CEACAM2 participates in the central regulation of brown adipogenesis, energy dissipation and sympathetic nervous tone in adipose tissues [40-42]. The underlying mechanism remains to be determined, but the sexually dimorphic phenotype is consistent with the well-documented sex difference with respect to obesity in rats and mice with VMH lesions, as reviewed by others [21].
Future studies using targeted deletion of Ceacam2 are under way to dissect the mechanisms involved in the sexually dimorphic central regulation of peripheral insulin action by CEACAM proteins. Nonetheless, the current study emphasises a novel role for CEACAM2 in the hypothalamic autonomic control of brown adipogenesis and energy dissipation.
Supplementary Material
Acknowledgments
The authors thank A.M. DeAngelis and T. Dai from the Najjar Laboratory at the University of Toledo College of Medicine for their assistance in genotyping, and Z. Zhang from the Kim Laboratory at Pennsylvania State University College of Medicine for his assistance in metabolic phenotyping. We also thank J. Kalisz at the Najjar Laboratory for excellent technical assistance in the generation, genotyping and maintenance of Cc2−/− null mice, in addition to the carrying out of routine RNA analyses and the oestrogen assay. We especially thank D. Accili from Columbia University Diabetes Center for frequent scientific discussions and critical reading of the manuscript. This work was supported by grants from the National Institutes of Health as follows: R01DK054254 and R01DK083850 (to S.M. Najjar), R01-DK80756 and U24-DK59635 (to J.K. Kim), HL084207 (to K. Rahmouni) and R00 HD056491 (to J. W. Hill). Grants were also received from the United States Department of Agriculture (USDA 38903-19826, to S.M. Najjar) and the American Diabetes Association (to J.K. Kim and K. Rahmouni).
Abbreviations
- BAT
Brown adipose tissue
- Cc2−/−
Ceacam2 homozygous null mutant mice
- Cc2+/+
Wild-type littermate mice
- CEACAM
Carcino-embryonic antigen-related cell adhesion molecule
- FATP1
Fatty acid transport protein 1
- α-IRβ
β-Subunit of the insulin receptor
- MRS
1H-Magnetic resonance spectroscopy
- SNA
Sympathetic nerve activity
- UCP
Uncoupling protein
- VMH
Ventromedial nucleus of the hypothalamus
- V̇CO2
CO2 production
- V̇O2
Oxygen consumption
- WAT
White adipose tissue
Footnotes
Contribution statement PRP researched data, contributed to conception and design, and to analysis and interpretation of data, and also wrote the manuscript. SKRam, MKK, CKR, SG, JSM, JWH, DYJ, DAM, GH and SJL researched and analysed data, and contributed to the drafting of the article. REB contributed to data analysis, discussion and reviewed/edited the manuscript. SKRah researched data and reviewed/edited the manuscript. JKK researched data and contributed to discussion and revision of the manuscript. SMN oversaw the work, including its conception and design, contributed to analysis and interpretation of data, and also reviewed/edited the manuscript. All authors approved the final version of the manuscript.
Electronic supplementary material The online version of this article (doi:10.1007/s00125-011-2388-x) contains peer-reviewed but unedited supplementary material, which is available to authorised users.
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Contributor Information
P. R. Patel, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
S. K. Ramakrishnan, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
M. K. Kaw, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
C. K. Raphael, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
S. Ghosh, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
J. S. Marino, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
G. Heinrich, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
S. J. Lee, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
R. E. Bourey, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Internal Medicine at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
J. W. Hill, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
D. Y. Jung, Department of Cellular and Molecular Physiology, Pennsylvania State University College of Medicine, Hershey, PA, USA Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA, USA.
D. A. Morgan, Department of Internal Medicine, University of Iowa Carver College of Medicine, Iowa City, IA, USA
J. K. Kim, Department of Cellular and Molecular Physiology, Pennsylvania State University College of Medicine, Hershey, PA, USA Program in Molecular Medicine, University of Massachusetts Medical School, Worcester, MA, USA.
S. K. Rahmouni, Department of Internal Medicine, University of Iowa Carver College of Medicine, Iowa City, IA, USA
S. M. Najjar, Center for Diabetes and Endocrine Research, College of Medicine and Life Sciences, University of Toledo, Health Science Campus, 3000 Arlington Avenue, Mail Stop 1009, Toledo, OH 43614, USA, sonia.najjar@utoledo.edu Department of Physiology and Pharmacology at the College of Medicine and Life Sciences, University of Toledo, Health Science Campus, Toledo, OH, USA.
References
- 1.Öbrink B. CEA adhesion molecules: multifunctional proteins with signal-regulatory properties. Curr Opin Cell Biol. 1997;9:616–626. doi: 10.1016/S0955-0674(97)80114-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poy MN, Yang Y, Rezaei K, et al. CEACAM1 regulates insulin clearance in liver. Nat Genet. 2002;30:270–276. doi: 10.1038/ng840. [DOI] [PubMed] [Google Scholar]
- 3.DeAngelis AM, Heinrich G, Dai T, et al. Carcinoembryonic antigen-related cell adhesion molecule 1: a link between insulin and lipid metabolism. Diabetes. 2008;57:2296–2303. doi: 10.2337/db08-0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu E, Dubois MJ, Leung N, et al. Targeted disruption of carcinoembryonic antigen-related cell adhesion molecule 1 promotes diet-induced hepatic steatosis and insulin resistance. Endocrinology. 2009;150:3503–3512. doi: 10.1210/en.2008-1439. [DOI] [PubMed] [Google Scholar]
- 5.Ergun S, Kilik N, Ziegeler G, et al. CEA-related cell adhesion molecule 1: a potent angiogenic factor and a major effector of vascular endothelial growth factor. Mol Cell. 2000;5:311–320. doi: 10.1016/s1097-2765(00)80426-8. [DOI] [PubMed] [Google Scholar]
- 6.Horst AK, Bickert T, Brewig N, et al. CEACAM1+ myeloid cells control angiogenesis in inflammation. Blood. 2009;113:6726–6736. doi: 10.1182/blood-2008-10-184556. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Simpson JF, Glackin C, Riethorf L, Wagener C, Shively JE. Expression of biliary glycoprotein (CD66a) in normal and malignant breast epithelial cells. Anticancer Res. 1998;18:3203–3212. [PubMed] [Google Scholar]
- 8.Lin SH, Pu YS. Function and therapeutic implication of CCAM cell-adhesion molecule in prostate cancer. Semin Oncol. 1999;26:227–233. [PubMed] [Google Scholar]
- 9.Laurie NA, Comegys MM, Carreiro MP, et al. Carcinoembryonic antigen-related cell adhesion molecule 1a-4L suppression of rat hepatocellular carcinomas. Cancer Res. 2005;65:11010–11017. doi: 10.1158/0008-5472.CAN-04-2841. [DOI] [PubMed] [Google Scholar]
- 10.Leung N, Turbide C, Balachandra B, Marcus V, Beauchemin N. Intestinal tumor progression is promoted by decreased apoptosis and dysregulated Wnt signaling in Ceacam1−/− mice. Oncogene. 2008;27:4943–4953. doi: 10.1038/onc.2008.136. [DOI] [PubMed] [Google Scholar]
- 11.Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nat Rev Immunol. 2006;6:433–446. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- 12.Najjar SM. Regulation of insulin action by CEACAM1. Trends Endocrinol Metab. 2002;13:240–245. doi: 10.1016/s1043-2760(02)00608-2. [DOI] [PubMed] [Google Scholar]
- 13.Nedellec P, Dveksler GS, Daniels E, et al. Bgp2, a new member of the carcinoembryonic antigen-related gene family, encodes an alternative receptor for mouse hepatitis viruses. J Virol. 1994;68:4525–4537. doi: 10.1128/jvi.68.7.4525-4537.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Han E, Phan D, Lo P, et al. Differences in tissue-specific and embryonic expression of mouse Ceacam1 and Ceacam2 genes. Biochem J. 2001;355:417–423. doi: 10.1042/0264-6021:3550417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zebhauser R, Kammerer R, Eisenried A, McLellan A, Moore T, Zimmermann W. Identification of a novel group of evolutionarily conserved members within the rapidly diverging murine Cea family. Genomics. 2005;86:566–580. doi: 10.1016/j.ygeno.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Robitaille J, Izzi L, Daniels E, Zelus B, Holmes KV, Beauchemin N. Comparison of expression patterns and cell adhesion properties of the mouse biliary glycoproteins Bbgp1 and Bbgp2. Eur J Biochem. 1999;264:534–544. doi: 10.1046/j.1432-1327.1999.00660.x. [DOI] [PubMed] [Google Scholar]
- 17.Heinrich G, Ghosh S, Deangelis AM, et al. Carcinoembryonic antigen-related cell adhesion molecule 2 controls energy balance and peripheral insulin action in mice. Gastroenterology. 2010;139:644–652. doi: 10.1053/j.gastro.2010.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sudo M, Minokoshi Y, Shimazu T. Ventromedial hypothalamic stimulation enhances peripheral glucose uptake in anesthetized rats. Am J Physiol. 1991;261:E298–E303. doi: 10.1152/ajpendo.1991.261.3.E298. [DOI] [PubMed] [Google Scholar]
- 19.Haque MS, Minokoshi Y, Hamai M, Iwai M, Horiuchi M, Shimazu T. Role of the sympathetic nervous system and insulin in enhancing glucose uptake in peripheral tissues after intrahypothalamic injection of leptin in rats. Diabetes. 1999;48:1706–1712. doi: 10.2337/diabetes.48.9.1706. [DOI] [PubMed] [Google Scholar]
- 20.Minokoshi Y, Okano Y, Shimazu T. Regulatory mechanism of the ventromedial hypothalamus in enhancing glucose uptake in skeletal muscles. Brain Res. 1994;649:343–347. doi: 10.1016/0006-8993(94)91085-5. [DOI] [PubMed] [Google Scholar]
- 21.King BM. The rise, fall, and resurrection of the ventromedial hypothalamus in the regulation of feeding behavior and body weight. Physiol Behav. 2006;87:221–244. doi: 10.1016/j.physbeh.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 22.Shiuchi T, Haque MS, Okamoto S, et al. Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle via sympathetic nervous system. Cell Metab. 2009;10:466–480. doi: 10.1016/j.cmet.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 23.Park SY, Cho YR, Kim HJ, et al. Mechanism of glucose intolerance in mice with dominant negative mutation of CEACAM1. Am J Physiol Endocrinol Metab. 2006;291:E517–E524. doi: 10.1152/ajpendo.00077.2006. [DOI] [PubMed] [Google Scholar]
- 24.Hong EG, Ko HJ, Cho YR, et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes. 2009;58:2525–2535. doi: 10.2337/db08-1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramadori G, Fujikawa T, Fukuda M, et al. SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell metabol. 2010;12:78–87. doi: 10.1016/j.cmet.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Z, Liu X, Morgan DA, et al. Neuronal receptor activity-modifying protein 1 promotes energy expenditure in mice. Diabetes. 2011;60:1063–1071. doi: 10.2337/db10-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seale P, Kajimura S, Yang W, et al. Transcriptional control of brown fat determination by PRDM16. Cell Metabol. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Jesus LA, Carvalho SD, Ribeiro MO, et al. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest. 2001;108:1379–1385. doi: 10.1172/JCI13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onai T, Kilroy G, York DA, Bray GA. Regulation of beta 3-adrenergic receptor mRNA by sympathetic nerves and glucocorticoids in BAT of Zucker obese rats. Am J Physiol. 1995;269:R519–R526. doi: 10.1152/ajpregu.1995.269.3.R519. [DOI] [PubMed] [Google Scholar]
- 30.Ravussin E, Kozak LP. Have we entered the brown adipose tissue renaissance? Obes Rev. 2009;10:265–268. doi: 10.1111/j.1467-789X.2008.00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenbaum M, Leibel RL. Adaptive thermogenesis in humans. Int J Obes. 2010;34(Suppl 1):S47–S55. doi: 10.1038/ijo.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chernogubova E, Hutchinson DS, Nedergaard J, Bengtsson T. Alpha1- and beta1-adrenoceptor signaling fully compensates for beta3-adrenoceptor deficiency in brown adipocyte norepinephrine-stimulated glucose uptake. Endocrinology. 2005;146:2271–2284. doi: 10.1210/en.2004-1104. [DOI] [PubMed] [Google Scholar]
- 33.Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology. 2008;149:2138–2148. doi: 10.1210/en.2007-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- 35.Cota D, Barrera JG, Seeley RJ. Leptin in energy balance and reward: two faces of the same coin? Neuron. 2006;51:678–680. doi: 10.1016/j.neuron.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 36.Koves TR, Ussher JR, Noland RC, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metabol. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 37.Wang H, Knaub LA, Jensen DR, et al. Skeletal muscle-specific deletion of lipoprotein lipase enhances insulin signaling in skeletal muscle but causes insulin resistance in liver and other tissues. Diabetes. 2009;58:116–124. doi: 10.2337/db07-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bray GA, Nishizawa Y. Ventromedial hypothalamus modulates fat mobilisation during fasting. Nature. 1978;274:900–902. doi: 10.1038/274900a0. [DOI] [PubMed] [Google Scholar]
- 39.Steffens AB, Scheurink AJ, Luiten PG, Bohus B. Hypothalamic food intake regulating areas are involved in the homeostasis of blood glucose and plasma FFA levels. Physiol Behav. 1988;44:581–589. doi: 10.1016/0031-9384(88)90322-8. [DOI] [PubMed] [Google Scholar]
- 40.Saito M, Minokoshi Y, Shimazu T. Brown adipose tissue after ventromedial hypothalamic lesions in rats. Am J Physiol. 1985;248:E20–E25. doi: 10.1152/ajpendo.1985.248.1.E20. [DOI] [PubMed] [Google Scholar]
- 41.Giordano A, Frontini A, Murano I, et al. Regional-dependent increase of sympathetic innervation in rat white adipose tissue during prolonged fasting. J Histochem Cytochem. 2005;53:679–687. doi: 10.1369/jhc.4A6566.2005. [DOI] [PubMed] [Google Scholar]
- 42.Song CK, Schwartz GJ, Bartness TJ. Anterograde trans-neuronal viral tract tracing reveals central sensory circuits from white adipose tissue. Am J Physiol Regul Integr Comp Physiol. 2009;296:R501–R511. doi: 10.1152/ajpregu.90786.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.