

Abstract
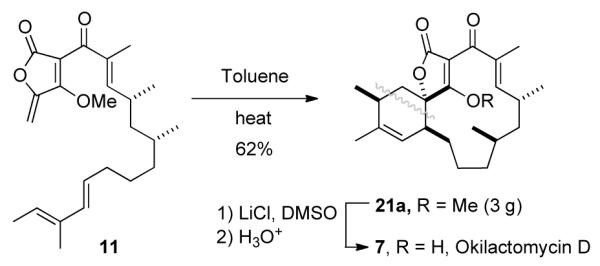
The spirotetronate okilactomycin D (7) has been efficiently synthesized by a route featuring a substrate-controlled, diastereoselective (8:1) intramolecular Diels-Alder (IMDA) reaction of 11. The assigned absolute configuration of (−)-7 was confirmed.
The spirotetronate polyketides (e.g., chlorothricolides, kijanimicin, and abyssomicins) comprise a class of natural product compounds that are characterized by the presence of a five-membered tetronic acid moiety spiro-linked to a cyclohexene ring (cf. 1, Figure 1). Their biological activities (which include antitumor, 1 antimicrobial,2 and cholesterol biosynthesis inhibition3) coupled with unusual architectures render them interesting targets for synthesis.
Figure 1.
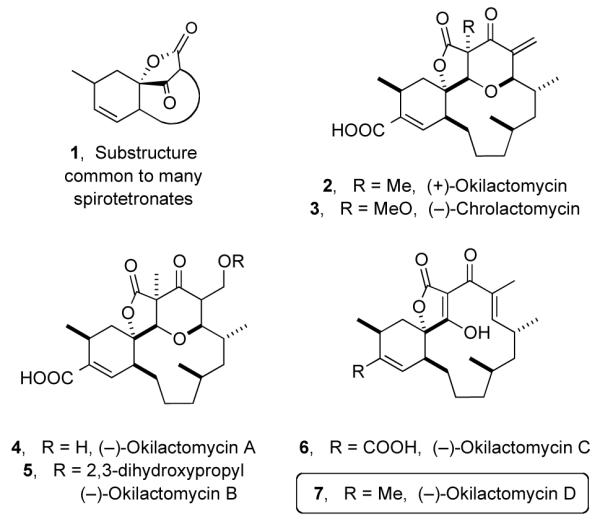
Structures of Okilactomycins and Chrolactomycin
The okilactomycins (2 and 4-7), a subclass of spirotetronates, contain either a tri- or tetracyclic skeleton. In 1987 Imai and co-workers reported the isolation and structural elucidation of okilactomycin (2) from Streptomyces griseoflavus.4 In 2001 Yamashita and co-workers described the isolation of the structurally related chrolactomycin (3) from Streptomyces sp. 569N-3.5 In 2009 Singh et al. reported the isolation of four new members of this class—namely, okilactomycins A, B, C, and D (4–7) [along with okilactomycin (2)] from the bacterium Streptomyces scabrisporus. 6 Whereas congeners 6 and 7 contain an intact acyltetronic acid structural subunit, 7 compounds 2-5 have a modified tetronate moiety. The structure of each of the new compounds 4-7 was assigned based upon spectroscopic analysis. The absolute configurations were assumed to be the same as that of okilactomycin (2), which had been established by the total syntheses achieved in the Smith and Scheidt laboratories.8 Notably, the sign of the specific rotation of each of 4-7 is negative, while that of okilactomycin itself (2) is positive, regardless of its producing organism (Streptomyces griseoflavus4 or Streptomyces scabrisporus6). Whether or not all members of the okilactomycin family are of the same antipodal series has interesting biosynthetic connotations.
With respect to biosynthesis, it has been proposed that some of the spirotetronate natural products derive from an intramolecular Diels-Alder like (IMDA) macrocyclization.9,10 This IMDA strategy enabled several remarkably efficient total synthesis of abyssomicin C (10).11 Cyclization of trienone 8 proceeded with high yield and diastereoselectivity, resulting in the construction of the macrocyclic spirotetronate 9 (Scheme 1).12 Conceivably, the macrocycle in okilactomycin D may be generated by a similar IMDA reaction from substrate 11. However, substrates 11 and 8 differ in both chain length (13- vs. 11-membered macrocycle) and electronic character of their diene/dienophile partners. We were interested in how such differences would influence both the rate and diastereoselectivity of this IMDA reaction and, hence, pursued the studies described here. We also note the possibility that 7 could serve as the biogenetic precursor to each of the other okilactomycins (2 and 4-6) by undergoing post-IMDA modifications. This thinking is consistent with the co-occurrence of 7 with these more highly oxidized okilactomycin members in Streptomyces scabrisporus.6
Scheme 1.
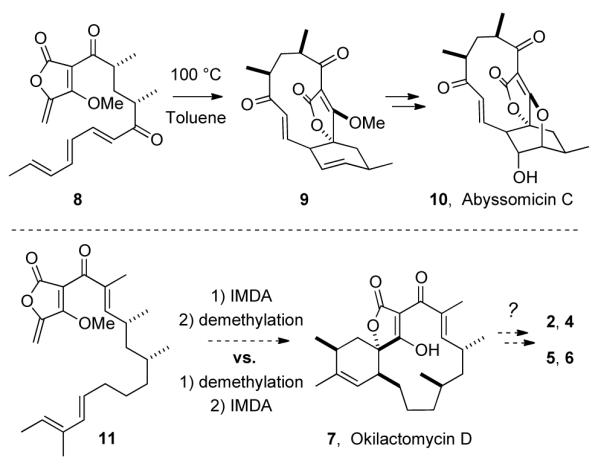
IMDAs in Reported Total Syntheses of Abyssomicin C (10)11 vs. Our Planned Okilactomycin D (7) Synthesis
We planned to assemble 11 from the four building blocks 12-15 shown in Scheme 2. Thus, we undertook coupling of the Grignard reagent derived from 14 with iodide 15, Wittig olefination of an elaborated aldehyde with ylide 13, and electrophilic net acylation of the lithium anion of methyl tetronate 12.
Scheme 2.
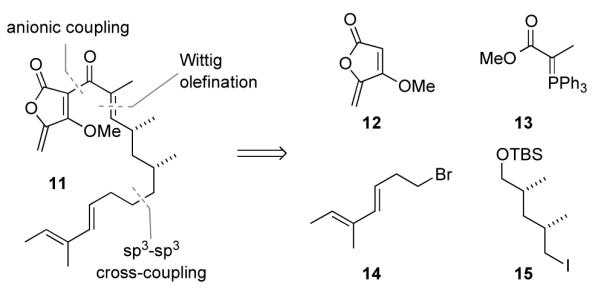
Retrosynthetic Plan for Accessing 11
The efficient construction of key intermediate 11 is summarized in Scheme 3. Cyclopropyl magnesium bromide (prepared from 16) was added to tiglic aldehyde, to give cyclopropyl alcohol 17, which, without purification, was treated with concentrated hydrobromic acid to provide the dienyl bromide 14 (ca. 16:1 ratio of 2E,4E- and 2Z,4E-isomers). 13 The Grignard reagent derived from 14 was cross coupled, under the action of Li2CuCl4, with known iodide (±)-1514 to give, following TBS ether cleavage, diene 18 (92% yield from 15) with an ca. 13:1 E/Z ratio at the terminal double bond.15 Swern oxidation gave aldehyde 19 in 85% yield. Wittig olefination by the stabilized ylide 13 16 and standard processing of the resulting enoate (DIBAL-H reduction and MnO2 oxidation) provided enal 20 in 89% yield over 3 steps. The protocol of Pattenden 17 (as successfully implemented in the abyssomicin C studies11) was used to effect anionic addition of lithiated methyl tetronate 1218 to 20. MnO2 oxidation yielded acyltetronate 11 in 55% yield over 2 steps (60% brsm). All the above reactions were performed at decagram scales.
Scheme 3.

Synthesis of Acyltetronate 11
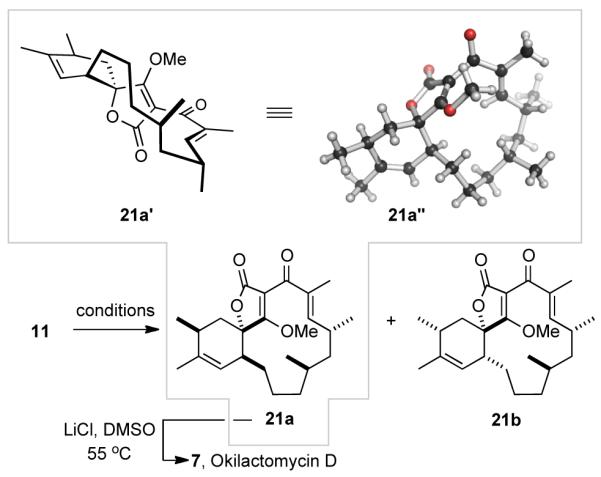
We were pleased to observe that a sample of 11 stored as a solution in CH2Cl2 at room temperature for several weeks had spontaneously undergone ca. 10% conversion to 21a (Table 1, entry 1). The relative configuration of this product was identical to that in okilactomycin D (7), as shown by subsequent single crystal X-ray crystallographic analysis (cf. Table 1 graphic, 21a’ and 21a“). The IMDA reaction of 11 was driven to completion by heating in toluene at 110 °C for 4 days.19 Careful chromatographic analysis and purification (MPLC on silica gel) provided pure O-methyl okilactomycin D (21a) as the dominant diastereomer in 62% yield, along with a mixture of three additional, coeluting, minor isomers. 20 Vapor diffusion crystallization (ethyl acetate vs. cyclohexane, Supporting Information) enabled isolation of the second most dominant component, 21b, from this mixture, and its relative configuration was also determined through X-ray crystallographic analysis. The major and minor isomers from these reactions–21a and 21b, respectively–arise from endo vs. exo (with respect to the endocyclic furanone alkene) addition of the diene to the dienophilic exo-methylene group.
Table 1.
IMDA Reaction of 11: Conditions and Outcomes
 | ||||
|---|---|---|---|---|
| entry | cond[a] | time | 21a:21b[b] | yield [%] (of 21a) |
| 1 | CH2Cl2, rt | 30 d | n.o.[c] | 10[d] |
| 2 | Toluene, 110 °C | 96 h | 8:1 | 62 |
| 3 | CH3OH/H2O, 90 °C | 6 h | 8:1 | 30-55 |
reactions were carried out at 0.01M concentration.
product ratios are determined by 1H NMR analysis.
not observed.
ca. 10% conversion based on 1H NMR analysis.
It is noteworthy that use of a protic solvent (2:1 MeOH/H2O) accelerated the rate of the IMDA reaction21 of 11. In situ 1H NMR monitoring indicated that the reaction in a 3:1 d4-methanol/D2O solution at 90 °C was ca. 30 times faster than that in d8-toluene at 110 °C. The reaction in this protic medium proceeded with a similar level of diastereoselectivity as in toluene, but with a lower yield of isolable material (Table 1, cf. entry 2 vs. entry 3).
To complete the total synthesis of okilactomycin D (7), the methyl ether 21a (200 mg) was dissolved in DMSO (15 mL) and LiCl (15 equiv) was added (Table 1 graphic). Once the mixture became homogenous, the solution was warmed to 55 °C for 48 h. Partitioning between water and ethyl acetate and washing the organic phase with brine resulted in isolation of the conjugate base of okilactomycin D (7), presumably as its sodium salt (see S-5 in Supporting Information). Alternatively, partitioning of the initial reaction mixture between 10% HCl (aq) and ethyl acetate cleanly gave okilactomycin D (7) directly as the neutral acyltetronic acid.
We then synthesized enantiomerically enriched 7, starting from the non-racemic 5-iodopentane derivative (+)-(2R,4S)-15.14c The resulting synthetic sample of okilactomycin D gave essentially identical 1H and 13C NMR spectral data and had the same sign of optical rotation as that of the natural sample22 {[α]Dsynthetic = −32 (c = 0.3, MeOH); lit.6 [α]Dnatural = −50 (c = ca. 0.1, MeOH)}, supporting the assigned absolute configuration of (−)-7 (cf. Figure 1).
In summary, we have demonstrated that the IMDA reaction of 11 is well-suited for construction of the macrocyclic ring in okilactomycins. This has enabled a scalable and efficient synthesis (ca. 17% overall yield) of (±)-okilactomycin D (7) comprising 17 steps (13 in its longest linear sequence) from commercially available starting materials. Synthesis of non-racemic (−)-7 corroborated the assigned absolute configuration of the natural material, thereby establishing that, indeed, okilactomycin [(+)-2] and okilactomycin D [(−)-7] share the same absolute configuration but differ in the sign of their optical rotation. Given the ready availability of ample quantities of late stage intermediates, we are studying other transforming reactions with an eye toward accessing additional members of the okilactomycin family.
Supplementary Material
Acknowledgment
This investigation was supported by a grant awarded by the National Cancer Institute (CA-76497) of the United States National Institutes of Health. We thank Gregory Rohde and Victor G. Young, Jr. (X-ray Crystallographic Facility, Department of Chemistry, Univeristy of Minnesota) for assistance with X-ray crystallographic analyses.
Footnotes
Supporting Information Available Experimental procedures and spectral data for all new compounds are provided. This material is available free of charge via the internet at http://pubs.acs.org.
references
- (1).Kang M, Jones BD, Mandel AL, Hammons JC, DiPasquale AG, Rheingold AL, La Clair JJ, Burkart MD. J. Org. Chem. 2009;74:9054–9061. doi: 10.1021/jo901826d. [DOI] [PubMed] [Google Scholar]
- (2).Igarashi Y, Ogura H, Furihata K, Oku N, Indananda C, Thanmchaipenet A. J. Nat. Prod. 2011;74:670–674. doi: 10.1021/np100727h. [DOI] [PubMed] [Google Scholar]
- (3).Kawashima A, Nakamura Y, Ohta Y, Akama T, Yamagishi M, Hanada K. J. Antibiot. 1992;45:207–212. doi: 10.7164/antibiotics.45.207. [DOI] [PubMed] [Google Scholar]
- (4).Imai HS, Suzuki KI, Morioka M, Numasaki Y, Kadota S, Nagai K, Sato T, Iwanami M, Saito T. J. Antibiot. 1987;40:1475–1482. doi: 10.7164/antibiotics.40.1475. [DOI] [PubMed] [Google Scholar]
- (5).Nakai R, Kakita S, Asai A, Chiba S, Akinaga S, Mizukami T, Yamashita Y. J. Antibiot. 2001;54:836–838. doi: 10.7164/antibiotics.54.836. [DOI] [PubMed] [Google Scholar]
- (6).Zhang C, Ondeyka J, Zink D, Basilio A, Vicente F, Salazar O, Genilloud O, Dorso K, Motyl M, Byrne K, Singh S. J. Antibiot. 2009;62:55–61. doi: 10.1038/ja.2008.8. [DOI] [PubMed] [Google Scholar]
- (7).The structure for 7 is arbitrarily (and for convenience) portrayed as an endocyclic enol; an internally hydrogen-bonded variant of that endocyclic enol (a rotamer about the C10-C11 bond) or E- or Z-exocyclic enols are also possible.
- (8).(a) Smith AB, Basu K, Bosanac T. J. Am. Chem. Soc. 2007;129:14872–14874. doi: 10.1021/ja077569l. [DOI] [PubMed] [Google Scholar]; (b) Smith AB, Bosanac T, Basu K. J. Am. Chem. Soc. 2009;131:2348–2358. doi: 10.1021/ja8084669. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tenenbaum JM, Morris WJ, Custar DW, Scheidt KA. Angew. Chem., Int. Ed. 2011;50:1–5. doi: 10.1002/anie.201102037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang H, White-Phillip J, Melançon C, Kwon H, Yu W, Liu H. J. Am. Chem. Soc. 2007;129:14670–14683. doi: 10.1021/ja0744854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jia X, Tian Z, Shao L, Qu X, Zhao Q, Tang J, Tang G, Liu W. Chem. Biol. 2006;13:575–585. doi: 10.1016/j.chembiol.2006.03.008. [DOI] [PubMed] [Google Scholar]
- (11).(a) Zapf CW, Harrison BA, Drahl C, Sorensen EJ. Angew. Chem. Int. Ed. 2005;44:6533–6537. doi: 10.1002/anie.200502119. [DOI] [PubMed] [Google Scholar]; (b) Snider BB, Zou Y. Org. Lett. 2005;7:4939–4941. doi: 10.1021/ol0518941. [DOI] [PubMed] [Google Scholar]; (c) Couladouros EA, Bouzas EA, Magos AD. Tetrahedron. 2006;62:5272–5279. [Google Scholar]
- (12).Other IMDA attempts have been reported too. For example: Takeda K, Shimotani A, Yoshii E. Heterocycles. 1992;34:2259–2261. Takeda K, Igarashi Y, Okazaki K, Yoshii E, Yamaguchi K. J. Org. Chem. 1990;55:3431–3434.
- (13).Jung ME, Miller SJ. Heterocycles. 1990;30:839–853. [Google Scholar]
- (14).Racemic 15 was synthesized in 3 steps (see Supporting Information) from the easily accessible14a,14b (or commercially available) cis-3,5-dimethyl glutaric anhydride. Enantiomerically pure 15 was prepared in 5 steps14c from this same anhydride. Racemic 15 was used to pioneer the initial route because of its easier availablity. Paquette LA, Boulet SL. Synthesis. 2002:888–894. Prusov E, Rohm H, Maier ME. Org. Lett. 2006;8:1025–1028. doi: 10.1021/ol052917e. Takagi R, Tsuyumine S, Nishitani H, Miyanaga W, Ohkata K. Aust. J. Chem. 2004;57:439–447.
- (15).Isomerization of the terminal double bond may derive from a cyclopropane ring-forming, bond rotation, ring-opening sequence. E.g., see: Tan Z, Negishi E. Org. Lett. 2006;8:2783–2785. doi: 10.1021/ol060856u.
- (16).Smonou I, Khan S, Foote CS, Elemes Y, Mavridis IM, Pantidou A, Orfanopoulos M. J. Am. Chem. Soc. 1995;117:7081–7087. [Google Scholar]
- (17).Clemo NG, Pattenden G. Tetrahedron Lett. 1982;23:585–588. [Google Scholar]
- (18).Montgomery LJ, Challis GL. Synlett. 2008:2164–2168. [Google Scholar]
- (19).Compound 11 has been demethylated under standard conditions (LiCl, DMSO, 50 °C, 4h). The resulting tetronate anion was found to be inert, even at elevated temperatures (sealed vial: d6-DMSO or CDCl3 at 100 °C, d8-PhMe at 150 °C, and CD3OD or D2O at 175 °C). On the other hand, the neutral tetronic acid decomposed (as evidenced by broadening over time of resonances throughout the 1H NMR spectrum) before giving any evidence of cyclization to a discrete compound.
- (20).It is relevant that the minor 2Z,4E-diene isomer (28-Z) was typically observed in the crude IMDA reaction product mixture, and it was isolated and characterized from a thermal reaction in toluene (Supporting Information). Thus, the E-isomer 28-E reacts faster than 28-Z, which is consistent with the expectation/assumption that this IMDA is a concerted process.
- (21).Blokzijl W, Blandamer MJ, Engberts JBFN. J. Am. Chem. Soc. 1991;113:4241–4246. [Google Scholar]
- (22).The sample of natural okilactomycin D was isolated after a final purification by HPLC using an eluent doped with trifluoroacetic acid. Thus, its structure is best formulated as 7, having the neutral acyltetronic acid. We observed that the 1H NMR spectrum of the sodium salt S-5 in CD3OD (or of the analogous cesium salt in CDCl3) contained a resonance for the enone β-proton (H6) that was ca. 0.6 (or 0.2) ppm upfield of that for the sample of neutral 7 (see Supporting Information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.