

Abstract
Legumes (Fabaceae or Leguminosae) are unique among cultivated plants for their ability to carry out endosymbiotic nitrogen fixation with rhizobial bacteria, a process that takes place in a specialized structure known as the nodule. Legumes belong to one of the two main groups of eurosids, the Fabidae, which includes most species capable of endosymbiotic nitrogen fixation 1. Legumes comprise several evolutionary lineages derived from a common ancestor 60 million years ago (Mya). Papilionoids are the largest clade, dating nearly to the origin of legumes and containing most cultivated species 2. Medicago truncatula (Mt) is a long-established model for the study of legume biology. Here we describe the draft sequence of the Mt euchromatin based on a recently completed BAC-assembly supplemented with Illumina-shotgun sequence, together capturing ~94% of all Mt genes. A whole-genome duplication (WGD) approximately 58 Mya played a major role in shaping the Mt genome and thereby contributed to the evolution of endosymbiotic nitrogen fixation. Subsequent to the WGD, the Mt genome experienced higher levels of rearrangement than two other sequenced legumes, Glycine max (Gm) and Lotus japonicus (Lj). Mt is a close relative of alfalfa (M. sativa), a widely cultivated crop with limited genomics tools and complex autotetraploid genetics. As such, the Mt genome sequence provides significant opportunities to expand alfalfa’s genomic toolbox.
Based on optical mapping, the eight pseudomolecules of assembly Mt3.5 span a physical distance of 375 million base pairs (Mb), while fluorescence in situ hybridization indicates they extend from pericentromeres almost to telomeric ends (Figures S1, S2). Altogether, Mt3.5 consists of 2,536 BACs (Tables S1, S2) with 273 physical gaps (including centromeres) (Table S3) and 101 internal sequencing gaps. The pseudomolecules contain 246 Mb of nonredundant sequence (Table S2) located entirely within the optical map (Figure S3). Another 146 unfinished BACs/BAC pools that cannot be placed on the optical map contribute 17.3 Mb. Regions not represented in pseudomolecules or unanchored BACs were captured through assembly of ~40x coverage Illumina sequencing, yielding 104.2 Mb of additional unique sequence. Though not directly tested, the Illumina sequence is expected to lie predominantly within the boundaries of pseudomolecules (See below). Based on EST alignments, the combined datasets capture ~94% of expressed genes, providing a highly informative, though still draft stage platform for analyzing the euchromatin of Mt.
Altogether there are 62,388 gene loci in Mt3.5 (Table S4, Figure S4) with 14,322 gene predictions annotated as transposons. Pseudomolecules and unassigned BACs contain a total of 44,124 gene loci, 177,271 retroelement-related regions, and 26,487 DNA transposons, while non-redundant Illumina assemblies contribute an additional 18,264 genes, 75,777 retrotransposon regions, and 8,476 DNA transposons (Tables S5-S9) along with 1,418 organellar insertions (Datafile S1). For pseudomolecules and unassigned BACs, this translates to 16.8 genes, 67.6 retrotransposons and 10.1 DNA transposons per 100 kbp. Within Illumina sequence assemblies, gene density (17.1 per 100 kbp) and retrotransposon density (72.2 per 100 kbp) are similar to pseudomolecules and unassigned BACs, while DNA transposon density is somewhat lower (8.2 per 100 kbp). Similarities in gene and transposon densities between BAC and Illumina sequences support the assertion that the Illumina sequence is euchromatic, though the possibility that some Illumina assemblies come from low copy regions within heterochromatin can not be excluded. Considering only the 47,845 genes with experimental or database support (Table S4), the average Mt gene is 2,211 bp in length, contains 4.0 exons, and has a CDS of 1,001 bp. These values are similar to those observed previously in Arabidopsis thaliana (At) (2,174 bp), Oryza sativa (3,403 bp) and Populus trichocarpa (Pt) (2,301 bp) 4-6.
Recent analyses of plant genomes indicate a shared whole genome hexaploidy (WGH) preceding the rosid-asterid split at 140-150 Mya 7. Duplication patterns and genomic comparisons strongly suggest an additional WGD ~58 Mya in the papilionoids 8, 9. Near the time of this WGD, papilionoids radiated into several clades, the largest of which split quickly into two subclades, the Hologalegina (including Mt and Lj) and the milletioids (including Gm and other phaseoloids) at ~54 Mya 2. We therefore compared Mt pseudomolecules with other sequenced plant genomes to learn more about shared synteny and genome duplication history.
There is significant macrosynteny among Mt, Lj and Gm (Figure 1, Figure S5a-b). Conserved blocks, sometimes as large as chromosome arms, span most euchromatin in all three genomes. A given Mt region is typically syntenic with one other Mt region as a result of the ~58 Mya WGD, usually in small blocks showing degraded synteny (Figure 2, Figure S6). A given Mt region is most similar to two Gm regions via speciation at ~54 Mya and the Glycine WGD at <13 Mya 10 and less similar to two other Gm regions resulting from the ~58 Mya and <13 Mya WGD events. A Mt region is likewise most similar to one Lj region via speciation at ~50 Mya and somewhat less similar to a second Lj region as a result of the ~58 Mya WGD. Finally, each Mt region and its homoeologue typically exhibit similarity to three Vitis vinifera (Vv) regions via the pre-Rosid WGH. Exceptions to these patterns could be due to gene losses, gains, or rearrangements specific to the Mt lineage, resulting in synteny being more evident between Mt and other genomes than in self-comparisons. Indeed, self-comparisons within Mt reveal few remnants of the legume specific WGD (Figure 2, Figures S6). While this seems paradoxical, it is probably explained by extensive gene fractionation between WGD-derived homoeologues in Mt. In Figure 3, two short regions on Mt01 and Mt03 resulting from the ~58 Mya WGD are displayed beside microsyntenic regions of Gm and Vv. As expected, many genes are microsyntenic between Mt and Gm (ranging from 7/19 between Mt03 and Gm14 to 10/20 between Mt01 and Gm17). Between the two Mt homoeologues, however, only 6 out of 33 genes (or collapsed gene families) are microsyntenic, with a homoeologue missing from one or the other duplicate (Table S10). Apparently, there have been many more changes, large and small, in Mt than in Gm since the legume WGD. This is borne out by the fact that synteny blocks in Mt are one-third the length of those remaining from the papilionoid WGD in Gm (524 kb vs. 1503 kb) with the average number of homologous gene pairs per block correspondingly lower (12.4 vs. 31.0).
Figure 1. Circos diagram illustrating syntentic relationships between Medicago, Glycine, Lotus and Vitis.
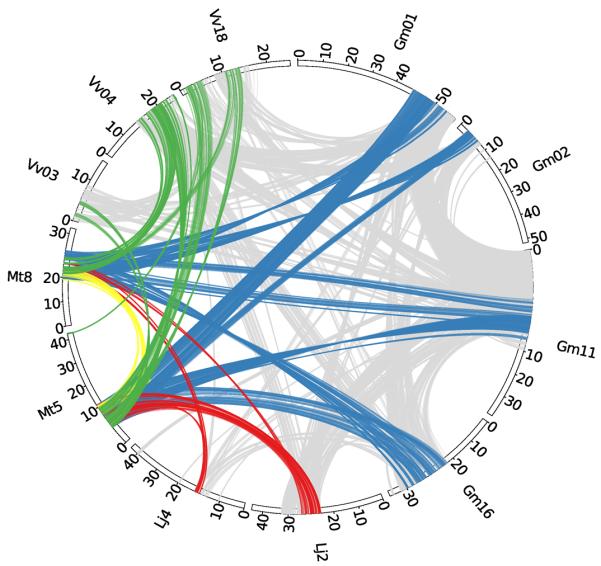
Homologous gene pairs were identified for all pairwise comparisons between Mt, Gm, Lj and Vv genomes. Syntenic regions associated with the ancestral WGD events were identified by visually inspection of correponding dot-plots. The large Mt5–Mt8 synteny block (yellow) was found to have two syntenic regions in Lj (red), four syntenic regions in Gm (blue) and three in Vv (green).
Figure 2. Circos diagram illustrating the Medicago WGD and selected gene families.
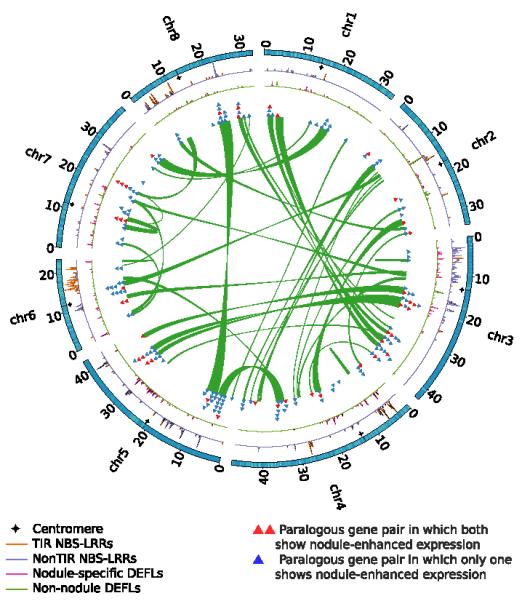
The 963 WGD-derived paralogous gene pairs were examined for overlap with the nodule-enhanced gene list (Datafile S2). Resulting gene pairs were joined and plotted as either blue circles (only one of the duplicates is nodule-enhanced) or red (both nodule enhanced). Gene densities of NBS-LRRs, NCRs and other defensin-like proteins are plotted against chromosome position. Density was calculated using a sliding window (100 kb window with 50 kb steps).
Figure 3. Microsynteny comparison between Medicago homoeologues and corresponding regions of Glycine and Vitis.
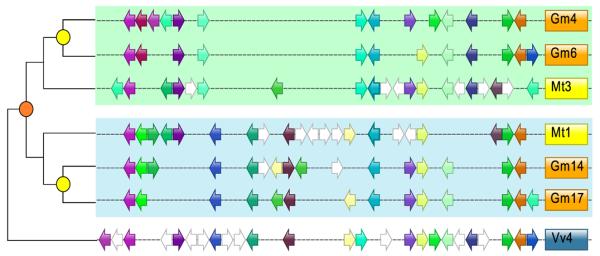
Microsyntenic genome segments are centered around Medtr3g104510/Medtr1g015890 (Table S10), a duplicated region derived from the ~58 Mya WGD event noted in orange. The <13 Mya Gm-specific WGD is colored yellow. Orthologous/paralogous gene pairs are indicated through use of a common color. White arrows represent genes with no syntenic homologue(s) in this genome region. Gray arrow indicates a Mt gene model with syntenic homologue in soybean, but no soybean gene model in the current annotation (www.phytozome.net/soybean).
The Mt genome also has undergone high rates of local gene duplication. The ratio of related genes within local clusters compared to all genes in families is 0.339 in Mt, 3.1-fold higher than in Gm and 1.6-fold higher than in At or Pt. (“Local clusters” are defined as genes in a family all within 100 gene models of one another.) The excess of local gene duplications in Mt is observed genome-wide and affects many families. There are 2.63 times as many gene families with local duplications in Mt compared with Gm (2,980 vs. 1,131), an excess that also is seen in detailed comparisons of syntenic regions in Mt and Gm. We examined 16.3 Mbp of Mt05 showing synteny to two large regions of Gm01 plus homoeologous blocks on Gm02, Gm09 and Gm11. In these regions, 25.8% of Mt genes are locally duplicated compared with just 8.0% in Gm. Local gene duplications and losses have contributed both to synteny disruptions (Figure 3, Figure S7) and to high gene count (62,388) in Mt — a value nearly as high as the 65,781 total gene models in Gm despite its additional (<13 Mya) WGD. Local gene duplications are evident in certain gene families, such as F-box genes, which have undergone dramatic expansions (Figure S8, Table S11). Mt also has experienced higher rates of base substitution compared to other plant genomes (Figure S9). Assuming 58 Mya as the date of the legume WGD, then the rate of synonymous substitutions per site per year (ss/s/yr) in Mt is 1.08 × 10−8, 1.8 times faster than estimates in other vascular plants 11. Higher rates of mutation and greater levels of rearrangement in Mt following the papilionoid duplication may have been driven by factors including short generation times, high selfing rates or small effective population sizes, though these characteristics are not unique to Mt.
Legumes and actinorhizal species are capable of forming a specialized organ, the root nodule, a highly differentiated structure hosting nitrogen-fixing symbionts. Phylogenetic studies suggest that nodulation may have evolved multiple times in the Fabidae, but the observation that all nodulating species are contained within this single clade implies a predisposition to nodulate evolved in their common ancestor 12. It is unknown whether nodulation with rhizobia preceded the divergence of the three legume subfamilies or evolved on multiple occassions 13. Nevertheless, rhizobial nodulation and the 58 Mya WGD are features common to most papilionoid legumes and both occurred early in the emergence of the group 2. Given that WGDs generate genetic redundancy that potentially facilitates the emergence of novel gene functions without compromising existing ones 14, we examined the Mt genome to ask whether the 58 Mya WGD might have played a role in the evolution of rhizobial nodulation in Mt and its relatives.
Nod factors are bacterial signalling molecules that initiate nodulation. Previous studies have shown that several of the plant components involved in the response to Nod factors also function in mycorrhizal signaling 15. However, some Nod factor receptors and transcription factors (TFs) have distinctly nodulation-specific functions. Among these nodulation-specific components, we found the Nod factor receptor, NFP, and the transcription factor, ERN1, each have paralogs, LYR1 and ERN2 respectively, that trace back to the papilionoid WGD based on genome location and Ks values (Figure S10, Datafile S2). Both sets of gene pairs also exhibit contrasting expression patterns and functional specialization. NFP and ERN1 are expressed predominantly in the nodule and are known to function in nodulation 16, 17, while LYR1 and ERN2 are highly expressed during mycorrhizal colonization (Figure S11). These observations indicate that two important nodulation-specific signaling components in Mt might have evolved from more ancient genes originally functioning in mycorrhizal signaling and then duplicated by the 58 Mya WGD. In the case of MtNFP/MtLYR1, this conclusion is supported by the observation that the apparent ortholog of NFP in the nodulating non-legume Parasponia andersonii functions in both nodule and mycorrhizal signaling 18. Thus, the 58 Mya WGD appears to have led to sub-functionalization of an ancestral gene participating in both interactions, resulting in two homoeologous genes that each performs just one of the original functions.
To further assess the contribution of the WGD to Mt nodulation, we analyzed expression of paralogous gene pairs using RNA-seq data from six organs (Supplemental Methods S5.1). A total of 963 WGD-derived gene pairs were found (Datafile S2) with 618 pairs (1,046 genes) having RNA-seq data for one or both homoeologue. We then determined the number of genes showing organ-enhanced expression (defined as genes with expression level in a single organ at least twice the level in any other) within the pseudomolecule and the WGD-derived gene sets (Table S12). In both cases, different organs contained markedly different numbers of genes with enhanced expression (Χ2 with 5 df, p = 1E-272), however the rank order among the organs was identical. Roots exhibited the largest number of genes with enhanced expression followed by flower, nodule, leaf, seed/pod and bud. Among gene pairs with nodule-enhanced expression, both paralogs were nodule-enhanced in eight pairs, while just a single paralog was nodule-enhanced in the other 43 pairs. This is consistent with nodulation predating the WGD and further sub- and neo-functionalization emerging afterwards. We went on to examine TFs since they can act as regulators of plant growth and development. A total of 3,692 putative TF genes were discovered (Datafile S3), representing 5.9% of all Mt gene models (Table S13). Of the 1,513 TF genes on pseudomolecules with RNA-seq data, 142 genes (9.4%) derived from the 58 Mya WGD (Figure S12, Datafile S4), consistent with previous observations indicating greater retention of TFs following polyploidy 19. Nodule-enhanced expression was significantly higher among TFs (92/1,513 or 6.1%) than among all pseudomolecule genes (1,111/23,478 or 4.7%) (Χ2 with 1 df, p = 0.024) (Table S12). Nodule-enhanced expression was even higher in WGD-derived TFs (11/142 or 7.7%), although this enrichment did not reach statistical significance (p = 0.113). As expected, ERN1 is found within this group of WGD-retained, nodule-enhanced TFs.
These results show that many paralogous genes retained from the 58 Mya WGD, especially signaling components and regulators, have undergone sub- or neo-functionalization, including several with specialized roles in nodulation. Nevertheless, separate phylogenetic analyses (Supplemental Methods S5.5) indicate that some nodule-related genes derive from the more ancient pre-Rosid WGH, with their nodule-related functions pre-dating the 58 Mya WGD (Datafile S5). Taken together, these results are consistent with a model where the capacity for primitive interaction with new symbionts derived from existing mycorrhizal machinery involving genes recruited from the pre-Rosid WGH. This capacity would have arisen early in the Fabidae clade and led to the appearance of nodulation in multiple lineages 13, 20. Later, the 58 Mya WGD would have resulted in additional genes, including NFP, ERN1 and the TFs described above, that went on to become specialized for nodule-related functions in the Papilionoideae.
Medicago contains additional amplified gene families, many nodulation-related and found in tandem clusters. Mt has nine symbiotic leghemoglobins, more than twice the number in Lj or Gm (Figure S13). Five of these genes are located in a tight cluster on Mt5. The Mt genome contains 593 nodule cysteine rich peptides (NCRs) (Datafile S6), a gene family restricted to Mt and its relatives 21. NCRs are noteworthy because they include members essential for terminal differentiation of rhizobia 22. NCRs are tightly clustered within the Mt genome (Figure 2), with 75% found in clusters of up to 11 members. The Mt genome also has 764 NBS-LRR genes (Datafile S7), more than other sequenced plant genomes to date 23-25, many with nodule-specific expression (Figure S14). Almost 90% of NBS-LRRs occur in clusters and genome regions showing limited macrosynteny to other species, such as Mt3 and Mt6, are locations of large NBS-LRR superclusters (Figure 2, Tables S14, S15). Finally, Mt secretes flavonoid signaling molecules to induce the nod genes of Sinorhizobium meliloti 26. In Mt, the corresponding biosynthetic pathway has expanded dramatically, with 28 Mt chalcone synthase genes in clusters of up to seven members compared to just four chalcone synthases in At 27 (Datafile S8). Mt has ten chalcone reductases compared to none in At 28 and Mt has 11 chalcone isomerase genes, including one cluster of seven members, compared to just one representative in At 29 (Figures S15, S16).
In summary, analysis of the Mt genome supports earlier studies indicating that the dramatic radiation of the legume family (at least the papilionoid subfamily) is partly attributed to the 58 Mya WGD 30. Our results suggest that the WGD early in papilionoid evolution allowed the emergence of critical components in Nod factor signaling and contributed to the complexity of rhizobial nodulation observed in this clade. As such, the WGD appears to have played a crucial role in the success of papilionoid legumes, enhancing their utility to humans.
METHODS SUMMARY
DNA sequencing
Six A17 BAC and one fosmid library were used to create Mt3.5 (Table S1). Most were processed by Sanger paired-end sequencing of 3-6 kb shotgun libraries. Sequences were downloaded in February/March 2009 with scaffolding performed by aligning all BAC and fosmid ends against contigs and then anchored and ordered primarily by optical mapping. Separately, 25 Gb of Illumina sequence was generated using short (375 nt) inserts plus 2.1 Gb from a 5 kb mate-pair library, then assembled using CLCbio (www.clcbio.com) and Soap (http://soap.genomics.org.cn/).
RNA sequencing
Five tissues were used for RNA-seq analysis with ~10 million Illumina 36 bp reads per library (Table S12). Three tissues were used for small RNA analysis with ~3 million reads per Illumina library (Figures S17-S18, Table S16, Datafile S9).
Supplementary Material
Acknowledgements
Funding support to N.D.Y, C.D.T, B.A.R. from The Noble Foundation and NSF-PGRP 0321460, 0604966; To N.D.Y. J.M., G.D.M. from NSF-PGRP 0820005; To C.D.T. from NSF-PGRP 0821966; To F.D., G.O., R.G., K.F.X.M., T.B., J.D., F.Q., J.R. from FP6 EU project GLIP/Grain Legumes FOOD-CT-2004-506223; To G.O., J.R. from BBSRC BBS/B/11524; To F.D., F.Q. from ANR project SEQMEDIC 2006-01122; To R.G. from Dutch Science Organization VIDI 864.06.007, ERA-PG FP-06.038A; To Y.V.D.P. from Belgian Federal Science Policy Office IUAP P6/25, Fund for Scientific Research Flanders, Institute for the Promotion of Innovation by Science and Technology in Flanders and Ghent University (MRP N2N); To D.R.C. from NSF IOS-0531408, IOS-0605251; To D.J.S., B.C.M., P.J.G. from USDA CSREES 2006-03567. We also thank Y.W. Nam for a BamHI BAC library used by Genoscope, Sunhee Park and Monica Accerbi for RNA isolation, Tim Paape for statistical consulting, and Maria Harrison for supplying myc infected and control root tissues used to make small RNA libraries.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Information. Medicago truncatula pseudomolecules are found at DDBJ/EMBL/GenBank as accessions CM001217-CM001224 and unanchored BACs as GL982851-GL982996. Illumina genome sequences are in the Short Read Archive under SRS150378, RNA-seq sequences under SRP008485, and small RNA sequences under GSM769273, GSM769274 and GSM769276. Pseudomolecule annotation and Illumina assemblies are available at ftp://ftp.jcvi.org/pub/data/m_truncatula/Mt3.5/.
Reprints and permissions information are available at www.nature.com/reprints.
The authors declare no competing financial interests.
This paper is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike license and is freely available to all readers at www.nature.com/nature.
References
- 1.Wang H, et al. Rosid radiation and the rapid rise of angiosperm-dominated forests. Proc. Natl. Acad. Sci. U. S. A. 2009;106:3853–3858. doi: 10.1073/pnas.0813376106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavin M, Herendeen PS, Wojciechowski MF. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the tertiary. Syst. Biol. 2005;54:575–594. doi: 10.1080/10635150590947131. [DOI] [PubMed] [Google Scholar]
- 3.Kulikova O, et al. Integration of the FISH pachytene and genetic maps of Medicago truncatula. Plant J. 2001;27:49–58. doi: 10.1046/j.1365-313x.2001.01057.x. [DOI] [PubMed] [Google Scholar]
- 4.The Arabidopsis Genome Initiative, I Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- 5.International Rice Genome Sequencing Project The map-based sequence of the rice genome. Nature. 2005;436:793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 6.Tuskan GA, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 7.Tang H, et al. Unraveling ancient hexaploidy through multiply-aligned angiosperm gene maps. Genome Res. 2008;18:1944–1954. doi: 10.1101/gr.080978.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeil BE, Schlueter JA, Shoemaker RC, Doyle JJ. Placing paleopolyploidy in relation to taxon divergence: a phylogenetic analysis in legumes using 39 gene families. Syst. Biol. 2005;54:441–454. doi: 10.1080/10635150590945359. [DOI] [PubMed] [Google Scholar]
- 9.Cannon SB, et al. Polyploidy did not predate the evolution of nodulation in all legumes. PLoS One. 2010;5:e11630. doi: 10.1371/journal.pone.0011630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmutz J, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463:178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 11.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 12.Soltis DE, et al. Chloroplast gene sequence data suggest a single origin of the predisposition for symbiotic nitrogen fixation in angiosperms. Proc. Natl. Acad. Sci. U. S. A. 1995;92:2647–2651. doi: 10.1073/pnas.92.7.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doyle JJ, Luckow MA. The rest of the iceberg. Legume diversity and evolution in a phylogenetic context. Plant Physiol. 2003;131:900–910. doi: 10.1104/pp.102.018150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeling M, Thomas BC. Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res. 2006;16:805–814. doi: 10.1101/gr.3681406. [DOI] [PubMed] [Google Scholar]
- 15.Oldroyd GE, Downie JA. Coordinating nodule morphogenesis with rhizobial infection in legumes. Annu. Rev. Plant. Biol. 2008;59:519–546. doi: 10.1146/annurev.arplant.59.032607.092839. [DOI] [PubMed] [Google Scholar]
- 16.Arrighi JF, et al. The Medicago truncatula lysin [corrected] motif-receptor-like kinase gene family includes NFP and new nodule-expressed genes. Plant Physiol. 2006;142:265–279. doi: 10.1104/pp.106.084657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Middleton PH, et al. An ERF transcription factor in Medicago truncatula that is essential for Nod factor signal transduction. Plant Cell. 2007;19:1221–1234. doi: 10.1105/tpc.106.048264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Op den Camp R, et al. LysM-type mycorrhizal receptor recruited for rhizobium symbiosis in nonlegume Parasponia. Science. 2011;331:909–912. doi: 10.1126/science.1198181. [DOI] [PubMed] [Google Scholar]
- 19.Thomas BC, Pedersen B, Freeling M. Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 2006;16:934–46. doi: 10.1101/gr.4708406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kistner C, Parniske M. Evolution of signal transduction in intracellular symbiosis. Trends Plant Sci. 2002;7:511–518. doi: 10.1016/s1360-1385(02)02356-7. [DOI] [PubMed] [Google Scholar]
- 21.Kato T, et al. Expression of genes encoding late nodulins characterized by a putative signal peptide and conserved cysteine residues is reduced in ineffective pea nodules. Mol. Plant-Microbe Interact. 2002;15:129–137. doi: 10.1094/MPMI.2002.15.2.129. [DOI] [PubMed] [Google Scholar]
- 22.Van de Velde W, et al. Plant peptides govern terminal differentiation of bacteria in symbiosis. Science. 2010;327:1122–1126. doi: 10.1126/science.1184057. [DOI] [PubMed] [Google Scholar]
- 23.Meyers BC, Kozik A, Griego A, Kuang H, Michelmore RW. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell. 2003;15:809–834. doi: 10.1105/tpc.009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang S, Zhang X, Yue JX, Tian D, Chen JQ. Recent duplications dominate NBS-encoding gene expansion in two woody species. Mol. Genet. Genomics. 2008;280:187–198. doi: 10.1007/s00438-008-0355-0. [DOI] [PubMed] [Google Scholar]
- 25.Zhou T, et al. Genome-wide identification of NBS genes in japonica rice reveals significant expansion of divergent non-TIR NBS-LRR genes. Mol. Genet. Genomics. 2004;271:402–415. doi: 10.1007/s00438-004-0990-z. [DOI] [PubMed] [Google Scholar]
- 26.Peters NK, Frost JW, Long SR. A plant flavone, luteolin, induces expression of Rhizobium meliloti nodulation genes. Science. 1986;233:977–980. doi: 10.1126/science.3738520. [DOI] [PubMed] [Google Scholar]
- 27.Winkel-Shirley B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001;126:485–493. doi: 10.1104/pp.126.2.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hegnauer R. Relevance of seed polysaccharides and flavonoids for the classification of the leguminosae: A chemotaxonomic approach. Phytochemistry. 1993;34:3. [Google Scholar]
- 29.Shirley BW, et al. Analysis of Arabidopsis mutants deficient in flavonoid biosynthesis. Plant J. 1995;8:659–671. doi: 10.1046/j.1365-313x.1995.08050659.x. [DOI] [PubMed] [Google Scholar]
- 30.Singer SR, et al. Venturing beyond beans and peas: what can we learn from Chamaecrista? Plant Physiol. 2009;151:1041–1047. doi: 10.1104/pp.109.144774. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.