

Abstract
Oncogenic c-Myc is known to balance excessive proliferation by apoptosis that can be triggered by p53-dependent and p53-independent signaling networks. Here, we provide evidence that the “BH3-only” pro-apoptotic Bcl-2 family members Bmf and Bad are potent antagonists of c-Myc-driven B cell lymphomagenesis. Tumor formation was preceded by accumulation of preneoplastic pre-B and immature IgM+ B cells in hematopoietic organs of Eμ-myc/bmf−/− mice, whereas Eμ-myc/bad−/− mice showed an increase of pre-B cells limited to the spleen. While loss of Bad had no impact on the tumor immunophenotype, Bmf-deficiency favored the development of IgM+ B cell over pre-B cell tumors. This phenomenon was due to a strong protection of immature IgM+ B cells from oncogene-driven apoptosis caused by loss of bmf and c-Myc-induced repression of Bmf expression in premalignant pre-B cells. Steady-state levels of B cell apoptosis were also reduced in the absence of Bad, in support of its role as a sentinel for trophic factor-deprivation. Loss of Bmf reduced the pressure to inactivate p53, whereas Bad-deficiency did not, identifying Bmf as a novel component of the p53-independent tumor suppressor pathway triggered by c-Myc.
Keywords: apoptosis, tumorigenesis, BH3-only proteins, c-Myc
Introduction
Defects in the ‘Bcl-2 regulated’ (‘intrinsic’ or ‘mitochondrial’) apoptosis pathway have been associated with cancer development, progression and drug-resistance. This apoptotic pathway is initiated when developmental cues, cytotoxic or oncogenic stress trigger activation of pro-apoptotic Bcl-2 family members of the BH3-only subgroup, such as Bim or Puma. This leads to activation of Bax/Bak proteins, the second pro-apoptotic subgroup of the Bcl-2 family, either by binding and neutralizing the function of Bcl-2-like pro-survival family members, including Bcl-2, Bcl-xL and Mcl-1, that sequester these molecules, or by their direct interaction with Bax 1,2. Subsequent oligomerization and pore formation by Bax and Bak causes mitochondrial outer membrane permeabilization (MOMP), allowing the release of apoptogenic molecules, including cytochrome c and smac/DIABLO, which promote activation of the proteolytic caspase cascade resulting in apoptotic cell death 1,2.
Eμ-Myc transgenic mice develop aggressive immature pre-B and IgM+ B cell lymphomas and are a potent model to study the molecular basis of c-Myc-driven malignancies 3,4. Disease pathogenesis in these mice resembles in certain aspects that of Burkitt lymphoma inasmuch that overexpression of c-Myc causes excessive proliferation of B cells, although of different developmental stages. This is initially balanced by massive apoptosis, until second genetic lesions, most commonly loss of p53 signaling or overexpression of Bcl-2 or Bcl-xL, blunts this response 5,6. Loss of p53 impedes c-Myc-driven apoptosis by inefficient induction of effectors of the intrinsic apoptosis pathway such as Puma 7,8, whereas overexpression of Bcl-2 or Bcl-xL not only blocks the proapoptotic potential of Puma but also that of another critical p53-independent sentinel of oncogenic stress, i.e. Bim 9,10. Consistently, loss of either BH3-only protein facilitates c-myc-driven lymphomagenesis in mice 10-12 and loss or decreased expression of Bim or Puma have been described in a number of human cancers 13. Consistent with their role as tumor suppressors in c-Myc-induced oncogenesis, Bim and Puma were found silenced in a portion of human Burkitt lymphoma 11,14. It is, however, currently unclear if also other BH3-only proteins can be engaged by c-Myc to prevent malignant transformation and if their absence may contribute to the pathogenesis of this disease.
Loss of the BH3-only protein Bmf in mice induces polyclonal B cell hyperplasia that is associated with decreased sensitivity of Bmf-deficient B cells to apoptosis-induction, although the physiological trigger during B cell development remains undefined 15. Consistently, Bmf has been implicated in cell death induction of primary chronic lymphocytic B cell leukemia (B-CLL) in humans 16 but also other tumor entities such as oral and esophageal squamous cell carcinoma cells 17. In contrast, mice deficient for Bad show normal lymphocyte development and number, but impaired B cell function 18. Serum deprivation rendered Bad-proficient, but not Bad-deficient mouse embryonic fibroblasts (MEF) more susceptible to the effects of death receptor ligation and Bad deficient MEF were more resistant to the combined effect of IGF-1 withdrawal and etoposide treatment. Bad-deficient mice were also reported to develop diffuse large B-cell lymphomas, with an incidence of about 20%, albeit late in life (latency > 15 months) 18, but evidence for a role of Bad in human lymphoid malignancies is currently lacking 19. However, loss-of-function mutations in the BH3-domain of Bad were reported in colon carcinoma patients and higher levels of Bad protein expression have been associated with better outcome in androgen dependent prostate cancer and in breast cancer 20-22. The pro-apoptotic potential of Bad is thought to be regulated in part by phosphorylation, leading to its cytoplasmatic sequestration and inactivation, which can be mediated by the lipid-activated protein kinase AKT/PKB 23, central to a signaling pathway frequently hyperactivated in human cancers 24.
To investigate the contribution of the BH3-only proteins Bad and Bmf in c-Myc-driven B cell lymphomagenesis we crossed bmf−/− and bad−/− mice with transgenic mice expressing the c-myc oncogene under control of the Eμ heavy chain enhancer and compared it to the effects observed in response to loss of bim, a well established tumor suppressor in this model system of c-Myc-driven malignant disease.
Material and methods
Mice
All animal experiments were approved by the Austrian Ministry for Education, Research and Culture. The generation and genotyping of the bmf−/−, bim−/−, bad−/−, and Eμ-myc transgenic mice have been described 4,15,18,25. All mice used were on an inbred C57BL/6 genetic background.
Cell culture and reagents
FACS-sorted pre-B, immature and mature IgM+ B cells were cultured in DMEM (PAA) supplemented with 10% FCS (PAA), 250μM L-glutamine (Gibco) and 50μM 2-mercaptoethanol. Isolated lymphoma cells were cultured on supporting irradiated NIH-3T3 cells. Source of reagents: Etoposide, Dexamethasone, Paclitaxel, 5-aza-2′-deoxycytidine (all from Sigma-Aldrich), Bortezomib/Velcade® (M. Ausserlechner, Dept. of Pediatrics, Innsbruck), ABT-737 (Steve Elmore, Abbott Pharmaceuticals), or SAHA (R.W. Johnstone, Peter MacCallum Cancer Center, Melbourne). Daudi, Raji and Ramos BL cell lines were maintained in RPMI 1640 medium (PAA) supplemented with 10% FCS and 250μM L-glutamine.
Flow cytometric analysis and cell sorting
The monoclonal antibodies used, and their specificities, are as follows: RA3-6B2, anti-B220; R2/60, anti-CD43; II/41, anti-IgM; 11/26C, anti-IgD; MB19-1, anti-CD19; 53-7.3, anti-CD5; AA4.1, anti-CD93; D7 anti-Sca-1; GK1.5, anti-CD4; H57-597, anti-TCRβ (all eBioscience); 53-6.7, anti-CD8; (all Becton Dickinson). Biotinylated antibodies were detected using streptavidin-RPE (DAKO) or streptavidin-PE-Cy7 (Becton Dickinson). HIB19, anti-human CD19 (eBioscience) was used for sorting B cells from peripheral blood. Sorting of cells was performed using a FACSVantage cell sorter (Becton Dickinson). In vivo BrdU-labeling was performed as previously described 26.
Immunoblotting
Western blotting was performed as previously described 15. Membranes were probed with rat anti-p19/ARF (5-C3-1), rabbit anti-p53 antiserum (FL-393) (Santa Cruz Biotechnology), monoclonal antibodies to Bcl-xL (54H6) (Cell Signaling), rabbit anti-Mcl-1 (Rockland), hamster anti-mouse Bcl-2 (3F11) and rat anti-mouse Bmf mAb (17A9) and rat anti-human Bmf (9G10) (a gift from A. Strasser). Equal loading of proteins was confirmed by probing filters with antibodies specific for β-actin (Sigma), GAPDH (Sigma) or MAPK (Cell Signaling). Horseradish peroxidase (HRP)-conjugated sheep anti-rat Ig antibodies (Jackson Research) rabbit anti-hamster antibodies (Southern Biotechnology) goat anti-rabbit or rabbit anti-mouse antibodies (DAKO) served as secondary reagents and the enhanced chemiluminiscence (ECL; Amersham) system was used for detection.
Cell viability assay
The percentage of viable cells in culture was determined by staining cell suspensions with 1μg/ml 7-AAD (Sigma) plus FITC-coupled Annexin-V (Beckton Dickinson) and analyzing the samples in a FACScan (Becton Dickinson).
Primary patient material
Material from Burkitt lymphoma cases (3, 8, 25, 35 and 45 years old male and one 18 years old female), diagnosed between 1991 and 2006 were collected from the tumor bank of the Institute of Pathology at the University Hospital of Basel. All cases fulfilled morphological and phenotypical criteria of Burkitt lymphoma and showed on revision c-myc rearrangements as assessed by a dual-color, break-apart probe from Vysis/Abott, Downers Grove, IL, USA; order no. 05J91-001. Five tumors were of primary extra nodal origin (each one of tonsillar, epidural- and cubital origin, and two of ileo-coecal origin), while one was primary nodal. Retrieval of tissue was according to the regulations of the local institutional review board and data safety laws.
Lentiviral transduction
Lentiviral transduction of BL cell lines with expression vectors encoding BMF-specific shRNA was performed as previously described 27.
Bisulfite modification and BMF DNA methylation analysis and quantitative analysis of BMF mRNA levels
See supplemental information
Statistical analysis
Estimation of statistical differences between groups was carried out using the unpaired Student t-test or ANOVA analysis, where appropriate. Comparison of tumor onset was performed using a log-rank test and the χ2-test was used for comparison of frequency distributions. P-values of <0.05 were considered to indicate statistically significant differences.
Results
Loss of Bmf or Bad accelerates the onset of lymphoma in Eμ-myc transgenic mice
To explore whether Bmf or Bad can act as tumor suppressors in oncogene-driven B cell lymphoma development, mice lacking the individual BH3-only proteins were crossed with Eμ-myc transgenic mice. For comparison, we also generated a cohort of mice expressing c-Myc on a bim+/− or bim−/− background 10. Cohorts of Eμ-myc transgenic mice lacking one or both alleles of either Bad or Bmf were monitored until onset of overt disease. Eμ-myc mice lacking one allele of bad did not contract disease significantly faster than wild type (wt) Eμ-myc mice (p=0.18, Fig 1A), while Eμ-myc/bad−/− mice showed a shortened survival (p<0.001, Fig 1A). Loss of one allele of bmf lead to a slight acceleration of lymphoma onset as compared to Eμ-myc mice (p<0.05, Fig 1B), which was further enhanced by loss of the second bmf allele (p<0.01, Fig1B). Interestingly, loss of both alleles of bmf or bad accelerated c-Myc-driven tumorigenesis significantly less efficiently than loss of one allele of bim (p<0.01 and p<0.0001 respectively, Figs. 1A, B). Notably, Eμ-myc mice deficient for both, Bad and Bmf, did not contract disease earlier than single-mutant animals expressing the transgene (Fig. 1C). Taken together, this demonstrates that both Bmf and Bad possess tumor suppressor potential, but are overall less potent than Bim, at least in this disease model. Furthermore, Bmf and Bad may act in a redundant manner in this process, or at different stages of B cell development.
Figure 1. Loss of bad or bmf accelerates c-myc-induced lymphomagenesis.

(A) Tumor free survival of Eμ-myc (n=29, median survival 138 days), Eμ-myc/bad+/− (n=47, median survival 100 days), Eμ-myc/bad−/− (n=29, median survival 78 days) and Eμ-myc/bim+/− (n=32, median survival 67 days). Lymphomas occurred significantly earlier in Eμ-myc/bad−/− than in wt Eμ-myc animals (p<0.001). (B) Tumor free survival of Eμ-myc, Eμ-myc/bmf+/− (n=30, median survival 100 days), Eμ-myc/bmf−/− (n=30, median survival 87 days) and Eμ-myc/bim+/−. Lymphomas occurred significantly earlier in Eμ-myc/bmf+/− (p<0.05) and Eμ-myc/bmf−/− (p<0.01) than in wt Eμ-myc animals. (C) Tumor free survival of Eμ-myc/bad−/−bmf+/+, Eμ-myc/bad−/−bmf+/− (n=16) and Eμ-myc/bad−/−bmf−/− mice (n=8). (D) Distributions of pro/pre-B (light grey), mixed (dark grey), IgM+ (black) and B220+CD4+ (white) lymphomas occurring in mice of the indicated genotypes. The distribution of lymphoma phenotypes was significantly different in Eμ-myc/bmf−/− compared to Eμ-myc animals (p<0.01, χ2-test). (E) Kaplan-Meier analysis of IgM+ and mixed lymphomas (upper panel) and of pre-B lymphomas (lower panel) of Eμ-myc (black solid line), Eμ-myc/bad+/− (light grey, dashed line), Eμ-myc/bad−/− (light grey, solid line), Eμ-myc/bmf+/− (dark grey dotted line) and Eμ-myc/bmf−/− (dark grey solid line) mice. IgM+ B lymphomas arose significantly earlier in Eμ-myc/bmf−/− (p<0.0001) and Eμ-myc/bad−/− (p<0.01) than in wt Eμ-myc mice. Pre-B lymphomas were not significantly accelerated by loss of either bad or bmf.
Loss of Bmf preferentially promotes development of IgM+ tumors in Eμ-myc transgenic mice
Tumors developing in Eμ-myc mice normally have a CD19+IgM− pre-B cell or an immature CD19+IgM+ B cell phenotype 4. Consistent with previously published data, immunophenotyping of the lymphomas revealed a frequency of ~60% pre-B cell tumors in the wt Eμ-myc mice (16/28), ~30% of all cases (8/28) were immature IgM+ B cell lymphomas and the remaining tumors displayed a mixed (pre-B/IgM+) phenotype (Fig. 1D). In strong contrast, Eμ-myc/bmf−/− mice developed predominantly IgM+ B cells lymphomas (20/30; 67%) and only 3/30 tumors (10%) were of pro/pre-B cell origin (Fig. 1D). The tumor spectrum observed in the Eμ-myc/bmf+/− mice was intermediate between wt and bmf−/− Eμ-myc mice with 10/29 cases (34%) being pro/pre-B cell lymphomas, demonstrating a clear gene-dosage effect (Fig. 1D). Similar observations were made in Eμ-myc mice lacking bim whereas the immunophenotype of bad+/− and bad−/− lymphomas mirrored that of wt Eμ-myc mice (Fig. 1D). Further analysis revealed that the observed acceleration of tumorigenesis was mainly due to an earlier onset of IgM+ lymphomas in all genotypes tested (Fig. 1E), as previously noted in Eμ-myc mice lacking Bim 10. Importantly, although not further reducing tumor latency, loss of Bad over Bmf again facilitated the development of pre-B tumors upon c-Myc overexpression (Fig. 1D). Together, this indicates that although Bmf and Bad are able to engage the same pro-survival molecules in vitro 30, the activities of both proteins are regulated differently during normal B cell maturation and/or in response to oncogenic stress in vivo.
Interestingly, Eμ-myc/bmf−/− and Eμ-myc/bmf+/− mice also showed an increased frequency of lymphomas that lacked expression of the B cell markers CD19 and IgM, but expressed B220, CD4, CD5, AA4.1 and Sca-1 (3/30 and 4/29, respectively, vs. 1/30 Eμ-myc mice, Fig. 1D). The immunophenotype of these lymphomas resembles that of lymphomas observed in Eμ-myc/Eμ-bcl-2 and Eμ-myc/Eμ-bcl-x double-transgenic mice 31,32, pointing towards a role for Bmf in apoptosis of early hematopoietic progenitors.
Bmf deficient Eμ-myc transgenic mice bear higher tumor load
A closer evaluation of the hematopoietic compartment of diseased mice revealed that loss of Bmf favored the development of leukemia in Eμ-myc transgenic mice. Compared to the mean white blood cell count of diseased Eμ-myc mice we observed a >4-fold increase in the number of circulating leukocytes in diseased transgenic mice lacking Bmf (p<0.0001) (Fig. 2A). In addition, Eμ-myc/bmf−/− mice had a significantly more pronounced splenomegaly than wt or Bad-deficient tumor mice (Fig. 2B). Taken together loss of Bmf significantly increased the tumor load of ill mice and favored development of IgM+ leukemia, similar to findings made previously in Eμ-myc/bim−/− mice 10 and recapitulated here (Figs. 2A, B).
Figure 2. Loss of bmf enhances the severity of Eμ-myc lymphomas.

(A) Numbers of total leukocytes in the blood of moribund mice of the indicated genotypes. The leukocyte counts were significantly higher bmf−/− Eμ-myc mice than in wt Eμ-myc mice (mean leukocyte count in Eμ-myc/bmf−/− mice was 263±251×106/ml versus 58±67×106/ml in Eμ-myc mice). Diamonds represent individual blood count of mice and bars the corresponding means (B) Spleen weights of moribund mice of the indicated genotypes. Loss of bmf but not loss of bad, lead to a significant increase in spleen size in moribund mice (mean spleen weight 0.599±0.253g in Eμ-myc/bmf−/−, vs. 0.395±0.140g in Eμ-myc or 0.428±0.160g in Eμ-myc/bad−/−). Diamonds represent individual spleen weights in mice of the indicated genotypes with bars indicating the corresponding means. * p<0.01, **p<0.001, ***p<0.0001 compared to Eμ-myc.
Loss of Bmf or Bad causes an accumulation of B cells in pre-leukemic mice
Young, healthy Eμ-myc transgenic mice display an expanded population of pre-B cells and immature IgM+ transitional (T1) B cells, caused by c-Myc-driven proliferation, leading to the accumulation of these cells in secondary lymphoid organs. In contrast, the number of mature B cells was reduced in Eμ-myc mice (Fig. 3A), as previously reported 4. The expansion of these premalignant cells is dampened for a limited period of time by c-Myc-induced apoptosis. While the increased numbers of pro/pre-B cells in the bone marrow were comparable between Eμ-myc, Eμ-myc/bmf−/− and Eμ-myc/bad−/− mice, we observed that the total B cell number in the spleens was significantly increased in Eμ-myc mice lacking Bad or Bmf (Fig. 3A). Interestingly, Eμ-myc/bad−/− mice showed increased pre-B cell numbers in the spleen when compared to Eμ-myc mice, while loss of Bmf favored the accumulation of more mature T1 B cells in Eμ-myc transgenic animals (Fig. 3A). Most striking, however, loss of Bmf caused an up to 10-fold increase in B cells of all differentiation stages in peripheral blood of premalignant Eμ-myc transgenic mice (7.3±3.5×106/ml vs. 75.5±8.0×106/ml), whereas loss of Bad had no such effect (Fig. 3A). This suggested that loss of either BH3-only protein facilitated B cell survival upon oncogenic stress, albeit at different developmental stages leading to the accumulation of the Eμ-myc transgenic B cells in premalignant animals.
Figure 3. Loss of Bmf enhances the survival of premalignant Eμ-myc B lymphocytes.

For analysis of pre-leukemic cells, lack of transplantable tumor cells was confirmed by injecting 2×106 spleen cells into wt C57BL/6 recipients followed for at least 2 months. (A) Cell number and B cell subset composition determined by cell counting and flow cytometric analysis of bone marrow (2 femora), spleen and blood from 4-week-old mice of the indicated genotypes. Data represent means ± SD from 3-4 mice per genotype. * p<0.05, ** p<0.01, *** p<0.001. Total B cells (CD19+), Pro/pre-B (CD19+IgM−CD43−), T1 (IgMhighCD21+), T2 (IgMhighCD21+CD23+) and mature (IgM+D+) B cells. (B) Pre-B cells (CD19+IgM−CD43−) sorted from bone marrow and immature (IgMhighIgDlow) as well as mature B cells (IgDhigh) sorted from spleens from 4-week-old mice of the indicated genotypes, were cultured up to 48h ex-vivo. Percentages of surviving cells were determined by Annexin-V/PI staining. Data represent means ± SEM from 3-4 independent experiments for each genotype. * p<0.05, ** p<0.01, *** p<0.001 compared to Eμ-myc, # p<0.05, ### p<0.001 compared to wt. (C) Freshly isolated splenocytes from 4-week-old mice of the indicated genotypes were immediately stained with anti-CD19-PE together with FITC-Annexin-V plus 7-AAD and analyzed by flow cytometry. Percentages of apoptotic cells in the CD19+ gate were determined. Data represent means ± SD from 3 independent experiments for each genotype. * p<0.05, ** p<0.01. (D) Four hours after in vivo labeling, the percentage of BrdU+ CD19+IgM− pro/pre-B cells in the bone marrow and of mature BrdU+CD19+IgM+ B cells in the spleen was evaluated by combined cell-surface and intracellular antigen staining. Data represent means ± SD from 2 experiments for each genotype.
To confirm our hypothesis, we isolated pre-B cells from the bone marrow as well as immature and mature B cells from the spleens of premalignant Eμ-myc transgenic mice either deficient or proficient for Bad or Bmf and assessed cell survival after in vitro culture in the absence of supporting cytokines. Eμ-myc transgenic pre-B and B cells died very rapidly, when compared to non-transgenic cells in vitro. Loss of Bad or Bmf did confer some minor protection to non-transgenic pre-B cells in culture (bad−/− p<0.05 and bmf−/− p<0.001 compared to wt at 48h) but mature B cells of both genotypes died as fast as wt cells (Fig. 3B). Notably, Bad deficiency failed to confer protection to any of the subsets from Eμ-myc transgenic mice. However, while Bmf-deficient pre-B cells were not protected from c-Myc-induced apoptosis, both immature and mature B cells lacking bmf survived oncogenic stress significantly better than their wt counterparts. In fact, the immature Eμ-myc-transgenic B cells were most efficiently protected from c-Myc-driven apoptosis by loss of Bmf and survived almost as well as non-transgenic B cells (Fig. 3B, lower panel). This contrasts observations made in bim−/− mice where loss of Bim protected pre-B and B cells potently from spontaneous and c-Myc-induced apoptosis 10.
Although the cells from Eμ-myc/bad−/− mice did not survive better than those from wt Eμ-myc mice when cultured in vitro, loss of Bad led to an accumulation of premalignant B cells in young Eμ-myc transgenic mice (Fig. 3A,B). This could be due to the absence of signals in vitro that would otherwise activate Bad in Eμ-myc transgenic cells to limit transformation in vivo. We therefore quantified the rate of steady-state levels of B cell apoptosis in spleens and lymph nodes of these mice by immediate Annexin-V/PI staining. Consistent with our hypothesis, steady-state levels of apoptosis of pre-B and B cells were lower in Eμ-myc mice lacking Bad when compared to Eμ-myc controls (Fig. 3C). In addition in vivo BrdU incorporation studies confirmed that the rates of proliferation did not differ between any of the cell and genotypes analyzed (Fig. 3D). It remains possible, however, that the survival advantage of Bad-deficient B cells observed in vivo is not cell-autonomous and that non cell-autonomous effects also contribute to tumor formation in bmf−/− mice. Once established, Bad-deficient tumors grow in wt and Bad-deficient hosts with equal kinetics, as do Bmf-deficient tumors in wt or Bmf-deficient hosts (suppl. Fig. 1), arguing against non cell-autonomous effects.
Loss of Bmf reduces, but does not eliminate, the pressure to lose p53 function
Due to the strong pro-apoptotic drive of c-Myc, tumors that develop in Eμ-myc mice frequently show aberrations in the p19ARF/Mdm2/p53 pathway 6. Western blotting for p53 and p19ARF, where high levels of the protein are indicative for non-functional p53 due to absence of the p53-induced negative feedback-loop on ARF expression, was used to investigate the status of this pathway in lymphomas lacking Bad or Bmf. High levels of ARF were detected in 9/38 (24%) of wt Eμ-myc; 8/24 (33%) of bad+/− Eμ-myc and 5/22 (23%) of bad−/−Eμ-myc lymphomas (Fig. 4A). In contrast, only 2/22 (9%) bmf+/−, 3/27 (11%) bmf−/− and 0/7 bad−/−bmf−/− Eμ-myc lymphomas, respectively, were deficient of functional p53 in this type of analysis (Fig. 4A and not shown). Consistent with previous findings 10, 0/6 bim−/− and only 1/9 bim+/− lymphomas analyzed showed increased ARF levels (not shown).
Figure 4. Loss of Bmf, but not Bad, reduces the pressure to lose p53.

(A) Representative Western blot analysis of p19/ARF and p53 expression as well as (B) Bcl-2, Bcl-x and Mcl-1 expression in lymphoma lysates derived from wt, bmf−/− and bad−/− Eμ-myc mice. Membranes were reprobed using anti-actin or anti-GAPDH antibodies as a loading control.
As c-Myc-driven transformation is facilitated either by inactivation of the p53 pathway or by overexpression of Bcl-2 or Bcl-xL 5 we tested whether up-regulation of a pro-survival Bcl-2 family member was preferred over loss of p53 during the transformation of Eμ-myc/bmf−/− B cells. Therefore, lymphomas derived from mice of the different genotypes were analyzed by Western blot for the expression of Bcl-2, Bcl-xL and Mcl-1. The three proteins were expressed in variable levels among the lymphomas, independent of their immunophenotype, but we failed to detect increased frequencies of overexpression of pro-survival Bcl-2 proteins in Bmf-deficient over wt or Bad-deficient c-Myc-driven lymphomas (Fig. 4B and data not shown).
Deregulated expression of Bmf in the presence of c-Myc
Myc is known to regulate the expression of several members of the Bcl-2 family. In premalignant Eμ-myc transgenic pre-B and B cells Bcl-2 and Bcl-xL are repressed 5, while Bim and Puma proteins are induced 10-12. To test whether the expression of Bmf or Bad were also deregulated on an Eμ-myc transgenic background, premalignant pre-B and B cells from wt and Eμ-myc mice were FACS-sorted and subjected to Western blot analysis. Surprisingly, although different isoforms of Bmf were expressed at high levels in wt pre-B cells 15, they were barely detectable in the Eμ-myc transgenic pre-B cells, whereas the protein levels were similar in IgM+ wt and Eμ-myc B cells (Fig. 5A). This observation might also explain why loss of Bmf favored the development of immature IgM+ over pre-B tumors. Consistent with the pattern of expression in the premalignant cells, Bmf levels were also very low or lacking in all pre-B cell tumors analyzed and was detected in 8/10 IgM+ tumors (Fig. 5B,C). Notably, loss of Bmf expression was also observed in 1/6 IgM+ Eμ-myc/bmf+/− tumors tested (Fig. 5C), suggesting that its loss or silencing may be a recurrent event in Myc-driven B cell lymphomagenesis. In contrast, both isoforms described for Bad 18 were present in wt and c-Myc transgenic premalignant cells (Fig. 5A) and one or the other isoform was expressed in Eμ-myc tumor samples (Fig. 5B). Tumors arising in Eμ-myc/bad+/− animals tested positive for Bad protein expression in 9/9 cases analyzed suggesting retention of the second allele (Fig. 5C). Furthermore, we aimed to investigate whether the Bad protein found in premalignant Eμ-myc transgenic B cells was hypo-phosphorylated, indicative of its activation 33. However, the weak signal that we obtained using different antibody against phospho-Bad was also observed in cell extracts from Bad-deficient mice (not shown).
Figure 5. Expression of Bmf and Bad in (pre)-malignant Eμ-myc and wt B lymphocytes.
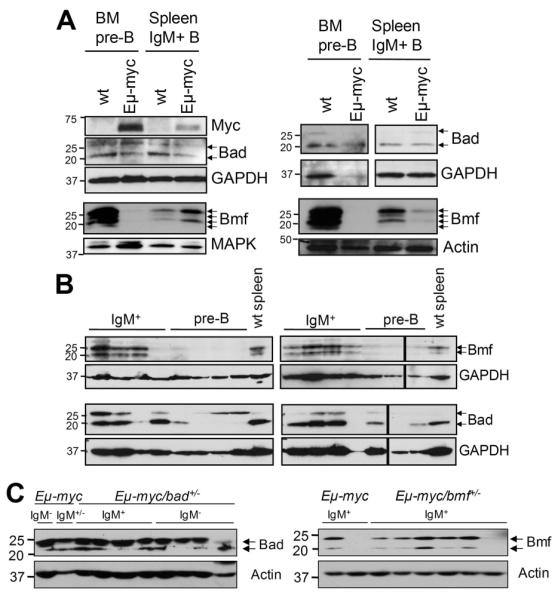
Wt and Eμ-myc FACS-sorted pre-B cells from the bone marrow and IgM+ B cells from the spleens of 4-week-old mice were analyzed for expression of (A) c-Myc, Bmf or Bad. Representative blots from two independent cell sorts are shown. Membranes were reprobed using anti-MAPK, anti-actin as a loading control. (B) Representative immunoblots assessing expression of Bmf and Bad in Eμ-myc-driven tumors. Membranes were reprobed using anti-GAPDH antibody as a loading control. (C) Representative immunoblots assessing LOH in Eμ-myc-driven tumors derived from bmf+/− or bad+/− animals. Membranes were reprobed using anti-actin antibody as a loading control.
Loss of Bim, but not loss of Bad or Bmf confers drug-resistance phenotypes in c-myc dependent lymphomas
Activation of Bmf or Bad has been reported to be required for cell death induced by certain anticancer agents, including inhibitors of histone-deacetylases or tyrosine-kinases as well as glucocorticoids 15,17,34. Therefore, we investigated if absence of Bad or Bmf would confer drug-resistance phenotypes to c-myc-dependent lymphomas. Freshly harvested tumor samples were cultivated for 24h in the absence or presence of graded doses of the glucocorticoid dexamethasone, the DNA-damaging drug etoposide, the HDAC-inhibitor SAHA, the proteasome inhibitor bortezomib, the microtubule-stabilizing agent paclitaxel, or the BH3-mimetic ABT-737. Surprisingly, neither loss of Bmf nor Bad conferred significant drug-resistance in vitro. This observation was independent of the immunophenotypes of the investigated tumor samples (not shown). In contrast, loss of Bim delayed tumor cell apoptosis triggered by etoposide, dexamethasone, paclitaxel, or, as reported before, SAHA 35, but not cell death caused by proteasome inhibition or treatment with ABT-737 (Fig. 6).
Figure 6. Loss of Bim but not Bad or Bmf protects Eμ-myc lymphomas from drug-induced apoptosis in vitro.

Freshly isolated Eμ-myc lymphoma cells were cultured on supporting irradiated NIH-3T3 cells in the presence of chemotherapeutic agents at the indicated concentrations for 24h. Cell death was determined by AnnexinV/7-AAD staining in CD19+ tumor cells. Specific cell death relative to cells cultured without the addition of any drugs was calculated. Values represent means ± SD of 4 animals/genotype. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Low-level expression of BMF in Burkitt lymphoma
In human B-cell chronic lymphocytic leukemia (CLL), Bmf is expressed at significant levels and further induced upon serum-deprivation 16,36. Furthermore, gene chip analysis demonstrated presence of BMF mRNA in acute lymphoblastic leukemia (ALL) 37, suggesting that BMF protein is expressed in human tumors that are the histogenetic equivalent to the lymphomas that arise in Eμ-myc transgenic mice. However, ALL and CLL usually do not associate with Myc-overexpression. In order to assess whether deregulated c-Myc might correlate with Bmf expression, we quantified its protein levels in three frequently studied Burkitt lymphoma (BL) cell lines i.e. Daudi, Ramos and Raji, as well as in six biopsy samples from patients diagnosed with BL. We also assessed expression of Bad as well as Bim, since the latter protein is reportedly lost or inactivated frequently in human BL 14. Protein lysates from FACS-sorted CD19+ B cells derived from the peripheral blood of healthy donors, known to express significant levels of BMF mRNA 16, were included for comparison. While Bad and Bim protein were found expressed at comparable levels in all samples analyzed, Bmf isoforms were only expressed at significant levels in healthy CD19+ B cells, but barely detectable in the three cell lines and primary tumor tissues (Fig. 7A). Since BMF contains a predicted CpG island (http://cpgislands.usc.edu/cpg.aspx) in its promoter region between position −688 to +492 in relation to the predicted transcription start site that may subject it to methylation-dependent silencing 17, we investigated if inhibition of DNA-methyltransferases by addition of 5′-aza-2′-deoxycytidine would suffice to restore Bmf expression and apoptosis in BL cell lines. Indeed, mRNA levels and protein were significantly induced in the BL lines (Fig. 7B, suppl. Fig. 2), suggesting that demethylation directly triggers transcription of the BMF gene. However, Methylight-PCR analysis covering seven methylation-sensitive CpGs failed to reveal evidence for direct promoter methylation in all cell lines, a finding confirmed by bisulfite sequencing, covering 43 additional putative methylation-sensitive sites in the promoter region and exon 1 of the tree cell lines (not shown). In search of other conditions that could induce Bmf in BL cells, we also found Bmf induction accompanied by cell death in the three cell lines after SAHA treatment or serum deprivation (Fig 7C and suppl. Fig. 3). Lentiviral knock-down of Bmf in Ramos cells had no significant effect on apoptosis induced by SAHA (Fig 7D), demonstrating that Bmf alone is not rate-limiting for apoptosis induction under these conditions, similar to our findings in Eμ-myc tumors derived from Bmf-deficient mice (Fig.6). Knock down of Bmf could however delay serum deprivation-induced cell death in these cells (Fig 7D), indicating that at least under certain conditions Bmf can be decisive in the regulation of cell death of Burkitt lymphoma cells.
Figure 7. Bmf expression is absent in Burkitt lymphoma cells but can be restored upon demethylation.

(A) Western blot analysis of Bmf, Bim and Bad in CD19+ cells derived from peripheral blood of healthy donors (#1 and #2), primary Burkitt lymphoma samples (BL1-BL6) and BL cell lines Daudi, Raji and Ramos. (B) Western blot analysis of Bmf levels in BL cell lines after inhibition of DNA-methyltransferases with 5-aza-2′-deoxycytidine (5μM) for the indicated times. (C) Western blot analysis of Bmf levels in Ramos cells after serum deprivation or inhibition of with SAHA (2μM) for the indicated times. (D) Cell death determined by AnnexinV/7-AAD staining in Ramos cells expressing either an shRNA against Bmf (white bars) or an unspecific shRNA (black bars) after serum deprivation or treatment with 2 or 4μM SAHA for the indicated times. Values represent mean±SE of 3 independent experiments. (E) Efficiency of knock down was confirmed by Western blot analysis of Bmf levels in cells treated with 2μM SAHA for 48 hours (left panel) or cells deprived of serum for 48 hours (right panel).
Discussion
Using the Eμ-myc transgenic mouse model of B cell lymphomagenesis we found that the BH3-only proteins Bad and Bmf are so far unrecognized antagonists of c-myc-driven tumor formation. Our findings extend the list of BH3-only proteins that can act as tumor suppressors in this disease model, next to Bim and Puma 10-12. Notably, other members of this family i.e. Noxa or Bid do play only redundant or no role in regulating Myc-induced lymphomagenesis [12,13 and R.W. Johnstone, pers. communication], highlighting the importance of understanding the contribution of individual Bcl-2 family proteins to oncogene-driven transformation.
Strikingly, loss of Bmf, but not Bad, caused a strong shift in the observed tumor spectrum and Eμ-myc bmf−/− animals presented with heavily increased tumor load and leukemia-like phenotype (Fig. 1,2). Onset of disease was preceded by an increased accumulation of premalignant pre-B- and immature B cells in different lymphoid organs in the absence of Bad or Bmf, exceeding numbers observed in Eμ-myc transgenic mice (Fig. 3A), which was not due to differences in proliferation capacity between genotypes (Fig. 3C). This indicated that loss of either BH3-only protein enhanced the survival of pre-leukemic cells in the presence of oncogenic c-Myc, increasing the pool of cells that can acquire a second oncogenic lesion that overcomes c-Myc-induced apoptosis, or that loss of Bmf or Bad may represent such secondary lesion, allowing transformation. We believe that loss of Bmf constitutes a genetic lesion that directly facilitates transformation by blocking c-Myc-driven B cell apoptosis (Fig. 3B). This is supported by our observations that although the size of the population “at-risk” for a second lesion is increased in the absence of Bmf up to ten-fold (Fig. 3A), the frequency of tumors that inactivate the p53-pathway actually dropped (Fig. 4A). This was not simply due to the shift towards the development of IgM+ lymphomas, seen when Bmf is absent (Fig. 1D), since p53-inactivation occurs as frequently in IgM+ as in pre-B wt Eμ-myc lymphomas (3/17 in pro/pre-B lymphomas and 3/13 in IgM+ lymphomas tested). While a preference of tumors to lose ARF over p53 in the absence of Bmf can currently not be excluded it is intriguing that some tumors arising in Eμ-myc and Eμ-myc/bmf+/− mice lose Bmf protein expression (Fig. 5B,C). The molecular basis of this phenomenon, however, awaits detailed investigation. Reduced selection pressure against p53 has also been reported in Eμ-myc tumors lacking bim or bax, but not noxa, while studies on puma revealed contradictory results10-12,38. Since neither Bim nor Bmf are regulated by p53 directly, they presumably act in the same Bax-dependent, but p53-independent apoptosis pathways, engaged by c-Myc.
Notably, Eμ-myc/bmf−/− mice developed mainly IgM+ lymphomas correlating with the fact that loss of Bmf protected immature IgM+ B cells most potently from c-Myc-induced apoptosis (Fig. 3B). The degree of protection provided by loss of Bmf to pre-B cells was minor and in line with the observation that c-Myc overexpressing wt pre-B cells showed a strong reduction of Bmf protein expression (Fig. 5A). A similar tumor immunophenotype was observed in Eμ-myc mice lacking Bim, as previously suggested by others 10 or in mice deficient for the tyrosine kinases Btk and Tec, that both trigger maturation and proliferation of developing pre-B cells and B cells after successful (pre)-BCR rearrangement 39. In btk−/−tec−/− double-knockout mice, pre-B cell development is essentially completely blocked, but efficiently restored by introduction of the Eμ-myc transgene. Interestingly, ~75% of tumors developing in these mice express IgM on their surface 39. It is unclear why IgM+ tumors preferentially develop in these mice, but maybe Btk and Tec-dependent maturation signals are required to maintain BH3-only protein expression and checkpoint function in developing pre-B cells.
Surprisingly, although loss of Bad also facilitated c-myc-driven lymphomagenesis, it did not cause a shift in tumor spectrum or promote a leukemia phenotype, as did loss of Bmf or Bim (Figs.1,2). Furthermore, loss of Bad preferentially facilitated the accumulation of premalignant pre-B cells (Fig. 3A), suggesting that it limits the survival of B cell precursors upon oncogenic stress only in a very narrow developmental window. Alternatively, loss of Bad may facilitate tumor formation by allowing the survival of cells under conditions where trophic factors are limiting, such as during rapid c-Myc-driven proliferation, as previously also suggested by others 40.
This effect may not even need to be B cell autonomous. Regardless of the mechanism, this increases the number of cells “at-risk” for secondary oncogenic lesions in the absence of Bad. Consistently, we did not observe a change in the percentage of bad−/− tumors that had inactivated the p53 pathway. Also, mice lacking Bmf and Bad simultaneously did not contract disease significantly earlier than single knockout mice expressing the Eμ-myc transgene, but in contrast to Bmf-deficient Eμ-myc mice, they developed pre-B as well as IgM+ B-cell lymphomas again (Fig. 1,2). It is interesting to note that loss of one allele of bim appears even more potent in accelerating tumorigenesis than loss of both alleles of bmf or bad (Fig. 1). This probably relates to the fact that loss of Bim potently protected both pre-B and B cells alike from death induced by Myc-overexpression 10 whereas loss of Bmf could only protect IgM+ B cells and Bad-deficiency appeared to delay Myc-driven B cell death only poorly, as suggested by our combined in vitro and in vivo results (Fig. 2). Also, Bim-levels are induced upon c-Myc overexpression 10, while neither Bad nor Bmf levels were found increased in pre-malignant B cells or tumors, suggesting an auxiliary and more cell type restricted role for these two proteins in Myc-induced killing. The broader efficacy of Bim may also be related to the fact that it can neutralize all Bcl-2 prosurvival homologues with comparable efficiency and/or its potential to activate Bax directly, while Bad and Bmf appear to bind and neutralize Bfl1/A1 and Mcl1 inefficiently and cannot trigger direct Bax activation 1,30.
Along that line, our screen for drug-resistance in Bad- or Bmf-deficient c-myc-driven lymphomas, in contrast to those lacking Bim, did not reveal any resistant phenotypes although a number of drugs have been tested that depend at least in part on Bad or Bmf for killing untransformed cells 15,18. Notably, in contrast to observations in squamous cell carcinomas and primary lymphocytes 15,17, but consistent with studies in CLL 41 and our own findings in BL-lines, loss of Bmf did not confer drug-resistance to HDAC-inhibition in vitro (Fig. 7; suppl. Fig.3), suggesting redundancy with other BH3-only proteins. Nonetheless, it will be interesting to see if loss of Bmf may affect the efficacy of such anticancer drugs or combinatorial treatment in vivo. Of note, tumor cells of all genotypes were equally responsive to the BH3-mimetic ABT-737, but killing was only achieved when high concentrations of the drug were applied, in line with recent findings from Whitecross and colleagues 43. This observation may be related to the fact that all lymphomas expressed significant levels of Mcl-1 (Fig. 4B), rendering tumor cells more resistant to this drug 44. Our results also indicate that the reported drug-resistance observed in Eμ-myc driven lymphomas expressing myr-AKT is presumably not due to repression of Bad function, but may depend on additional anti-apoptotic effects exerted by the AKT-pathway, e.g. repression of Bim and/or Puma 45,46.
Finally, since we failed to find evidence for direct regulation of BMF gene expression by c-Myc in promoter reporter studies or promoter methylation in human BL-lines, we speculate that oncogenic signals, such as the one provided by c-Myc, can down-modulate Bmf expression in mice and men by alternative means, e.g. by induction of miRNAs 47. Notably, miR-125b and miR-221 were recently shown to bind to the 3′UTR of the BMF mRNA in human glioma and hepatocellular carcinoma cell lines, respectively 48,49. The relevance of Bmf levels for lymphoma formation and/or progression driven by aberrant expression of c-Myc in humans remains to be investigated in full detail.
Supplementary Material
Acknowledgements
We thank K. Rossi, B. Rieder and M. Saurwein for animal husbandry, C. Soratroi and I. Gaggl for excellent technical assistance and G. Böck for cell-sorting. Furthermore, we thank A. Strasser, S. Rosenberg, S. Korsmeyer, R. Kofler and R.W. Johnstone for mice and/or reagents and W. Parson for DNA fingerprinting. This work was supported by the Association for International Cancer Research (AICR), Grant # 06-440, the Austrian Science Fund (SFB021) and ONCOTYROL.
Abbreviations
- Bcl-2
B cell lymphoma 2
- BH
Bcl-2 homology
- BL
Burkitt lymphoma
- Bmf
Bcl-2 modifying factor
- Bad
Bcl-2 antagonist of cell death
- Bim
Bcl-2 interacting mediator of cell death
- Puma
p53-upregulated mediator of apoptosis
- HDAC
histone deacetylase
- SAHA
suberoylanilide hydroxamic acid
- PI
propidium iodide
- MEF
mouse embryonic fibroblasts
Footnotes
The authors have no conflicting financial interests.
Disclosure of interests: The authors have no conflict of interest
References
- 1.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 3.Adams JM, Harris AW, Pinkert CA, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 4.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Em-myc transgenic mouse: a model for high-incidence spontaneous lymphoma and leukemia of early B cells. Journal of Experimental Medicine. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eischen CM, Woo D, Roussel MF, Cleveland JL. Apoptosis triggered by myc-induced suppression of Bcl-XL or Bcl-2 Is bypassed during lymphomagenesis. Molecular and Cellular Biology. 2001;21:5063–5070. doi: 10.1128/MCB.21.15.5063-5070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL. Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes and Development. 1999;13:2658–2669. doi: 10.1101/gad.13.20.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hermeking H, Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994;265:2091–2043. doi: 10.1126/science.8091232. [DOI] [PubMed] [Google Scholar]
- 8.Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 9.O’Connor L, Strasser A, O’Reilly LA, et al. Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO Journal. 1998;17:384–395. doi: 10.1093/emboj/17.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci U S A. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garrison SP, Jeffers JR, Yang C, et al. Selection against PUMA gene expression in Myc-driven B-cell lymphomagenesis. Mol Cell Biol. 2008;28:5391–5402. doi: 10.1128/MCB.00907-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Michalak EM, Jansen ES, Happo L, et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 2009;16:684–696. doi: 10.1038/cdd.2008.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frenzel A, Grespi F, Chmelewskij W, Villunger A. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis. 2009;14:584–596. doi: 10.1007/s10495-008-0300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109:271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- 15.Labi V, Erlacher M, Kiessling S, et al. Loss of the BH3-only protein Bmf impairs B cell homeostasis and accelerates gamma irradiation-induced thymic lymphoma development. J Exp Med. 2008;205:641–655. doi: 10.1084/jem.20071658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morales AA, Olsson A, Celsing F, Osterborg A, Jondal M, Osorio LM. Expression and transcriptional regulation of functionally distinct Bmf isoforms in B-chronic lymphocytic leukemia cells. Leukemia. 2004;18:41–47. doi: 10.1038/sj.leu.2403183. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Adachi M, Kawamura R, Imai K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2006;11:1349–1357. doi: 10.1038/sj.cdd.4401686. [DOI] [PubMed] [Google Scholar]
- 18.Ranger AM, Zha J, Harada H, et al. Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100:9324–9329. doi: 10.1073/pnas.1533446100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labi V, Erlacher M, Kiessling S, Villunger A. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death Differ. 2006;13:1325–1338. doi: 10.1038/sj.cdd.4401940. [DOI] [PubMed] [Google Scholar]
- 20.Cannings E, Kirkegaard T, Tovey SM, Dunne B, Cooke TG, Bartlett JM. Bad expression predicts outcome in patients treated with tamoxifen. Breast Cancer Res Treat. 2007;102:173–179. doi: 10.1007/s10549-006-9323-8. [DOI] [PubMed] [Google Scholar]
- 21.Teo K, Gemmell L, Mukherjee R, Traynor P, Edwards J. Bad expression influences time to androgen escape in prostate cancer. BJU Int. 2007;100:691–696. doi: 10.1111/j.1464-410X.2007.07001.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee JW, Soung YH, Kim SY, et al. Inactivating mutations of proapoptotic Bad gene in human colon cancers. Carcinogenesis. 2004;25:1371–1376. doi: 10.1093/carcin/bgh145. [DOI] [PubMed] [Google Scholar]
- 23.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not Bcl-xL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 24.Downward J. PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol. 2004;15:177–182. doi: 10.1016/j.semcdb.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 25.Bouillet P, Metcalf D, Huang DCS, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 26.Hamrouni A, Olsson A, Wiegers GJ, Villunger A. Impact of cellular lifespan on the T cell receptor repertoire. Eur J Immunol. 2007;37:1978–1985. doi: 10.1002/eji.200636632. [DOI] [PubMed] [Google Scholar]
- 27.Ploner C, Rainer J, Niederegger H, et al. The BCL2 rheostat in glucocorticoid-induced apoptosis of acute lymphoblastic leukemia. Leukemia. 2008;22:370–377. doi: 10.1038/sj.leu.2405039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eads CA, Danenberg KD, Kawakami K, et al. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weisenberger DJ, Campan M, Long TI, et al. Analysis of repetitive element DNA methylation by MethyLight. Nucleic Acids Res. 2005;33:6823–6836. doi: 10.1093/nar/gki987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 31.Swanson PJ, Kuslak SL, Fang W, et al. Fatal acute lymphoblastic leukemia in mice transgenic for B cell-restricted bcl-xL and c-myc. J Immunol. 2004;172:6684–6691. doi: 10.4049/jimmunol.172.11.6684. [DOI] [PubMed] [Google Scholar]
- 32.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 33.Datta SR, Dudek H, Tao X, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 34.She QB, Solit DB, Ye Q, O’Reilly KE, Lobo J, Rosen N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell. 2005;8:287–297. doi: 10.1016/j.ccr.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lindemann RK, Newbold A, Whitecross KF, et al. Analysis of the apoptotic and therapeutic activities of histone deacetylase inhibitors by using a mouse model of B cell lymphoma. Proc Natl Acad Sci U S A. 2007;104:8071–8076. doi: 10.1073/pnas.0702294104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackus WJ, Kater AP, Grummels A, et al. Chronic lymphocytic leukemia cells display p53-dependent drug-induced Puma upregulation. Leukemia. 2005;19:427–434. doi: 10.1038/sj.leu.2403623. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt S, Rainer J, Riml S, et al. Identification of glucocorticoid-response genes in children with acute lymphoblastic leukemia. Blood. 2006;107:2061–2069. doi: 10.1182/blood-2005-07-2853. [DOI] [PubMed] [Google Scholar]
- 38.Eischen CM, Roussel MF, Korsmeyer SJ, Cleveland JL. Bax loss impairs Myc-induced apoptosis and circumvents the selection of p53 mutations during Myc-mediated lymphomagenesis. Molecular and Cellular Biology. 2001;21:7653–7662. doi: 10.1128/MCB.21.22.7653-7662.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Habib T, Park H, Tsang M, et al. Myc stimulates B lymphocyte differentiation and amplifies calcium signaling. J Cell Biol. 2007;179:717–731. doi: 10.1083/jcb.200704173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobsen KA, Prasad VS, Sidman CL, Osmond DG. Apoptosis and macrophage-mediated deletion of precursor B cells in the bone marrow of Em-myc transgenic mice. Blood. 1994;84:2784–2794. [PubMed] [Google Scholar]
- 41.Inoue S, Riley J, Gant TW, Dyer MJ, Cohen GM. Apoptosis induced by histone deacetylase inhibitors in leukemic cells is mediated by Bim and Noxa. Leukemia. 2007;21:1773–1782. doi: 10.1038/sj.leu.2404760. [DOI] [PubMed] [Google Scholar]
- 42.Labi V, Grespi F, Baumgartner F, Villunger A. Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ. 2008;15:977–987. doi: 10.1038/cdd.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Whitecross KF, Alsop AE, Cluse LA, et al. Defining the target specificity of ABT-737 and synergistic antitumor activities in combination with histone deacetylase inhibitors. Blood. 2009;113:1982–1991. doi: 10.1182/blood-2008-05-156851. [DOI] [PubMed] [Google Scholar]
- 44.van Delft MF, Wei AH, Mason KD, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.You H, Pellegrini M, Tsuchihara K, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stahl M, Dijkers PF, Kops GJ, et al. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. Journal of Immunology. 2002;168:5024–5031. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 47.Chang TC, Yu D, Lee YS, et al. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008;40:43–50. doi: 10.1038/ng.2007.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia HF, He TZ, Liu CM, et al. MiR-125b expression affects the proliferation and apoptosis of human glioma cells by targeting Bmf. Cell Physiol Biochem. 2009;23:347–358. doi: 10.1159/000218181. [DOI] [PubMed] [Google Scholar]
- 49.Gramantieri L, Fornari F, Ferracin M, et al. MicroRNA-221 targets Bmf in hepatocellular carcinoma and correlates with tumor multifocality. Clin Cancer Res. 2009;15:5073–5081. doi: 10.1158/1078-0432.CCR-09-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.