

Abstract
Context
Circulating concentration of lipoprotein(a) (Lp[a]), a large glycoprotein attached to a low-density lipoprotein–like particle, may be associated with risk of coronary heart disease (CHD) and stroke.
Objective
To assess the relationship of Lp(a) concentration with risk of major vascular and nonvascular outcomes.
Study Selection
Long-term prospective studies that recorded Lp(a) concentration and subsequent major vascular morbidity and/or cause-specific mortality published between January 1970 and March 2009 were identified through electronic searches of MEDLINE and other databases, manual searches of reference lists, and discussion with collaborators.
Data Extraction
Individual records were provided for each of 126 634 participants in 36 prospective studies. During 1.3 million person-years of follow-up, 22 076 first-ever fatal or nonfatal vascular disease outcomes or nonvascular deaths were recorded, including 9336 CHD outcomes, 1903 ischemic strokes, 338 hemorrhagic strokes, 751 unclassified strokes, 1091 other vascular deaths, 8114 nonvascular deaths, and 242 deaths of unknown cause. Within-study regression analyses were adjusted for within-person variation and combined using meta-analysis. Analyses excluded participants with known preexisting CHD or stroke at baseline.
Data Synthesis
Lipoprotein(a) concentration was weakly correlated with several conventional vascular risk factors and it was highly consistent within individuals over several years. Associations of Lp(a) with CHD risk were broadly continuous in shape. In the 24 cohort studies, the rates of CHD in the top and bottom thirds of baseline Lp(a) distributions, respectively, were 5.6 (95% confidence interval [CI], 5.4-5.9) per 1000 person-years and 4.4 (95% CI, 4.2-4.6) per 1000 person-years. The risk ratio for CHD, adjusted for age and sex only, was 1.16 (95% CI, 1.11-1.22) per 3.5-fold higher usual Lp(a) concentration (ie, per 1 SD), and it was 1.13 (95% CI, 1.09-1.18) following further adjustment for lipids and other conventional risk factors. The corresponding adjusted risk ratios were 1.10 (95% CI, 1.02-1.18) for ischemic stroke, 1.01 (95% CI, 0.98-1.05) for the aggregate of nonvascular mortality, 1.00 (95% CI, 0.97-1.04) for cancer deaths, and 1.00 (95% CI, 0.95-1.06) for nonvascular deaths other than cancer.
Conclusion
Under a wide range of circumstances, there are continuous, independent, and modest associations of Lp(a) concentration with risk of CHD and stroke that appear exclusive to vascular outcomes.
Lipoprotein(a)(Lp[a]) is a low density lipoprotein (LDL)– like particle synthesized by the liver that consists of an apolipoprotein B100 (apo B100) molecule covalently linked to a very large glycoprotein known as apolipoprotein(a) (apo[a]).1-3 The physiological and vascular effects of the particle remain uncertain, but Lp(a) has been shown to enter the arterial intima of humans4; in vitro and animal studies have reported that Lp(a) can promote thrombosis, inflammation, and foam cell formation.5-7
Many prospective epidemiological studies have reported positive associations of baseline Lp(a) concentration with coronary heart disease (CHD) risk.8-10 A literature-based meta-analysis of published data from 31 prospective studies reported a relative risk of 1.5 (95% confidence interval [CI], 1.3-1.6) in a comparison of people in the top third vs those in the bottom third of the Lp(a) distribution (corresponding to mean values in these categories of approximately 50 vs 5 mg/dL).10 However, such reviews8-10 have been insufficiently detailed to enable reliable assessment of the nature of any independent association with CHD and have not addressed possible associations with ischemic stroke11 and nonvascular outcomes. In particular, Lp(a) concentration is believed to be correlated with some lipid markers,12,13 but published studies have not adjusted for them in a consistent way.
It has been suggested that Lp(a) is associated with CHD only at very high concentrations,14,15 but this suggestion is controversial,16 indicating that studies with greater power than hitherto are needed to characterize the shape of any dose-response relationship reliably.
The objective of this report is to produce reliable estimates of associations of Lp(a) with CHD, stroke, and non-vascular mortality, incorporating adjustment for potential confounding by risk factors. The present study differs from previous reports on Lp(a) in several important ways that enhance its scientific value and reliability. First, it is large and comprehensive. Second, harmonization of individual records allows a consistent approach to adjustment for lipids and other potential confounders. Third, correction for within-person variation (regression dilution)17,18 in Lp(a) concentration and in potential confounders has been made by use of serial measurements in a subset of participants. Fourth, individual records are available for each participant, allowing detailed analyses under different circumstances (such as by age or at different lipid levels). Fifth, individuals with known preexisting CHD and stroke are excluded, limiting any effects of clinically evident disease on Lp(a) concentration (ie, reverse causality). Given the substantial variations in average Lp(a) levels across available studies, we emphasize that the current analyses compare participants only within each contributing study.
METHODS
Study Design
Details of study selection, data collection, and harmonization procedures in the Emerging Risk Factors Collaboration (ERFC) have been described previously.19 Studies were identified through electronic searches of databases, scanning of the reference lists of relevant articles (including previously published reviews), and discussion with collaborators of the ERFC (FIGURE 1). Electronic searches, not limited to the English language, were performed in MEDLINE and EMBASE for studies published between January 1970 and March 2009 using terms related to Lp(a) (eg, lipoprotein[a], Lp[a], apo[a], apolipoprotein[a]) and cardiovascular disease outcomes (eg, cardiovascular disease, coronary heart disease, myocardial infarction, stroke).
Figure 1.
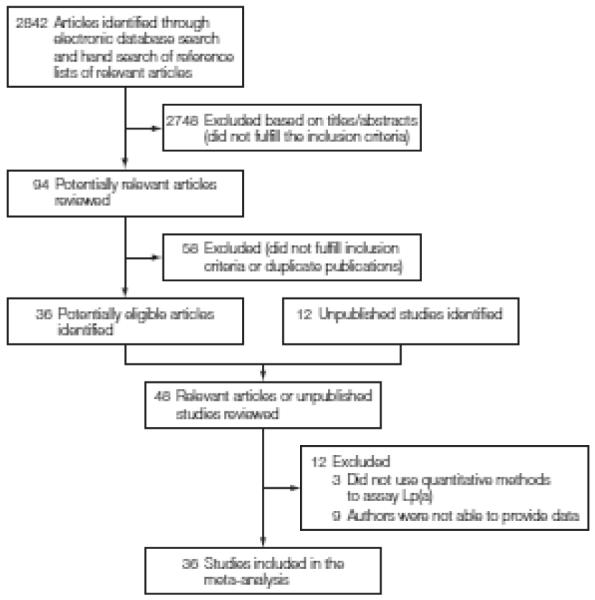
Literature Search and Study Selection
Studies were considered for inclusion if they had baseline information on age, sex, Lp(a), and several conventional vascular risk factors; if they did not select participants on the basis of having previous cardiovascular disease; used quantitative Lp(a) assay methods; recorded cause-specific mortality and/or major vascular morbidity using accepted criteria; and had accrued more than 1 year of follow-up.
Thirty-six eligible prospective studies,10,15,16,20-52 including 12 that had not previously published their findings (references 21, 24, 29, 31, 32, 38-42, 47, 50), were included. These studies involved a total of 126 634 individuals who had no known prior history of CHD (ie, myocardial infarction [MI] or angina, which was defined in each study) or stroke at the initial (baseline) examination. The contributing studies comprise about 90% of relevant incident CHD cases identified in known Western studies (TABLE 1); several smaller studies (collectively comprising about 10% of relevant known incident CHD cases) could not supply data.53-61 A few studies62-64 could not be included because they did not use quantitative assay methods.
Table 1.
Characteristics of 36 Prospective Studies Contributing Data to the Current Analysis
| No. of Events |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sourcea | Participants, No./Male, No. |
Age at Survey, Mean (SD), y |
Lp(a), Median (IQR), mg/dL |
Median Follow-up (5th-95th Percentile) |
Nonfatal MI/GHD Death |
CHD Death |
Nonfatal MI |
Fatal MI |
Fatal/Nonfatal Stroke |
Non- CVD Death |
||
| Ische- mic |
Hemor- rhagic |
Unclas- sified |
||||||||||
| Cohort Studies | ||||||||||||
| AFTCAPS42C | 902/745 | 59 (7.1) | 7.6 (3.3-17.9) | 5.7 (4.5-6.8) | 21 | 1 | 20 | 1 | 3 | 0 | 0 | 7 |
|
| ||||||||||||
| ARIC,20 2001 | 14033/6087 | 54 (5.7) | 18.3 (6.9-43.8) | 14.1 (5.0-15.7) | 850 | 190 | 660 | 114 | 431 | 52 | 16 | 947 |
|
| ||||||||||||
| ATTICA21C | 1508/777 | 51 (11.1) | 11.4 (4.9-25.2) | 5.0 (5.0-5.0) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 16 |
|
| ||||||||||||
| BRUN,22 1999 | 798/385 | 58 (11.4) | 88 (4.4-21.6) | 15.3 (3.9-15.5) | 53 | 31 | 22 | 19 | 24 | 14 | 0 | 120 |
|
| ||||||||||||
| CHARL24C | 165/165 | 70 (7.5) | 10.4 (3.4-22.3) | 6.3 (1.2-7.5) | 19 | 3 | 16 | 2 | 0 | 2 | 7 | 15 |
|
| ||||||||||||
| CHS 1,23 2003 | 3860/1480 | 72 (5.2) | 12.6 (4.8-22.2) | 12.1 (2.0-12.9) | 592 | 212 | 380 | 212 | 367 | 62 | 36 | 797 |
|
| ||||||||||||
| COPEN,16 2008 | 7487/3144 | 59 (13.6) | 19.1 (6.9-42.6) | 7.4 (2.4-8.9) | 283 | 36 | 247 | 0 | 184 | 39 | 94 | 525 |
|
| ||||||||||||
| DUBBO,25 2002 | 2008/842 | 68 (6.7) | 11.0 (5.0-27.8) | 14.1 (1.8-14.9) | 273 | 56 | 217 | 0 | 73 | 19 | 81 | 315 |
|
| ||||||||||||
| EAS,26 2001 | 637/323 | 64 (5.6) | 92 (3.7-25.4) | 15.1 (2.3-15.6) | 54 | 25 | 29 | 18 | 0 | 2 | 34 | 123 |
|
| ||||||||||||
| FINRISK 92,27 2005 | 2201/1022 | 54 (62) | 12.2 (4.5-31.7) | 11.8 (4.4-11.9) | 92 | 21 | 71 | 10 | 45 | 18 | 0 | 114 |
|
| ||||||||||||
| FRAMOFF,28 1996 | 2850/1316 | 54 (9.8) | 16.7 (7.1-36.6) | 12.0 (5.7-14.4) | 109 | 12 | 97 | 0 | 52 | 6 | 0 | 182 |
|
| ||||||||||||
| GOH29C | 638/307 | 71 (6.7) | 17.5 (10.0-37.0) | 3.9 (0.3-6.9) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
|
| ||||||||||||
| GRIPS,30 1997 | 5784/5784 | 48 (5.1) | 9.0 (4.0-25.0) | 9.8 (4.3-10.0) | 299 | 0 | 299 | 0 | 0 | 0 | 103 | 158 |
|
| ||||||||||||
| KIHD31C | 1996/1996 | 53 (5.3) | 9.6 (3.8-22.1) | 19.2 (2.9-23.1) | 386 | 11 | 375 | 6 | 104 | 34 | 3 | 239 |
|
| ||||||||||||
| NHANES 332C | 4496/1923 | 54 (15.7) | 23.0 (9.0-46.0) | 7.5 (3.9-9.0) | 107 | 107 | 0 | 38 | 0 | 0 | 46 | 321 |
|
| ||||||||||||
| NPHS II,33 2001 | 2375/2375 | 57 (3.4) | 10.9 (4.3-29.3) | 8.3 (3.5-10.4) | 157 | 13 | 139 | 16 | 23 | 7 | 17 | 97 |
|
| ||||||||||||
| PRIME,34 2002 | 7441/7441 | 55 (2.9) | 10.0 (5.0-30.0) | 5.2 (5.0-7.3) | 115 | 13 | 102 | 10 | 24 | 3 | 3 | 92 |
|
| ||||||||||||
| PROCAM,35 1996 | 3198/2255 | 43 (10.4) | 4.0 (2:0-13.0) | 17.4 (5.3-18.6) | 94 | 23 | 71 | 3 | 12 | 4 | 2 | 98 |
|
| ||||||||||||
| QUEBEC,36 1998 | 2012/2012 | 56 (6.9) | 19.0 (7.8-47.3) | 5.3 (4.3-5.6) | 53 | 5 | 48 | 4 | 0 | 0 | 9 | 45 |
|
| ||||||||||||
| SHS,37 2002 | 3837/1515 | 56 (8.0) | 3.0 (1.1-6.7) | 12.5 (2.1-14.3) | 416 | 133 | 283 | 62 | 8 | 8 | 177 | 750 |
|
| ||||||||||||
| TARFS38C | 1400/667 | 54 (10.5) | 10.1 (4.2-21.6) | 2.2 (1.2-4.5) | 3 | 3 | 0 | 3 | 0 | 0 | 3 | 12 |
|
| ||||||||||||
| ULSAM39C | 1866/1866 | 51 (4.5) | 8.3 (3.4-22.3) | 27.1 (5.9-35.8) | 485 | 124 | 361 | 60 | 164 | 42 | 30 | 457 |
|
| ||||||||||||
| WHITE 240C | 7903/5467 | 49 (6.0) | 21.0 (12.0-46.0) | 7.6 (3.8-8.2) | 170 | 23 | 147 | 18 | 1 | 0 | 3 | 86 |
|
| ||||||||||||
| WHS,15 2006 | 27 791/0 | 55 (7.1) | 10.6 (4.4-32.8) | 10.2 (8.4-10.8) | 227 | 10 | 217 | 4 | 229 | 25 | 1 | 540 |
|
| ||||||||||||
| WOSCOPS,43 2000 | 4617/4617 | 55 (5.6) | 17.0 (7.0-50.0) | 5.0 (2.8-6.0) | 299 | 60 | 239 | 0 | 0 | 0 | 61 | 83 |
|
| ||||||||||||
| ZUTE41C | 305/305 | 75 (4.5) | 12.3 (5.8-28.7) | 9.1 (1.1-10.1) | 42 | 13 | 29 | 9 | 1 | 1 | 25 | 65 |
| Subtotal | 112 108/54 816 | 55 (9.5) | 12.9 (5.0-32.7) | 9.7 (3.6-15.7) | 5199 | 1130 | 4069 | 614 | 1750 | 338 | 751 | 6204 |
|
| ||||||||||||
| Nested Case-Control Studies (Individually Matched) | ||||||||||||
| BUPA,44 1994 | 1505/1505 | 53 (7.2) | 19.2 (8.7-47.7) | 23.7 (4.5-26.9) | 208 | 208 | 0 | 170 | 0 | 0 | 0 | 173 |
|
| ||||||||||||
| FIA,45 1998 | 1492/1073 | 55 (7.6) | 26.5 (11.8-45.0) | 3.7 (0.5-8.6) | 519 | 118 | 401 | 118 | 0 | 0 | 0 | 0 |
|
| ||||||||||||
| FLETCHER,45 2007 | 689/541 | 57 (14.3) | 20.7 (7.2-59.5) | 5.6 (2.2-6.4) | 140 | NA | NA | 0 | 0 | 0 | 0 | 0 |
|
| ||||||||||||
| HPFS47C | 726/726 | 63 (8.3) | 13.0 (5.6-37.3) | 7.7 (3.0-8.5) | 220 | 35 | 185 | 9 | 0 | 0 | 0 | 18 |
|
| ||||||||||||
| MRFIT,48 2001 | 736/736 | 47 (5.6) | 3.4 (1.2-9.3) | 7.1 (6.0-73) | 246 | 19 | 227 | 13 | 0 | 0 | 0 | 5 |
|
| ||||||||||||
| NHS,49 2005 | 705/0 | 60 (6.5) | 9.5 (43-23.2) | 8.0 (1.4-8.8) | 234 | 27 | 207 | 27 | 0 | 0 | 0 | 10 |
|
| ||||||||||||
| Subtotal | 5353/4581 | 55 (9.6) | 16.0 (5.5-40.5) | 7.0 (1.3-25.9) | 1567 | 407b | 1020b | 337 | 0 | 0 | 0 | 206 |
|
| ||||||||||||
| Nested Case-Control Studies (Frequency-Matched) | ||||||||||||
| BRHS50C | 1561/1561 | 52 (5.3) | 6.5 (3.4-16.6) | 20.3 (3.7-23.6) | 461 | 169 | 292 | 122 | 0 | 0 | 0 | 221 |
|
| ||||||||||||
| GOTO 33,51 1993 | 128/128 | 51 (0.2) | 10.2 (4.2-32.0) | 12.8 (1.7-13.1) | 16 | 7 | 9 | 4 | 0 | 0 | 0 | 7 |
|
| ||||||||||||
| REYK,10 2008 | 6179/4359 | 55 (9.0) | 9.3 (2.9-22.8) | 20.3 (3.3-33.5) | 1850 | 810 | 1040 | 228 | 0 | 0 | 0 | 1476 |
|
| ||||||||||||
| USPHS,52 1993 | 805/805 | 60 (9.0) | 9.5 (3.8-24.1) | NA | 243 | 22 | 221 | 22 | 153 | 0 | 0 | 0 |
|
| ||||||||||||
| Subtotal | 8673/6853 | 55 (8.6) | 3.7 (3.2-21.8) | 20.1 (3.4-32.9) | 2570 | 1008 | 1562 | 376 | 153 | 0 | 0 | 1704 |
|
| ||||||||||||
| Total | 126 634/65 755 | 55 (9.4) | 12.6 (4.9-32.1) | 9.3 (3.5-21.3) | 9336 | 2545b | 6651b | 1327 | 1903 | 338 | 751 | 8114 |
Abbreviations: CHD, coronary heart disease; IQR, interquartile range; Lp(a), lipoprotein(a); MI, myocardial infarction; MA, data not available; non-CVD, nonvascular.
eAppendix 3 lists the study acronyms.
Numbers sum to less than the total of CHD events because 1 study46 did not provide seperate data on CHD death and nonfatal MI.
Studies that had not previously published their findings on LP(a) and vascular risk.
Concomitant information was available on Lp(a), age, sex, systolic blood pressure, smoking habits, history of diabetes, body mass index, triglycerides, and total cholesterol in 106 645 participants from 30 studies. A total of 96 113 participants from 26 studies had concomitant data on all the preceding characteristics plus high-density lipoprotein (HDL) cholesterol. To measure Lp(a), 2 studies used in-house assays, 32 used commercially available assays, and 2 did not specify the assay used. Twenty-one studies used enzyme-linked immunosorbent assay methods, 9 immunoturbimetry or nephelometry, 3 immunoradiometry, and 1enzyme immunodiffusion (eTable 1; available at http://www.jama.com). Twenty-four studies used assays insensitive to apo(a) isoforms.
In registering fatal outcomes, all contributing studies used International Classification of Diseases coding to at least 3 digits and ascertainment was based on death certificates. Twenty-eight of the 36 contributing studies also involved medical records, autopsy findings, and other supplementary sources to help classify deaths (eTable 2). Twenty-nine studies used standard definitions of MI based on Monitoring Trends and Determinants in Cardiovascular Disease (MONICA) or World Health Organization criteria. Twenty-five studies reported diagnosis of strokes on the basis of typical clinical features and characteristic changes on brain imaging, and most attempted to provide attribution of stroke subtype.
Statistical Analyses
Details of the statistical methods are provided in eAppendixes 1 and 2. Normal distributions were achieved by taking natural logarithms (loge) of Lp(a). The pooled standard deviation across studies in baseline loge Lp(a) concentration was 1.25, which corresponds to about a 3.5-fold difference (ie, e1.25) on the original scale of Lp(a) measurement in milligrams per deciliter. The primary disease outcome was CHD (ie, first-ever MI or fatal CHD), with subsidiary analyses of stroke by subtype and all cardiovascular deaths. Analyses involved a 2-stage approach with estimates of association calculated separately within each study before pooling across studies by random-effects meta-analysis. Parallel analyses using fixed-effect models yielded very similar results (eFigure 1).
For the 26 studies analysed as prospective cohort studies, hazard ratios were calculated using Cox proportional hazard regression models stratified by sex (and, where appropriate, by study group). The assumptions of the proportionality of hazards for loge Lp(a) levels were satisfied. Each participant contributed only either the first nonfatal outcome or death recorded at age 20 years or older (ie, deaths preceded by nonfatal CHD or stroke were not included in the analyses).
For the 10 “nested” case-control studies within prospective cohorts, odds ratios were calculated using either conditional or unconditional logistic regression models, as appropriate. Hazard ratios and odds ratios were assumed to approximate the same relative risk and are collectively described as risk ratios (RRs).
To assess the shape of association, study-specific RRs calculated within overall quantiles (eg, tenths) of baseline Lp(a) levels were combined by multivariate random-effects meta-analysis and plotted against mean usual loge Lp(a) levels within each quantile. Ninety-five percent CIs were estimated from the floated variances that reflect the amount of information underlying each group (including the reference group).65 When associations were approximately log-linear, regression coefficients were calculated to estimate the RR associated with a 3.5-fold (ie, 1-SD) higher Lp(a). Risk ratios were adjusted progressively for age, sex, and several other conventional risk factors, with evidence of association indicated by the Wald χ2 statistic.66 Heterogeneity between studies was assessed by the I2 statistic.67,68 (I2 is a measure of consistency across studies: the percentage of variance in estimated loge RRs that is attributable to between study variation as opposed to sampling variation. Values of I2 close to 0 indicate lack of evidence of heterogeneity.) Diversity at the study level (such as differences by study design or laboratory methods) was investigated by grouping studies by recorded characteristics and by meta-regression. Non-HDL cholesterol (calculated by subtraction of HDL cholesterol from total cholesterol) was used as the principal marker of cholesterol content in proatherogenic lipoproteins (eAppendix 2).
Because most characteristics in epi-demiological studies are measured with some error and are subject to fluctuations within individuals over time, correction for such regression dilution— ideally, both in levels of Lp(a) and in potential confounding factors—can help avoid biases that may exaggerate or obscure associations.18,69 Regression dilution ratios for each characteristic were calculated by regressing serial measurements, taken from participants in the ERFC, on the established baseline vascular risk factors listed above plus baseline levels of Lp(a) and duration of follow-up (eAppendix 1).18,69
Correction for within-person variation in Lp(a) and in potential confounders was achieved by use of conditional expectations of long-term average (ie, “usual”) levels of Lp(a) and error-prone confounders predicted from these regression calibration models, and used in assessments of associations with disease risk, as previously described.70-72 Regression calibration models allowed variability in Lp(a) to vary by its baseline levels. Analyses were performed using Stata software, release 10 (StataCorp, College Station, Texas), involving 2-sided statistical tests, a significance level of P<.05, and 95% CIs.
This study was approved by the Cambridgeshire Ethics Review Committee and was conducted and analyzed independently from its funders.
RESULTS
Mean age at entry of participants was 57 (SD, 8) years and 48% were women; 47% were European and 50% North American. During 1.3 million person-years at risk (mean, 10.2 years to first outcome), there were 9336 CHD outcomes, 1903 ischemic strokes, 338 hemorrhagic strokes, 751 unclassified strokes, 1091 other vascular deaths, 8114 nonvascular deaths, and 242 deaths of unknown cause (Table 1).
As expected, mean Lp(a) concentration varied across studies, but values were as diverse within groups of studies that used similar assay methods as across studies that used different methods (eFigure 2). The overall median of Lp(a) at baseline was 12.6 (interquartile range, 4.9-32.1) mg/dL. (To convert to μmol/L, multiply by 0.0357.) Blacks had more than a 100% higher Lp(a) concentration than whites (TABLE 2). Racial groups were examined separately in subanalyses.
Table 2.
Summary of Available Data and Correlates of Lp(a) Levels
| Summary of Available Data |
Correlates of Lp(a) |
||||
|---|---|---|---|---|---|
| No. of Studies |
No. of Participants |
Mean (SD) or % |
Pearson Correlation r (95% CI)a |
Percentage Difference (96% CI) in Lp(a) Levels per 1 SD Higher or Compared With Reference Category of Correlateb |
|
| Loge Lp(a), mg/dLc | 36 | 126 634 | 2.37 (1.25) | ||
|
| |||||
| Age at survey, y | 36 | 126 634 | 57 (8) | 0.01 (0.00 to 0.02) | 2 (0 to 3) |
|
| |||||
| Sex | 36 | 126 634 | |||
|
| |||||
| Male | 34 | 66 250 | 52 | Reference | |
|
| |||||
| Female | 21 | 60 384 | 48 | 12 (8 to 16) | |
|
| |||||
| Race | 26 | 91 706 | |||
|
| |||||
| White | 26 | 85 046 | 93 | Reference | |
|
| |||||
| Black | 11 | 6223 | 7 | 119 (84 to 161) | |
|
| |||||
| Smoking status | 35 | 122 994 | |||
|
| |||||
| Never/former | 35 | 89 658 | 73 | Reference | |
|
| |||||
| Current | 34 | 33 336 | 27 | 0 (−2 to 3) | |
|
| |||||
| History of diabetes | 36 | 121 027 | |||
|
| |||||
| No | 35 | 113 991 | 94 | Reference | |
|
| |||||
| Yes | 34 | 7036 | 6 | −11 (−17 to −4) | |
|
| |||||
| Systolic blood pressure, mm Hg | 35 | 120 643 | 134 (18) | 0.01 (−0.01 to 0.02) | 1 (0 to 2) |
|
| |||||
| Body mass indexd | 35 | 123 740 | 26 (5) | −0.02 (−0.04 to 0.00) | −4 (−6 to −1) |
|
| |||||
| Lipid markers, mg/dL | |||||
| Total cholestrol | 36 | 126 128 | 228 (42) | 0.12 (0.10 to 0.13) | 16 (14 to 18) |
|
| |||||
| HDL-C | 33 | 114889 | 49 (15) | 0.03 (0.02 to 0.04) | 4 (2 to 6) |
|
| |||||
| Non–HDL-C | 33 | 114876 | 178 (42) | 0.11 (0.09 to 0.13) | 14 (12 to 17) |
|
| |||||
| Loge triglyceridesc | 35 | 124 232 | 4.85 (0.51) | −0.05 (−0.07 to −0.02) | −6 (−9 to −3) |
|
| |||||
| Apolipoprotein AI | 21 | 91 480 | 151 (29) | 0.02 (0.00 to 0.04) | 1 (−1 to 4) |
|
| |||||
| Apolipoprotein B | 23 | 93 058 | 108 (28) | 0.11 (0.09 to 0.13) | 15 (11 to 18) |
|
| |||||
| Inflammatory markers | |||||
| Loge C-reactive protein, mg/Lc | 27 | 78 153 | 0.62 (1.12) | 0.03 (0.01 to 0.05) | 4 (2 to 6) |
|
| |||||
| Fibrinogen, mg/dL | 25 | 101 346 | 326 (78) | 0.08 (0.06 to 0.10) | 11 (8 to 15) |
SI conversions: To convert total cholesterol, HDL-C, and non-HDL-C to mmol/L, multiply by 0.0259; triglycerides to mmol/L, multiply by 0.0113; apolipoproteins to g/L, multiply by 0.01; C-reactive protein to nmol/L, multiply by 9.524; and fibrinogen to μmol/L, multiply by 0.0294.
Abbreviations: CI, confidence interval; HDL-C, high-density lipoprotein cholesterol; Lp(a), lipoprotein(a).
Pearson correlation coefficients between loge, Lp(a) and the row variables, pooled across studies using random-effects meta-analysis.
Percentage change in Lp(a) levels per 1-SD increase in the variable (or for categorical variables, the percentage difference in mean Lp[a] levels for the category vs the reference), adjusted for age and sex and allowing for random effects across studies.
Median (interquartile range) values were for Lp(a), 12.6 mg/dL (4.9-32.1 mg/dL); triglycerides, 120 mg/dL (86-173 mg/dL); and C-reactive protein, 1.75 mg/L (0.82-3.87 mg/L).
Body mass index is calculated as weight in kilograms divided by height in meters squared.
Correlates and Within-Person Variation Over Time
Lp(a) concentration was weakly correlated with several known or suspected risk factors: positively with total and non-HDL cholesterol, apo B100, and fibrinogen and inversely with loge triglycerides. Lp(a) levels were 12% (95% CI, 8%-16%) higher in women and 11% (95% CI, 4%-17%) lower in people with diabetes (Table 2). Repeat information on Lp(a) was available in 6597 participants from 7 studies (mean interval, 8.3 years) (eFigure 3). The regression dilution ratio of loge Lp(a), adjusted for age and sex, was 0.87 (95% CI, 0.81-0.93), which was considerably higher in these studies than those for total cholesterol (0.65; 95% CI, 0.62-0.65), HDL cholesterol (0.72; 95% CI, 0.70-0.75), loge tri-glycerides (0.63; 95% CI, 0.61-0.65), or systolic blood pressure (0.52; 95% CI, 0.49-0.55).
Associations With CHD
In analyses adjusted for age and sex only, there were continuous associations of Lp(a) with the risk of CHD, potentially consistent with either a curvilinear or a log-linear shape (FIGURE 2). Statistical tests of the compatibility of the data with a linear vs a quadratic model suggested a better fit with a curvilinear shape (P=.003) (eAppendix 1 and eTable 3). In analyses restricted to participants with complete information on relevant covariates, the RR for CHD per 3.5-fold higher Lp(a) level, adjusted for age and sex only, was 1.16 (95% CI, 1.11-1.22), and it was 1.13 (95% CI, 1.09-1.18) following further adjustment for systolic blood pressure, smoking, history of diabetes, and total cholesterol (TABLE 3). There was moderate heterogeneity among studies contributing to the fully adjusted CHD result (I2=49%; 95% CI, 22%-66%) (Table 3).
Figure 2. Risk Ratios for Coronary Heart Disease, Ischemic Stroke, or Nonvascular Death by Quantile of Usual Lp(a) Level.

Lp(a) indicates lipoprotein(a); MI myocardial infarction. Sizes of data markers are proportional to the inverse of the variance of the risk ratios. Confidence Intervals (CIs) were calculated using a floating absolute risk technique. Studies involving fewer than 10 cases of any outcome were excluded from the analysis of ttiat outcome.
aFurther adjustment for usual levels of systolic blood pressure, smoking status, history of diabetes, body mass index, and total cholesterol. The x- and y-axes are shown on a log scale. Lowest quantiles are referents.
Table 3.
Risk Ratios for Coronary Heart Disease and Ischemic Stroke per 3.5-Fold (1-SD) Higher Usual Lipoprotein(a) Levels With Progressive Adjustment for Usual Levels of Confoundersa
| Adjustments | Risk Ratio (95% CI) |
Wald χ2 |
I2 % (95% CI) |
|---|---|---|---|
| Coronary heart diseaseb | |||
| Age and sex only | 1.16 (1.11-1.22) | 46 | 57 (36-72) |
|
| |||
| Age and sex plus | |||
| Systolic blood pressure | 1.16 (1.11-1.21) | 43 | 57 (36-71) |
|
| |||
| Smoking status | 1.16 (1.11-1.21) | 42 | 57 (36-72) |
|
| |||
| History of diabetes | 1.17 (1.12-1.22) | 47 | 58 (37-72) |
|
| |||
| Body mass index | 1.17 (1.12-1.23) | 51 | 57 (36-71) |
|
| |||
| Total cholesterol | 1.13 (1.09-1.18) | 36 | 49 (22-66) |
|
| |||
| Ischemic strokec | |||
| Age and sex only | 1.11 (1.02-1.20) | 6 | 46 (0-72) |
|
| |||
| Age and sex plus | |||
| Systolic blood pressure | 1.09 (1.01-1.17) | 6 | 31 (0-64) |
|
| |||
| Smoking status | 1.09 (1.01-1.17) | 6 | 30 (0-64) |
|
| |||
| History of diabetes | 1.10 (1.02-1.17) | 7 | 26 (0-62) |
|
| |||
| Body mass index | 1.10 (1.03-1.18) | 8 | 25 (0-61) |
|
| |||
| Total cholesterol | 1.10 (1.02-1.18) | 7 | 30 (0-64) |
Abbreviaton: CI, confidence interval.
Analyses were restricted to participants with complete information on sex and all confounding variables. Risk ratios are stratified by sex and study group where appropriate. Studies with fewer than 10 cases of coronary heart disease or ischemic stroke outcomes were excluded from the analyses of that outcome.
For coronary heart disease, 106 645 individuals, 8362 cases, 30 studies.
For ischemic stroke, 69 539 individuals, 1684 cases, 13 studies.
Findings were broadly similar in sub-analyses of coronary death and nonfatal MI (FIGURE 3 and eFigure 4), adjusted for non-HDL and HDL cholesterol (instead of total cholesterol) and adjusted for fibrinogen, C-reactive protein, or apo AI and apo B100 (eTable 4). Because adjustment for total cholesterol may obscure associations of Lp(a) with disease risk because total cholesterol includes the cholesterol contained in Lp(a) particles, we conducted sensitivity analyses that corrected also for estimated Lp(a) cholesterol concentration,73 which gave a higher RR than without such correction (eTable 4).
Figure 3. Risk Ratios for Vascular and Nonvascular Outcomes per 3.5-Fold (1-SD) Higher Usual Lp(a) Level, Adjusted for Cardiovascular Risk Factors.

Lp(a) indicates lipoprotein(a); MI myocardial infarction, CI, confidence interval. Sizes of data markers are proportional to the inverse of the variance of the risk ratios. Risk ratios are adjusted for age, usual levels of systolic blood pressure, smoking status, history of diabetes, body mass index, and total cholesterol and are stratified, where appropriate, by sex and study group. Studies involving fewer than 10 cases of any outcome were excluded from the analysis of that outcome.
aSubtotals do not add to the total number of coronary heart disease outcomes because some nested case-control studies did not subdivide outcomes into coronary death or nonfatal MI.
The findings were qualitatively similar in analyses that excluded the first 5 years of follow-up (eFigure5), ignored regression dilution (eTable 5), and used fixed-effect models (eFigure 1). The RR, adjusted for several conventional risk factors, was 1.27 (95% CI, 1.17-1.38) in a comparison of those in the top third with those in the bottom third of baseline Lp(a) concentration (eTable5). In the cohort studies, the rates of CHD in the top and bottom thirds of baseline Lp(a) distributions, respectively, were 5.6 (95%CI, 5.4-5.9) per 1000 person-years and 4.4 (95%CI, 4.2-4.6) per 1000 person-years.
The RRs for CHD did not vary importantly by sex, non-HDL or HDL cholesterol, triglycerides, blood pressure, diabetes, or body mass index (FIGURE 4). There was no convincing evidence of major variations in RRs of studies using isoform-sensitive vs isoform-insensitive assays or with other features of study design recorded (eFigure 6). Subsidiary analyses restricted to people of European continental ancestry (>90% of the participants) yielded very similar findings to the overall findings described herein (data available from the authors on request), but comparisons of RRs between racial groups lacked power because data were limited on other races/ethnicities (eFigure 6). In a common set of participants, the adjusted RR for CHD per1-SD higher Lp(a) concentration was considerably weaker than the corresponding RR with non-HDL cholesterol (1.14 vs 1.66, respectively) (eFigure 7).
Figure 4. Risk Ratios for Coronary Heart Disease per 3.5-Fold (1-SD) Higher Usual Lp(a) Level, by Age and Thirds of Individual Characteristics.

Lp(a) indicates lipoprotein(a), HDL-C, high-density lipoprotein cholesterol; CI, confidence interval. Sizes of data markers are proportional to the inverse of the variance of the risk ratios. Risk ratios are adjusted for age, usual levels of systolic blood pressure, smoking status, history of diabetes, body mass index, and total cholesterol and are stratified, where appropriate, by sex and study group. Studies with fewer than 3 cases per stratum were excluded from analyses.
aBody mass index is calculated as weight in kilograms divided by height in meters squared.
bCorrection for the cholesterol content of Lp(a) was made by subtracting estimated Lp(a) cholesterol values from total cholesterol, Lp(a) cholesterol was estimated from Lp(a) total mass using the following equation: Lp(a)−cholesterol (mg/dL)=0.15×Lp(a) (mg/dL)+1.24.73
Associations With Stroke
In analyses adjusted for age and sex only, the shape of association of Lp(a) with the risk of ischemic stroke was indistinct (Figure2). Assuming a log-linear association with risk, the age-and-sex-only– adjusted RR for ischemic stroke was 1.11 (95% CI, 1.02-1.20) per 3.5-fold higher usual Lp(a) levels in analyses restricted to participants with complete information on relevant covariates (Table 3). The RR was 1.10 (95% CI, 1.02-1.18) following further adjustment for systolic blood pressure, smoking, history of diabetes, and total cholesterol (Table 3). There was no clear evidence of heterogeneity among studies contributing to ischemic stroke (I2=30%;,95%CI,0%-64%).The adjusted RRs per 3.5-fold higher usual Lp(a) levels were 1.01 (95%CI, 0.92-1.12) for unclassified stroke and 1.06 (95% CI, 0.901.26) for hemorrhagic stroke (Figure 3).
Associations With Nonvascular Mortality
The adjusted RR for the aggregate of nonvascular mortality was 1.01 (95% CI, 0.98-1.05) (Figure 3). The adjusted RRs were 1.00 (95% CI, 0.97-1.04) for all cancer deaths and 1.03 (95% CI, 0.97-1.09) for smoking-related cancer deaths. The adjusted RR for nonvascular deaths other than cancer was 1.00 (95% CI, 0.95-1.06). There were too few cases of particular types of cancer (or other nonvascular outcomes) to enable reliable analyses by subtype. Adjusted RRs for major vascular and nonvascular outcomes were qualitatively similar in analyses that included fatal outcomes without censoring previous nonfatal outcomes (eFigure 8).
COMMENT
Contrary to previous suggestions of steep threshold effects, the current analysis of 126 634 individuals has demonstrated broadly continuous associations of Lp(a) concentration with the risk of CHD. Because these associations were only slightly reduced after adjustment for long-term average levels of lipids and other established risk factors, it increases the likelihood that Lp(a) is an independent risk factor for CHD. Lipoprotein(a) concentration is, however, a relatively modest coronary risk factor, being only about one-quarter as strong overall as non–HDL cholesterol, although Lp(a) may become proportionally more important to CHD at very high concentrations owing to its potentially curvilinear risk relationship. Because associations of higher Lp(a) concentration with CHD are similar at different levels of non-HDL cholesterol, the absolute benefits of cholesterol lowering should be greater if Lp(a) concentration is high (or when absolute risk is high for some other reason).
Whereas previous literature-based reviews of Lp(a) have focused only on CHD,8-10 the current individual participant meta-analysis also investigated stroke subtypes and cause-specific mortality, including nonvascular deaths. Although current data in relation to Lp(a) concentration and stroke were somewhat sparser and less distinct than those for CHD, findings were broadly similar to those for CHD. In contrast, Lp(a) concentration was unrelated to the aggregate of nonvascular mortality, including cancer and noncancer deaths. Hence, Lp(a) appears to be more specifically associated with vascular outcomes than are a number of systemic markers of inflammation that have been strongly associated with both vascular and nonvascular outcomes.66,74,75 As a subsidiary finding, the current analyses convincingly demonstrate that Lp(a) concentration is more consistent within individuals over several years than are levels of total cholesterol, HDL cholesterol, or systolic blood pressure.
Recent large studies have reported highly significant associations of variants in or near the LPA gene (a locus known to strongly influence circulating Lp[a] concentration)76-78 with CHD risk.79,80 Together with the current findings of continuous, independent, and specific associations of Lp(a) concentration with vascular outcomes, available data are consistent with the existence of a causal relationship and increase priority for investigation of Lp(a) as a potential therapeutic target. Because the current findings show that Lp(a) concentration is a relatively modest risk factor for CHD, however, interventions capable of much more powerful and specific Lp(a) lowering than currently available may be required to demonstrate any vascular benefits in randomized trials.
Substantial modification of Lp(a) concentration has been difficult to achieve without pharmacological agents.81 Niacin and certain inhibitors of cholesteryl ester transfer protein can reduce Lp(a) by about 20% and about 40%, respectively.82 Contradictory findings have been reported about the effect of statins on Lp(a) concentration,83,84 and it remains uncertain whether statin use attenuates the CHD risk associated with Lp(a) concentration.2,85,86 Large randomized trials of niacin and cholesteryl ester transfer protein inhibitors in the secondary prevention of CHD are in progress.87 Such studies may not, however, enable causal inferences because, in addition to Lp(a) lowering, these agents increase HDL cholesterol and decrease LDL cholesterol and triglyceride concentrations. Similar considerations may apply to mipomersen, an antisense oligonucleotide directed at human apo B100 now in phase 2 clinical trials that has been shown to reduce circulating Lp(a) concentration by 70% in transgenic mice, as well as reducing LDL cholesterol, apo B100, and oxidized phospholipids.88,89
Even though the first epidemiological study of Lp(a) and CHD was reported in 1972,90 the investigation of this lipoprotein as a potential cardiovascular risk factor has been hampered by the lack of consistent approaches to its measurement. International reference material for Lp(a) laboratory standardization emerged only in 200091 and was accepted by the World Health Organization in 2003.92 Even with methods that use the same standard, however, there is significant variability in measured Lp(a) concentration if assays are sensitive to variation in numbers of repeat domains in apo(a).93,94 Hence, in 2003 an expert panel recommended use of assay systems not sensitive to apo(a) isoforms (eFigure 2B).83 Population differences can also contribute to variation in Lp(a) concentration, particularly since values differ substantially between individuals and are highly heritable.1,78,95 Nevertheless, pooled analyses of individual data from prospective studies should remain informative, provided that, as in the current study, analyses compare cases and noncases only within each study and explore potential diversity across groups of studies using similar assay methods.
Despite considerable scope for such diversity, it is notable that there is relatively moderate heterogeneity in RRs among the studies based in 15 different Western countries contributing to the current findings, an observation that supports the ability to generalize these data to such populations. Because more than 90% of the participants in the current study were of European continental ancestry, however, further studies are needed in non-white racial groups, particularly in black and South Asian populations, which have different Lp(a) concentrations.96,97 The RRs in the current analysis were not strongly different between studies using assays sensitive and insensitive to apo(a) isoforms (although there was, of course, some heterogeneity within each of these groups of studies). Although the findings did not differ appreciably in subgroups defined by the laboratory and population features recorded, further studies are needed that can explore in greater depth such potential sources of heterogeneity and joint effects with other lipid markers. For example, large studies are needed to assess whether Lp(a) particles with smaller-sized apo(a) isoforms confer even higher RRs for CHD55,98 (such assessment was not possible in the current study because it lacked concomitant data on apo[a] isoforms). Similarly, larger studies are needed to assess proposed synergy in the promotion of vascular disease through oxidative damage (again, this was not possible in the current study because the data set lacked concomitant information on oxidized LDL and lipoprotein-associated phospholipase A2).99-101
CONCLUSION
Under a wide range of circumstances, there are continuous, independent, and modest associations of Lp(a) concentration with the risk of CHD and stroke that appear exclusive to vascular outcomes.
Supplementary Material
Acknowledgments
Funding/Support: The ERFC Coordinating Centre is supported by a program grant from the British Heart Foundation (RG/08/014) and supported by grants from the UK Medical Research Council and the BUPA Foundation. Aspects of this work have been supported by unrestricted educational grants from GlaxoSmithKline and a grant from Merck Sharp and Dohme in relation to Lp(a). A variety of sources have supported recruitment, follow-up, and laboratory measurements in the 116 cohorts contributing to the ERFC. Investigators of several of these studies have contributed to a list naming some of these funding sources, available at http://www.phpc.cam.ac.uk/MEU/. Dr Erqou is supported by a Gates Cambridge Trust and Overseas Studentship Award. Drs Di Angelantonio and Thompson have been supported by UK Medical Research Council PhD studentships.
Role of the Sponsor: None of the sponsors had any role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; or preparation, review, or approval of the manuscript.
Footnotes
Authors/Writing Committee: The following members of the Emerging Risk Factors Collaboration take authorship responsibility for the study results: Sebhat Erqou, MD, Stephen Kaptoge, PhD, Philip L. Perry, MBChB, Emanuele Di Angelantonio, MD, University of Cambridge, Cambridge, England; Alexander Thompson, PhD, University of Cambridge; Ian R. White, MSc, MRC Biostatistics Unit, Cambridge, England; Santica M. Marcovina, PhD, University of Washington, Seattle; Rory Collins, FMedSci, University of Oxford, Oxford, England; Simon G. Thompson, DSc, MRC Biostatistics Unit; John Danesh, FRCP, University of Cambridge.
Author Contributions: Drs Erqou and Danesh had full access to all of the data in the study and take responsibility for the integrity of the data and accuracy of the analyses.
Study concept and design: Erqou, Kaptoge, Di Angelantonio, Thompson, White, Marcovina, Thompson, Danesh.
Acquisition of data: Erqou, Kaptoge, Perry, Di Angelantonio, Thompson, White, Collins, Thompson, Danesh.
Analysis and interpretation of data: Erqou, Kaptoge, Perry, Di Angelantonio, Thompson, White, Marcovina, Collins, Thompson, Danesh.
Drafting of the manuscript: Erqou, Di Angelantonio, Danesh.
Critical revision of the manuscript for important in-tellectual content: Erqou, Kaptoge, Perry, Di Angelantonio, Thompson, White, Marcovina, Collins, Thompson, Danesh.
Statistical analysis: Erqou, Kaptoge, Perry, Di Angelantonio, Thompson.
Obtained funding: Danesh.
Administrative, technical, or material support: Kaptoge, Thompson, White, Marcovina, Collins, Thompson, Danesh.
Study supervision: Thompson, Danesh.
Financial Disclosures: Dr Collins reports having received research funding from AstraZeneca, Bayer, British Heart Foundation, Cancer Research UK, European Union, Kadoorie Trust, Medical Research Council, Merck, Schering, Solvay, and UK Biobank. Dr Danesh reports having received research funding from the British Heart Foundation, BUPA Foundation, diaDexus, European Union, Evelyn Trust, GlaxoSmithKline, Medical Research Council, Merck Sharp and Dohme, Roche, and Wellcome Trust. No other disclosures were reported.
Investigators/Contributors: (Appendix 1 in Danesh et al19 lists the study acronyms.) AFTCAPS: Robert W. Tipping, MS, Merck Research Laboratories, United States; ALLHAT: Charles E. Ford, PhD, University of Texas School of Public Health, United States; Lara M. Simpson, PhD, University of Texas School of Public Health, United States; AMORIS: Göran Walldius, MD, Karolinska Institutet, Sweden; Ingmar Jungner, MD, Karolinska Institutet, Sweden; ARIC: Aaron R. Folsom, MD, University of Minnesota, United States; Lloyd Chambless, PhD, University of North Carolina, United States; ATTICA: Demosthenes Panagiotakos, MD, Harokopio University, Greece; Christos Pit-savos, MD, University of Athens, Greece; Christina Chrysohoou, MD, University of Athens, Greece; Christodoulos Stefanadis, MD, University of Athens, Greece; BIP: Uri Goldbourt, PhD, Sheba Medical Center, Israel; Michal Benderly, PhD, Sheba Medical Center, Israel; David Tanne, MD, Sheba Medical Center, Israel; BRHS: Peter Whin-cup, FRCP, University of London, England; S. Goya Wannamethee, PhD, University College London, England; Richard W. Morris, PhD, University College London, England; BRUN: Stefan Kiechl, MD, Medical University Innsbruck, Austria; Johann Willeit, MD, Medical University Innsbruck, Austria; Peter Santer, MD, Bruneck Hospital, Italy; Agnes Mayr, MD, Bruneck Hospital, Italy; BUPA: Nicholas Wald, FRS, Wolfson Institute of Preventive Medicine, England; BWHHS: Shah Ebrahim, DM, London School of Hygiene & Tropical Medicine, England; Debbie Lawlor, PhD, University of Bristol, England; CaPS: John Yarnell, MD, Queen’s University of Belfast, Northern Ireland; John Gallacher, PhD, Cardiff University, Wales; CASTEL: Edoardo Casiglia, MD, University of Padova, Italy; Valerie Tikhonoff, MD, University of Padova, Italy; CHARL: Paul J. Nietert, PhD, Medical University of South Carolina, United States; Susan E. Sutherland, PhD, Medical University of South Carolina, United States; David L. Bachman, MD, Medical University of South Carolina, United States; CHS: Mary Cushman, MD, University of Vermont, United States; Bruce M. Psaty, MD, University of Washington, United States; Russ Tracy, PhD, University of Vermont, United States (see http://chs-nhlbi.org for acknowledgments); COPEN: Anne Tybjærg-Hansen, MD, University of Copenhagen, Denmark; Børge G. Nordestgaard, MD, University of Copenhagen, Denmark; Ruth Frikke-Schmidt, MD, University of Copenhagen, Den-mark; Pia R. Kamstrup, MD, University of Copenhagen, Denmark; CUORE: Simona Giampaoli, MD, Istituto Superiore di Sanità, Italy; Luigi Palmieri, DrStat, Istituto Superiore di Sanità, Italy; Salvatore Panico, MD, Federico II University, Italy; Diego Vanuzzo, MD, Centre for Cardiovascular Prevention, Italy; Lorenza Pilotto, MD, Centre for Cardiovascular Prevention, Italy; DRECE: Agustın Gomez de la Camara, MD, Hospital 12 de Octubre, Spain; Juan A. Gomez Gerique, PhD, Hospital Marques de Valdecilla, Spain; DUBBO: Leon Simons, MD, University of New South Wales, Australia; John McCallum, DPhil, Victoria University, Australia; Yechiel Friedlander, PhD, Hebrew University, Israel; EAS: F. Gerry R. Fowkes, MBChB, University of Edinburgh, Scotland; Amanda Lee, PhD, University of Edinburgh, Scotland; Felicity B. Smith, PhD, University of Edinburgh, Scotland; EPESEBOS: James Taylor, MD, East Boston Neighborhood Health Center, United States; Jack M. Guralnik, MD, US National Institute on Aging, United States; Caroline L. Phillips, MS, US National Institute on Aging, United States; EPESEIOW: Robert B. Wallace, MD, University of Iowa, United States; Jack M. Guralnik, MD, US National Institute on Aging, United States; Caroline L. Phillips, MS, US National Institute on Aging, United States; EPESENCA: Dan G. Blazer, MD, Duke University Medical Centre, United States; Jack M. Guralnik, MD, US National Institute on Aging, United States; Caroline L. Phillips, MS, US National Institute on Aging, United States; EPESENHA: Caroline L. Phillips, MS, US National Institute on Aging, United States; Jack M. Guralnik, MD, US National Institute on Aging, United States; ESTHER: Hermann Brenner, MD, German Cancer Research Center, Germany; Elke Raum, MD, German Cancer Research Center, Germany; Heiko Müller, DrScHum, German Cancer Research Center, Germany; Dietrich Rothenbacher, MD, German Cancer Research Center, Germany; FIA: Jan-Hakan Jansson, MD, Umea University, Sweden; Patrik Wennberg, MD, Umea University, Sweden; FINE_FIN: Aulikki Nissinen, MD, National Institute for Health and Welfare, Finland; FINE_IT: Chiara Donfrancesco, DrStat, Istituto Superiore di Sanità, Italy; Simona Giampaoli, MD, Istituto Superiore di Sanità, Italy; FINRISK92, FINRISK97: Veikko Salomaa, MD, National Institute for Health and Welfare, Finland; Kennet Harald, MA, National Institute for Health and Welfare, Finland; Pekka Jousilahti, MD, National Institute for Health and Welfare, Finland; Erkki Vartiainen, MD, National Institute for Health and Welfare, Finland; FLETCHER: Mark Woodward, PhD, Mount Sinai School of Medicine, United States; FRAMOFF: Ralph B. D’Agostino, PhD, Boston University, United States; Philip A. Wolf, MD, Boston University School of Medicine, United States; Ramachandran S. Vasan, MD, Boston University School of Medicine, United States; Michael J. Pencina, PhD, Boston University, United States; GLOSTRUP: Else-Marie Bladbjerg, PhD, University of Southern Denmark, Denmark; Torben Jørgensen, MD, University of Copenhagen, Denmark; Lars Møller, MD, World Health Organization; Jørgen Jespersen DSc, University of Southern Denmark, Denmark; GOH: Rachel Dankner, MD, Gertner Institute for Epidemiology and Health Policy Research, Israel; Angela Chetrit, MSc, Gertner Institute for Epidemiology and Health Policy Research, Israel; Flora Lubin, RD, Gertner Institute for Epidemiology and Health Policy Research, Israel; GOTO33, GOTO43: Annika Rosengren, MD, Göteborg University, Sweden; Lars Wilhelmsen, MD, Göteborg University, Sweden; Georgios Lappas, Göteborg University, Sweden; Henry Eriksson, MD, Göteborg University, Sweden; GOTOW: Cecilia Björkelund, MD, Göteborg University, Sweden; Lauren Lissner, PhD, Göteborg University, Sweden; Calle Bengtsson, MD, Göteborg University, Sweden; GRIPS: Peter Cremer, MD, Klinikum der Universität München LMU, Germany; Dorothea Nagel, PhD, Uni-versity of Munich, Germany; HELSINAG: Reijo S. Tilvis, MD, Helsinki University Hospital, Finland; Timo E. Strandberg, MD, Oulu University Hospital, Finland; HONOL: Beatriz Rodriguez, MD, University of Hawaii, United States; HOORN: Jacqueline Dekker, PhD, VU University Medical Center, the Netherlands; G. Nijpels, MD, Vrije Universiteit Medical Center, the Netherlands; Coen D.A. Stehouwer, MD, Maastricht University Medical Centre, the Netherlands; HPFS: Eric Rimm, ScD, Harvard University, United States; Jennifer K. Pai, ScD, Brigham and Women’s Hospital, United States; IKNS: Shinichi Sato, MD, Osaka Medical Center for Health Science and Promotion, Japan; Hiroyasu Iso, MD, Osaka University, Japan; Akihiko Kitamura, MD, Osaka Medical Center for Health Science and Promotion, Japan; Hiroyuki Noda, MD, Osaka University, Japan; ISRAEL: Uri Goldbourt, PhD, Sheba Medical Center, Israel; KIHD: Jukka T. Salonen, MD, University of Kuopio, Finland; Kristiina Nyyssönen, PhD, University of Kuopio, Finland; Tomi-Pekka Tuomainen, MD, University of Kuopio, Finland; LASA: Dorly J. H. Deeg, PhD, VU University Medical Centre, the Netherlands; Jan L. Poppelaars, MA, VU University Medical Centre, the Netherlands; MALMO: Bo Hedblad, MD, Lund University, Sweden; Göran Berglund, MD, Lund University, Sweden; Gunnar Engström, MD, Lund University, Sweden; MCVDRFP: W. M. M. Verschuren, PhD, National Institute of Public Health and the Environment, the Netherlands; Anneke Blokstra, MSc, National Institute for Public Health and the Environment, the Netherlands; MOGERAUG1, MOGERAUG2, MOGERAUG3: Angela Döring, MD, German Research Center for Environmental Health, Germany; Wolfgang Koenig, MD, University of Ulm Medical Center, Germany; Christa Meisinger, MD, German Research Center for Environmental Health, Germany; Wilfried Mraz, PhD, University of Munich, Germany; MORGEN: W. M. M. Verschuren, PhD, National Institute of Public Health and the Environment, the Netherlands; Anneke Blokstra, MSc, National Institute for Public Health and the Environment, the Netherlands; H. Bas Bueno-de-Mesquita, PhD, National Institute for Public Health and the Environment, the Netherlands; MOSWEGOT: Lars Wilhelmsen, MD, Göteborg University, Sweden; Annika Rosengren, MD, Göteborg University, Sweden; Georgios Lappas, Göteborg University, Sweden; MRFIT: Lewis H. Kuller, MD, University of Pittsburgh, United States; Greg Grandits, MS, University of Minnesota, United States; NCS: Randi Selmer, PhD, Norwegian Institute of Public Health, Norway; Aage Tverdal, PhD, Norwegian Institute of Public Health, Norway; Wenche Nystad, PhD, Norwegian Institute of Public Health, Norway; NHANES I, NHANES II, NHANES III: R. F. Gillum, MD, Centers for Disease Control and Prevention, United States; Michael Mussolino, PhD, National Institutes of Health, United States; NHS: Eric Rimm, ScD, Harvard University, United States; Sue Hankinson, ScD, Harvard School of Public Health, United States; JoAnn E Manson, MD, Harvard Medical School, United States; Jennifer K. Pai, ScD, Brigham and Women’s Hospital, United States; NORTH KARELIA: Veikko Salomaa, MD, National Institute for Health and Welfare, Finland; Kennet Harald, MA, National Institute for Health and Welfare, Finland; Pekka Jousilahti, MD, National Institute for Health and Welfare, Finland; Erkki Vartiainen, MD, National Institute for Health and Welfare, Finland; NPHS II: Jackie A. Cooper, MSc, University College London, England; Kenneth A Bauer, MD, Harvard Medical School, United States; OSAKA: Shinichi Sato, MD, Osaka Medical Center for Health Science and Promotion, Japan; Akihiko Kitamura, MD, Osaka Medical Center for Health Science and Promotion, Japan; Yoshihiko Naito, MD, Mukogawa Women’s University, Japan; Hiroyasu Iso, MD, Osaka University, Japan; OSLO: Ingar Holme, PhD, Oslo University Hospital, Norway; Randi Selmer, PhD, Norwegian Institute of Public Health, Norway; Aage Tverdal, PhD, Norwegian Institute of Public Health, Norway; Wenche Nystad, PhD, Norwegian Institute of Public Health, Norway; OYABE: Hideaki Nakagawa, MD, Kanazawa Medical University, Japan; Katsuyuki Miura, MD, Shiga University of Medical Science, Japan; PARIS1: Pierre Ducimetiere, PhD, INSERM, France; Xavier Jouven, MD, INSERM, France; Gerald Luc, MD, University of Lille, France; PRHHP: Carlos J. Crespo, DrPH, Portland State University, United States; Mario R. Garcia Palmieri, MD, University of Puerto Rico, United States; PRIME: Philippe Amouyel, MD, Institut Pasteur de Lille, France; Dominique Arveiler, MD, Universite de Strasbourg, France; Alun Evans, MD, The Queens University of Belfast, Northern Ireland; Jean Ferrieres, MD, University of Toulouse, France; PROCAM: Helmut Schulte, PhD, Assmann-Stiftung für Prävention, Germany; Gerd Assmann FRCP, Assmann-Stiftung für Prävention, Germany; PROSPER: James Shepherd, MD, Glasgow Royal Infirmary, Scotland; Chris J. Packard, DSc, University of Glasgow, Scotland; Naveed Sattar, FRC Path, University of Glasgow, Scotland; Ian Ford, PhD, University of Glasgow, Scotland; QUEBEC: Bernard Cantin, MD, Institutde Cardiologie de Quebec, Ho pital Laval, Canada; Benoıt Lamarche, PhD, Laval University, Canada; Jean-Pierre Despres, PhD, Centre de Recherche de l’Institut Universitaire de Cardiologie et de Pneumologie de Quebec, Canada; Gilles R. Dagenais. MD, Institut Universitaire de Cardiologie et Pneumologie de Quebec, Canada; RANCHO: Elizabeth Barrett-Connor, MD, University of California, United States; Lori B. Daniels, MD, University of California, United States; Gail A. Laughlin, PhD, University of California, United States; REYK: Vilmundur Gudnason, MD, University of Iceland, Iceland; Thor Aspelund, PhD, University of Iceland, Iceland; Gunnar Sigurdsson, MD, University of Iceland, Iceland; Bolli Thorsson, MD, Icelandic Heart Association, Iceland; RIFLE: Maurizio Trevisan, MD, Nevada System of Higher Education, United States; ROTT: Jacqueline Witteman, PhD, Erasmus MC, the Netherlands; Isabella Kardys, MD, Erasmus MC, the Netherlands; Monique M. B. Breteler, MD, Erasmus MC, the Netherlands; Albert Hofman, MD, Erasmus MC, the Netherlands; SHHEC: Hugh Tunstall-Pedoe, MD, University of Dundee, Scotland; Roger Tavendale, PhD, University of Dundee, Scotland; Gordon Lowe, DSc, University of Glasgow, Scotland; Mark Woodward, PhD, Mount Sinai School of Medicine, United States; SPEED: Yoav Ben-Shlomo, PhD, University of Bristol, England; George Davey-Smith, MD, University of Bristol, England; SHS: Barbara V. Howard, PhD, Medstar Research Institute, United States; Ying Zhang, MD, University of Oklahoma Health Sciences Center, United States; Lyle Best, MD, Missouri Breaks Industries Research Inc, United States; Jason Umans, MD, Georgetown University Medical Center, United States; TARFS: Altan Onat, MD, Istanbul University, Turkey; TROMSØ: Inger Njølstad, MD, University of Tromsø, Norway; Ellisiv B. Mathiesen, MD, University of Tromsø, Norway; Maja-Lisa Løchen, PhD, University of Tromsø, Norway; Tom Wilsgaard, PhD, University of Tromsø, Norway; ULSAM: Erik Ingelsson, MD, Karolinska Institutet, Sweden; Johan Sundström, MD, Uppsala University, Sweden; Lars Lind, MD, Uppsala University Hospital, Sweden; Lars Lannfelt, MD, Uppsala University, Sweden; USPHS: J. Michael Gaziano, MD, Brigham and Women’s Hospital, United States; Meir Stampfer, MD, Harvard School of Public Health, United States; Paul M Ridker, MD, Brigham and Women’s Hospital, United States; USPHS2: J. Michael Gaziano, MD, Brigham and Women’s Hospital, United States; Paul M Ridker, MD, Brigham and Women’s Hospital, United States; VHMPP: Hanno Ulmer, PhD, Innsbruck Medical University, Austria; Günter Diem, MD, Agency for Preventive and Social Medicine, Austria; Hans Concin, MD, Agency for Preventive and Social Medicine, Austria; VITA: Alberto Tosetto, MD, San Bortolo Hospital, Italy; Francesco Rodeghiero, MD, San Bartolo Hospital, Italy; WHITEI: Michael Marmot, FMedSci, University College London, England; Robert Clarke, MD, University of Oxford, England; Rory Collins, FMedSci, University of Oxford, England; Astrid Fletcher, PhD, London School of Hygiene and Tropical Medicine, England; WHITE II: Eric Brunner, PhD, University College London, England; Martin Shipley, MSc, University College London, England; WHS: Paul M Ridker, MD, Brigham and Women’s Hospital, United States; Julie Buring, ScD, Brigham and Women’s Hospital, United States; WOSCOPS: James Shepherd, MD, Glasgow Royal Infirmary, Scotland; Stuart Cobbe, FMedSci, BHF Glasgow Cardiovascular Research Centre, Scotland; Ian Ford, PhD, University of Glasgow, Scotland; Michele Robertson, BSc, University of Glasgow, Scotland; XIAN: Yao He, MD, Chinese PLA General Hospital, China; ZARAGOZA: Alejandro Marin Ibanez, MD, San Jose Norte Health Centre, Spain; ZUTE: Edith Feskens, PhD, Wageningen University, the Netherlands; Daan Kromhout, PhD, Wageningen University, the Netherlands.
Investigators/Data Management Team: Matthew Walker, PhD, University of Cambridge, England; Sarah Watson, MMath, University of Cambridge, England.
Investigators/Coordinating Center: Rory Collins, FMedSci, University of Oxford, England; Emanuele Di Angelantonio, MD, University of Cambridge, England; Sebhat Erqou, MD, University of Cambridge, England; Stephen Kaptoge, PhD, University of Cambridge, England; Sarah Lewington, DPhil, University of Oxford, England; Lia Orfei, MSc, University of Cambridge, England; Lisa Pennells, MSc, University of Cambridge, England; Philip L. Perry, MBChB, University of Cambridge, England; Kausik K. Ray, MD, University of Cambridge, England; Nadeem Sarwar, PhD, University of Cambridge, England; Myriam Alexander, MPhil, University of Cambridge, England; Alexander Thompson, PhD, University of Cambridge, England; Simon G. Thompson, DSc, MRC Biostatistics Unit, England; Matthew Walker, PhD, University of Cambridge, England; Sarah Watson, MMath, University of Cambridge, England; Frances Wensley, MSc, University of Cambridge, England; Ian R. White, MSc, MRC Biostatistics Unit, England; Angela M. Wood, PhD, University of Cambridge, England; John Danesh, FRCP, University of Cambridge, England (principal investigator).
Additional Information: eAppendixes 1 through 3, eTables 1 through 5, and eFigures 1 through 8 are available online at http://www.jama.com.
Additional Contributions: Hannah Sneath and Angela Harper, University of Cambridge, provided secretarial support.
REFERENCES
- 1.Marcovina SM, Koschinsky ML. Lipoprotein(a) as a risk factor for coronary artery disease. Am J Cardiol. 1998;82(12A):57U–66U. doi: 10.1016/s0002-9149(98)00954-0. [DOI] [PubMed] [Google Scholar]
- 2.Anuurad E, Boffa MB, Koschinsky ML, Berglund L. Lipoprotein(a): a unique risk factor for cardiovascular disease. Clin Lab Med. 2006;26(4):751–772. doi: 10.1016/j.cll.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Hobbs HH, White AL. Lipoprotein(a): intrigues and insights. Curr Opin Lipidol. 1999;10(3):225–236. doi: 10.1097/00041433-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Nielsen LB, Gronholdt MLM, Schroeder TV, Stender S, Nordestgaard BG. In vivo transfer of lipoprotein(a) into human atherosclerotic carotid arterial intima. Arterioscler Thromb Vasc Biol. 1997;17(5):905–911. doi: 10.1161/01.atv.17.5.905. [DOI] [PubMed] [Google Scholar]
- 5.Boffa MB, Marcovina SM, Koschinsky ML. Lipo-protein(a) as a risk factor for atherosclerosis and thrombosis: mechanistic insights from animal models. Clin Biochem. 2004;37(5):333–343. doi: 10.1016/j.clinbiochem.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Poon M, Zhang X, Dunsky KG, Taubman MB, Harpel PC. Apolipoprotein(a) induces monocyte chemotactic activity in human vascular endothelial cells. Circulation. 1997;96(8):2514–2519. doi: 10.1161/01.cir.96.8.2514. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen LB, Juul K, Nordestgaard BG. Increased degradation of lipoprotein(a) in atherosclerotic com-pared with nonlesioned aortic intima-inner media of rabbits: in vivo evidence that lipoprotein(a) may con-tribute to foam cell formation. Arterioscler Thromb Vasc Biol. 1998;18(4):641–649. doi: 10.1161/01.atv.18.4.641. [DOI] [PubMed] [Google Scholar]
- 8.Craig WY, Neveux LM, Palomaki GE, Cleveland MM, Haddow JE. Lipoprotein(a) as a risk factor for ischemic heart disease: metaanalysis of prospective studies. Clin Chem. 1998;44(11):2301–2306. [PubMed] [Google Scholar]
- 9.Danesh J, Collins R, Peto R. Lipoprotein(a) and coronary heart disease: meta-analysis of prospective studies. Circulation. 2000;102(10):1082–1085. doi: 10.1161/01.cir.102.10.1082. [DOI] [PubMed] [Google Scholar]
- 10.Bennet A, Di Angelantonio E, Erqou S, et al. Lipoprotein(a) levels and risk of future coronary heart disease: large-scale prospective data. Arch Intern Med. 2008;168(6):598–608. doi: 10.1001/archinte.168.6.598. [DOI] [PubMed] [Google Scholar]
- 11.Smolders B, Lemmens R, Thijs V. Lipoprotein (a) and stroke: a meta-analysis of observational studies. Stroke. 2007;38(6):1959–1966. doi: 10.1161/STROKEAHA.106.480657. [DOI] [PubMed] [Google Scholar]
- 12.Dahlen GH, Guyton JR, Attar M, Farmer JA, Kautz JA, Gotto AM., Jr Association of levels of lipoprotein Lp(a), plasma lipids, and other lipoproteins with coronary artery disease documented by angiography. Circulation. 1986;74(4):758–765. doi: 10.1161/01.cir.74.4.758. [DOI] [PubMed] [Google Scholar]
- 13.Braeckman L, De Bacquer D, Rosseneu M, De Backer G. Determinants of lipoprotein(a) levels in a middle-aged working population. Eur Heart J. 1996;17(12):1808–1813. doi: 10.1093/oxfordjournals.eurheartj.a014796. [DOI] [PubMed] [Google Scholar]
- 14.Rifai N, Ma J, Sacks FM, et al. Apolipoprotein(a) size and lipoprotein(a) concentration and future risk of angina pectoris with evidence of severe coronary atherosclerosis in men: the Physicians’ Health Study. Clin Chem. 2004;50(8):1364–1371. doi: 10.1373/clinchem.2003.030031. [DOI] [PubMed] [Google Scholar]
- 15.Suk Danik J, Rifai N, Buring JE, Ridker PM. Lipoprotein(a), measured with an assay independent of apolipoprotein(a) isoform size, and risk of future cardiovascular events among initially healthy women. JAMA. 2006;296(11):1363–1370. doi: 10.1001/jama.296.11.1363. [DOI] [PubMed] [Google Scholar]
- 16.Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117(2):176–184. doi: 10.1161/CIRCULATIONAHA.107.715698. [DOI] [PubMed] [Google Scholar]
- 17.Clarke R, Shipley M, Lewington S, et al. Under-estimation of risk associations due to regression dilution in long-term follow-up of prospective studies. Am J Epidemiol. 1999;150(4):341–353. doi: 10.1093/oxfordjournals.aje.a010013. [DOI] [PubMed] [Google Scholar]
- 18.Wood AM, White I, Thompson SG, Lewington S, Danesh J, Fibrinogen Studies Collaboration Regression dilution methods for meta-analysis: assessing long-term variability in plasma fibrinogen among 27 247 adults in 15 prospective studies. Int J Epidemiol. 2006;35(6):1570–1578. doi: 10.1093/ije/dyl233. [DOI] [PubMed] [Google Scholar]
- 19.Danesh J, Erqou S, Walker M, et al. Emerging Risk Factors Collaboration The Emerging Risk Factors Collaboration: analysis of individual data on lipid, inflammatory and other markers in over 1.1 million participants in 104 prospective studies of cardiovascular diseases. Eur J Epidemiol. 2007;22(12):839–869. doi: 10.1007/s10654-007-9165-7. [DOI] [PubMed] [Google Scholar]
- 20.Sharrett AR, Ballantyne CM, Coady SA, et al. Atherosclerosis Risk in Communities Study Group Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2001;104(10):1108–1113. doi: 10.1161/hc3501.095214. [DOI] [PubMed] [Google Scholar]
- 21.Pitsavos C, Panagiotakos DB, Chrysohoou C, Stefanadis C. Epidemiology of cardiovascular risk factors in Greece: aims, design and baseline characteristics of the ATTICA study. BMC Public Health. 2003;3:32. doi: 10.1186/1471-2458-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kronenberg F, Kronenberg MF, Kiechl S, et al. Role of lipoprotein(a) and apolipoprotein(a) phenotype in atherogenesis: prospective results from the Bruneck study. Circulation. 1999;100(11):1154–1160. doi: 10.1161/01.cir.100.11.1154. [DOI] [PubMed] [Google Scholar]
- 23.Ariyo AA, Thach C, Tracy R, Cardiovascular Health Study Investigators Lp(a) lipoprotein, vascular disease, and mortality in the elderly. N Engl J Med. 2003;349(22):2108–2115. doi: 10.1056/NEJMoa001066. [DOI] [PubMed] [Google Scholar]
- 24.Keil JE, Loadholt CB, Weinrich MC, Sandifer SH, Boyle E., Jr Incidence of coronary heart disease in blacks in Charleston, South Carolina. Am Heart J. 1984;108(3 pt 2):779–786. doi: 10.1016/0002-8703(84)90671-9. [DOI] [PubMed] [Google Scholar]
- 25.Simons LA, Simons J, Friedlander Y, McCallum J. Risk factors for acute myocardial infarction in the elderly (the Dubbo study) Am J Cardiol. 2002;89(1):69–72. doi: 10.1016/s0002-9149(01)02168-3. [DOI] [PubMed] [Google Scholar]
- 26.Price JF, Lee AJ, Rumley A, Lowe GD, Fowkes FG. Lipoprotein(a) and development of intermittent claudication and major cardiovascular events in men and women: the Edinburgh Artery Study. Atherosclerosis. 2001;157(1):241–249. doi: 10.1016/s0021-9150(00)00719-x. [DOI] [PubMed] [Google Scholar]
- 27.Rajecki M, Pajunen P, Jousilahti P, Rasi V, Vahtera E, Salomaa V. Hemostatic factors as predictors of stroke and cardiovascular diseases: the FINRISK ’92 Hemostasis Study. Blood Coagul Fibrinolysis. 2005;16(2):119–124. doi: 10.1097/01.mbc.0000161565.74387.5b. [DOI] [PubMed] [Google Scholar]
- 28.Bostom AG, Cupples LA, Jenner JL, et al. Elevated plasma lipoprotein(a) and coronary heart disease in men aged 55 years and younger: a prospective study. JAMA. 1996;276(7):544–548. doi: 10.1001/jama.1996.03540070040028. [DOI] [PubMed] [Google Scholar]
- 29.Lubin F, Chetrit A, Lusky A, Modan M. Method-ology of a two-step quantified nutritional questionnaire and its effect on results. Nutr Cancer. 1998;30(1):78–82. doi: 10.1080/01635589809514645. [DOI] [PubMed] [Google Scholar]
- 30.Cremer P, Nagel D, Mann H, et al. Ten-year follow-up results from the Goettingen Risk, Incidence and Prevalence Study (GRIPS), I: risk factors for myocardial infarction in a cohort of 5790 men. Atherosclerosis. 1997;129(2):221–230. doi: 10.1016/s0021-9150(96)06030-3. [DOI] [PubMed] [Google Scholar]
- 31.Lakka HM, Lakka TA, Tuomilehto J, Sivenius J, Salonen JT. Hyperinsulinemia and the risk of cardiovascular death and acute coronary and cerebrovascular events in men: the Kuopio Ischaemic Heart Disease Risk Factor Study. Arch Intern Med. 2000;160(8):1160–1168. doi: 10.1001/archinte.160.8.1160. [DOI] [PubMed] [Google Scholar]
- 32.Gardner CD, Winkleby MA, Fortmann SP. Population frequency distribution of non-high-density lipoprotein cholesterol (Third National Health and Nutrition Examination Survey [NHANES III], 1988-1994) Am J Cardiol. 2000;86(3):299–304. doi: 10.1016/s0002-9149(00)00918-8. [DOI] [PubMed] [Google Scholar]
- 33.Seed M, Ayres KL, Humphries SE, Miller GJ. Lipoprotein(a) as a predictor of myocardial infarction in middle-aged men. Am J Med. 2001;110(1):22–27. doi: 10.1016/s0002-9343(00)00652-5. [DOI] [PubMed] [Google Scholar]
- 34.Luc G, Bard JM, Arveiler D, et al. PRIME Study Group Lipoprotein(a) as a predictor of coronary heart disease: the PRIME Study. Atherosclerosis. 2002;163(2):377–384. doi: 10.1016/s0021-9150(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 35.Assmann G, Schulte H, von Eckardstein A. Hypertriglyceridemia and elevated lipoprotein(a) are risk factors for major coronary events in middle-aged men. Am J Cardiol. 1996;77(14):1179–1184. doi: 10.1016/s0002-9149(96)00159-2. [DOI] [PubMed] [Google Scholar]
- 36.Cantin B, Gagnon F, Moorjani S, et al. Is lipoprotein(a) an independent risk factor for ischemic heart disease in men? the Quebec Cardiovascular Study. J Am Coll Cardiol. 1998;31(3):519–525. doi: 10.1016/s0735-1097(97)00528-7. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Hu D, Lee ET, et al. Lipoprotein(a) in American Indians is low and not independently associated with cardiovascular disease: the Strong Heart Study. Ann Epidemiol. 2002;12(2):107–114. doi: 10.1016/s1047-2797(01)00273-3. [DOI] [PubMed] [Google Scholar]
- 38.Onat A. Risk factors and cardiovascular disease in Turkey. Atherosclerosis. 2001;156(1):1–10. doi: 10.1016/s0021-9150(01)00500-7. [DOI] [PubMed] [Google Scholar]
- 39.Ingelsson E, Arnlov J, Sundstrom J, Zethelius B, Vessby B, Lind L. Novel metabolic risk factors for heart failure. J Am Coll Cardiol. 2005;46(11):2054–2060. doi: 10.1016/j.jacc.2005.07.059. [DOI] [PubMed] [Google Scholar]
- 40.Marmot MG, Smith GD, Stansfeld S, et al. Health inequalities among British civil servants: the Whitehall II study. Lancet. 1991;337(8754):1387–1393. doi: 10.1016/0140-6736(91)93068-k. [DOI] [PubMed] [Google Scholar]
- 41.Stehouwer CD, Weijenberg MP, van den Berg M, et al. Serum homocysteine and risk of coronary heart disease and cerebrovascular disease in elderly men: a 10-year follow-up. Arterioscler Thromb Vasc Biol. 1998;18(12):1895–1901. doi: 10.1161/01.atv.18.12.1895. [DOI] [PubMed] [Google Scholar]
- 42.Downs JR, Beere PA, Whitney E, et al. Design and rationale of the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) Am J Cardiol. 1997;80(3):287–293. doi: 10.1016/s0002-9149(97)00347-0. [DOI] [PubMed] [Google Scholar]
- 43.Gaw A, Brown EA, Docherty G, Ford I. Is lipoprotein (a)-cholesterol a better predictor of vascular disease events than total lipoprotein(a) mass? a nested case control study from the West of Scotland Coronary Prevention Study. Atherosclerosis. 2000;148(1):95–100. doi: 10.1016/s0021-9150(99)00259-2. [DOI] [PubMed] [Google Scholar]
- 44.Wald NJ, Law M, Watt HC, et al. Apolipoproteins and ischaemic heart disease: implications for screening. Lancet. 1994;343(8889):75–79. doi: 10.1016/s0140-6736(94)90814-1. [DOI] [PubMed] [Google Scholar]
- 45.Dahlen GH, Weinehall L, Stenlund H, et al. Lipoprotein(a) and cholesterol levels act synergistically and apolipoprotein A-I is protective for the incidence of primary acute myocardial infarction in middle-aged males: an incident case-control study from Sweden. J Intern Med. 1998;244(5):425–430. doi: 10.1046/j.1365-2796.1998.00422.x. [DOI] [PubMed] [Google Scholar]
- 46.Woodward M, Rumley A, Welsh P, Macmahon S, Lowe G. A comparison of the associations between seven hemostatic or inflammatory variables and coronary heart disease. J Thromb Haemost. 2007;5(9):1795–1800. doi: 10.1111/j.1538-7836.2007.02677.x. [DOI] [PubMed] [Google Scholar]
- 47.Pai JK, Pischon T, Ma J, et al. Inflammatory markers and the risk of coronary heart disease in men and women. N Engl J Med. 2004;351(25):2599–2610. doi: 10.1056/NEJMoa040967. [DOI] [PubMed] [Google Scholar]
- 48.Evans RW, Shpilberg O, Shaten BJ, Ali S, Kamboh MI, Kuller LH. Prospective association of lipoprotein(a) concentrations and apo(a) size with coronary heart disease among men in the Multiple Risk Factor Intervention Trial. J Clin Epidemiol. 2001;54(1):51–57. doi: 10.1016/s0895-4356(00)00260-2. [DOI] [PubMed] [Google Scholar]
- 49.Shai I, Rimm EB, Hankinson SE, et al. Lipoprotein (a) and coronary heart disease among women: beyond a cholesterol carrier? Eur Heart J. 2005;26(16):1633–1639. doi: 10.1093/eurheartj/ehi222. [DOI] [PubMed] [Google Scholar]
- 50.Shaper AG, Pocock SJ, Walker M, Cohen NM, Wale CJ, Thomson AG. British Regional Heart Study: cardiovascular risk factors in middle-aged men in 24 towns. Br Med J (Clin Res Ed) 1981;283(6285):179–186. doi: 10.1136/bmj.283.6285.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosengren A, Wilhelmsen L, Eriksson E, Risberg B, Wedel H. Lipoprotein (a) and coronary heart disease: a prospective case-control study in a general population sample of middle aged men. BMJ. 1990;301(6763):1248–1251. doi: 10.1136/bmj.301.6763.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ridker PM, Hennekens CH, Stampfer MJ. A pro-spective study of lipoprotein(a) and the risk of myocardial infarction. JAMA. 1993;270(18):2195–2199. [PubMed] [Google Scholar]
- 53.D’Angelo A, Ruotolo G, Garancini P, Sampietro F, Mazzola G, Calori G. Lipoprotein(a), fibrinogen and vascular mortality in an elderly northern Italian population. Haematologica. 2006;91(12):1613–1620. [PubMed] [Google Scholar]
- 54.Sweetnam PM, Bolton CH, Downs LG, et al. Apo-lipoproteins A-I, A-II and B, lipoprotein(a) and the risk of ischaemic heart disease: the Caerphilly study. Eur J Clin Invest. 2000;30(11):947–956. doi: 10.1046/j.1365-2362.2000.00725.x. [DOI] [PubMed] [Google Scholar]
- 55.Klausen IC, Sjol A, Hansen PS, et al. Apolipoprotein (a) isoforms and coronary heart disease in men: a nested case-control study. Atherosclerosis. 1997;132(1):77–84. doi: 10.1016/s0021-9150(97)00071-3. [DOI] [PubMed] [Google Scholar]
- 56.Coleman MP, Key TJ, Wang DY, et al. A prospective study of obesity, lipids, apolipoproteins and ischaemic heart disease in women. Atherosclerosis. 1992;92(2-3):177–185. doi: 10.1016/0021-9150(92)90276-m. [DOI] [PubMed] [Google Scholar]
- 57.Jauhiainen M, Koskinen P, Ehnholm C, et al. Lipoprotein (a) and coronary heart disease risk: a nested case-control study of the Helsinki Heart Study participants. Atherosclerosis. 1991;89(1):59–67. doi: 10.1016/0021-9150(91)90007-p. [DOI] [PubMed] [Google Scholar]
- 58.Schaefer EJ, Lamon-Fava S, Jenner JL, et al. Lipoprotein(a) levels and risk of coronary heart disease in men: the Lipid Research Clinics Coronary Primary Prevention Trial. JAMA. 1994;271(13):999–1003. doi: 10.1001/jama.1994.03510370051031. [DOI] [PubMed] [Google Scholar]
- 59.Alfthan G, Pekkanen J, Jauhiainen M, et al. Relation of serum homocysteine and lipoprotein(a) concentrations to atherosclerotic disease in a prospective Finnish population based study. Atherosclerosis. 1994;106(1):9–19. doi: 10.1016/0021-9150(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 60.Gaw A, Murray HM, Brown EA, PROSPER Study Group Plasma lipoprotein(a) [Lp(a)] concentrations and cardiovascular events in the elderly: evidence from the prospective study of pravastatin in the elderly at risk (PROSPER) Atherosclerosis. 2005;180(2):381–388. doi: 10.1016/j.atherosclerosis.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 61.Wild SH, Fortmann SP, Marcovina SM. A prospective case-control study of lipoprotein(a) levels and apo(a) size and risk of coronary heart disease in Stanford Five-City Project participants. Arterioscler Thromb Vasc Biol. 1997;17(2):239–245. doi: 10.1161/01.atv.17.2.239. [DOI] [PubMed] [Google Scholar]
- 62.Nguyen TT, Ellefson RD, Hodge DO, Bailey KR, Kottke TE, Abu-Lebdeh HS. Predictive value of electrophoretically detected lipoprotein(a) for coronary heart disease and cerebrovascular disease in a com-munity-based cohort of 9936 men and women. Circulation. 1997;96(5):1390–1397. doi: 10.1161/01.cir.96.5.1390. [DOI] [PubMed] [Google Scholar]
- 63.Bostom AG, Gagnon DR, Cupples LA, et al. A prospective investigation of elevated lipoprotein (a) detected by electrophoresis and cardiovascular disease in women: the Framingham Heart Study. Circulation. 1994;90(4):1688–1695. doi: 10.1161/01.cir.90.4.1688. [DOI] [PubMed] [Google Scholar]
- 64.Dahlen G. Lipoprotein (a) as a risk factor for atherosclerotic diseases. Arctic Med Res. 1988;47(Suppl 1):458–461. [PubMed] [Google Scholar]
- 65.Easton DF, Peto J, Babiker A. Floating absolute risk: an alternative to relative risk in survival and case-control analysis avoiding an arbitrary reference group. Stat Med. 1991;10(7):1025–1035. doi: 10.1002/sim.4780100703. [DOI] [PubMed] [Google Scholar]
- 66.Danesh J, Lewington S, Thompson SG, et al. Fi-brinogen Studies Collaboration Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294(14):1799–1809. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 67.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21(11):1539–1558. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 68.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Knuiman MW, Divitini ML, Buzas JS, Fitzgerald PE. Adjustment for regression dilution in epidemiological regression analyses. Ann Epidemiol. 1998;8(1):56–63. doi: 10.1016/s1047-2797(97)00107-5. [DOI] [PubMed] [Google Scholar]
- 70.Rosner B, Willett WC, Spiegelman D. Correction of logistic regression relative risk estimates and confidence intervals for systematic within-person measurement error. Stat Med. 1989;8(9):1051–1069. doi: 10.1002/sim.4780080905. [DOI] [PubMed] [Google Scholar]
- 71.Rosner B, Spiegelman D, Willett WC. Correction of logistic regression relative risk estimates and confidence intervals for measurement error: the case of multiple covariates measured with error. Am J Epidemiol. 1990;132(4):734–745. doi: 10.1093/oxfordjournals.aje.a115715. [DOI] [PubMed] [Google Scholar]
- 72.Fibrinogen Studies Collaboration Correcting for multivariate measurement error by regression calibration in meta-analyses of epidemiological studies. Stat Med. 2009;28(7):1067–1092. doi: 10.1002/sim.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tate JR, Rifai N, Berg K, et al. International Federation of Clinical Chemistry standardization project for the measurement of lipoprotein(a), I: evaluation of the analytical performance of lipoprotein(a) assay systems and commercial calibrators. Clin Chem. 1998;44(8 pt 1):1629–1640. [PubMed] [Google Scholar]
- 74.Andresdottir MB, Sigfusson N, Sigvaldason H, Gudnason V. Erythrocyte sedimentation rate, an independent predictor of coronary heart disease in men and women: the Reykjavik Study. Am J Epidemiol. 2003;158(9):844–851. doi: 10.1093/aje/kwg222. [DOI] [PubMed] [Google Scholar]
- 75.Gillum RF, Mussolino ME, Madans JH. Counts of neutrophils, lymphocytes, and monocytes, cause-specific mortality and coronary heart disease: the NHANES-I epidemiologic follow-up study. Ann Epidemiol. 2005;15(4):266–271. doi: 10.1016/j.annepidem.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 76.Boerwinkle E, Leffert CC, Lin J, Lackner C, Chiesa G, Hobbs HH. Apolipoprotein(a)geneaccountsforgreater than 90% of the variation in plasma lipoprotein(a) concentrations. J Clin Invest. 1992;90(1):52–60. doi: 10.1172/JCI115855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mooser V, Scheer D, Marcovina SM, et al. The Apo(a) gene is the major determinant of variation in plasma Lp(a) levels in African Americans. Am J Hum Genet. 1997;61(2):402–417. doi: 10.1086/514851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boomsma DI, Knijff P, Kaptein A, et al. The effect of apolipoprotein(a)-, apolipoprotein E-, and apolipoprotein A4-polymorphisms on quantitative lipoprotein(a) concentrations. Twin Res. 2000;3(3):152–158. doi: 10.1375/136905200320565436. [DOI] [PubMed] [Google Scholar]
- 79.Tregouët DA, Konig IR, Erdmann J, et al. Wellcome Trust Case Control Consortium. Cardiogenics Consortium Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41(3):283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 80.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(22):2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 81.Kostner KM, Kostner GM. Factors affecting plasma lipoprotein(a) levels: role of hormones and other nongenetic factors. Semin Vasc Med. 2004;4(2):211–214. doi: 10.1055/s-2004-835380. [DOI] [PubMed] [Google Scholar]
- 82.Insull W, Jr, McGovern ME, Schrott H, et al. Efficacy of extended-release niacin with lovastatin for hypercholesterolemia: assessing all reasonable doses with innovative surface graph analysis. Arch Intern Med. 2004;164(10):1121–1127. doi: 10.1001/archinte.164.10.1121. [DOI] [PubMed] [Google Scholar]
- 83.Marcovina SM, Koschinsky ML, Albers JJ, Skarlatos S. Report of the National Heart, Lung, and Blood Institute Workshop on Lipoprotein(a) and Cardiovascular Disease: recent advances and future directions. Clin Chem. 2003;49:1785–1796. doi: 10.1373/clinchem.2003.023689. [DOI] [PubMed] [Google Scholar]
- 84.Kostner GM, Gavish D, Leopold B, Bolzano K, Weintraub MS, Breslow JL. HMG CoA reductase inhibitors lower LDL cholesterol without reducing Lp(a) levels. Circulation. 1989;80(5):1313–1319. doi: 10.1161/01.cir.80.5.1313. [DOI] [PubMed] [Google Scholar]
- 85.Maher VM, Brown BG, Marcovina SM, Hillger LA, Zhao XQ, Albers JJ. Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein(a) JAMA. 1995;274(22):1771–1774. [PubMed] [Google Scholar]
- 86.Berg K, Dahlen G, Christophersen B, Cook T, Kjekshus J, Pedersen T. Lp(a) lipoprotein level predicts survival and major coronary events in the Scandinavian Simvastatin Survival Study. Clin Genet. 1997;52(5):254–261. doi: 10.1111/j.1399-0004.1997.tb04342.x. [DOI] [PubMed] [Google Scholar]
- 87.Treatment of HDL to reduce the incidence of vas-cular events HPS2-THRIVE. http://clinicaltrials.gov/ct2/show/NCT00461630. Accessed July 1, 2009.
- 88.Merki E, Graham MJ, Mullick AE, et al. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118(7):743–753. doi: 10.1161/CIRCULATIONAHA.108.786822. [DOI] [PubMed] [Google Scholar]
- 89.Kastelein JJ, Wedel MK, Baker BF, et al. Potent reduction of apolipoprotein B and low-density lipo-protein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation. 2006;114(16):1729–1735. doi: 10.1161/CIRCULATIONAHA.105.606442. [DOI] [PubMed] [Google Scholar]
- 90.Dahlen G, Ericson C, Furberg C, Lundkvist L, Svardsudd K. Studies on an extra pre-beta lipoprotein fraction. Acta Med Scand Suppl. 1972;531:1–29. [PubMed] [Google Scholar]
- 91.Marcovina SM, Albers JJ, Scanu AM, et al. Use of a reference material proposed by the International Federation of Clinical Chemistry and Laboratory Medicine to evaluate analytical methods for the determination of plasma lipoprotein(a) Clin Chem. 2000;46(12):1956–1967. [PubMed] [Google Scholar]
- 92.Dati F, Tate JR, Marcovina SM, Steinmetz A, International Federation of Clinical Chemistry and Laboratory Medicine. IFCC Working Group for Lipoprotein(a) Assay Standardization First WHO/IFCC international reference reagent for lipoprotein(a) for immunoassay—Lp(a) SRM 2B. Clin Chem Lab Med. 2004;42(6):670–676. doi: 10.1515/CCLM.2004.114. [DOI] [PubMed] [Google Scholar]
- 93.McLean JW, Tomlinson JE, Kuang WJ, et al. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987;330(6144):132–137. doi: 10.1038/330132a0. [DOI] [PubMed] [Google Scholar]
- 94.Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP. Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a) Clin Chem. 1995;41(2):246–255. [PubMed] [Google Scholar]
- 95.Barlera S, Specchia C, Farrall M, et al. PROCARDIS Consortium Multiple QTL influence the serum Lp(a) concentration: a genome-wide linkage screen in the PROCARDIS study. Eur J Hum Genet. 2007;15(2):221–227. doi: 10.1038/sj.ejhg.5201732. [DOI] [PubMed] [Google Scholar]
- 96.Paultre F, Pearson TA, Weil HF, et al. High levels of Lp(a) with a small apo(a) isoform are associated with coronary artery disease in African American and white men. Arterioscler Thromb Vasc Biol. 2000;20(12):2619–2624. doi: 10.1161/01.atv.20.12.2619. [DOI] [PubMed] [Google Scholar]
- 97.Anand SS, Yusuf S, Vuksan V, et al. Differences in risk factors, atherosclerosis, and cardiovascular disease between ethnic groups in Canada: the Study of Health Assessment and Risk in Ethnic Groups (SHARE) Lancet. 2000;356(9226):279–284. doi: 10.1016/s0140-6736(00)02502-2. [DOI] [PubMed] [Google Scholar]
- 98.Holmer SR, Hengstenberg C, Kraft HG, et al. Association of polymorphisms of the apolipoprotein(a) gene with lipoprotein(a) levels and myocardial infarction. Circulation. 2003;107(5):696–701. doi: 10.1161/01.cir.0000048125.79640.77. [DOI] [PubMed] [Google Scholar]
- 99.Kiechl S, Willeit J, Mayr M, et al. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler Thromb Vasc Biol. 2007;27(8):1788–1795. doi: 10.1161/ATVBAHA.107.145805. [DOI] [PubMed] [Google Scholar]
- 100.Tsimikas S, Brilakis ES, Miller ER, et al. Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353(1):46–57. doi: 10.1056/NEJMoa043175. [DOI] [PubMed] [Google Scholar]
- 101.Tsimikas S, Tsironis LD, Tselepis AD. New insights into the role of lipoprotein(a)-associated lipo-protein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2007;27(10):2094–2099. doi: 10.1161/01.ATV.0000280571.28102.d4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.