

Abstract
Spinal muscular atrophies (SMA, also known as hereditary motor neuropathies) and hereditary motor and sensory neuropathies (HMSN) are clinically and genetically heterogeneous disorders of the peripheral nervous system. Here we report that mutations in the TRPV4 gene cause congenital distal SMA, scapuloperoneal SMA, HMSN 2C. We identified three missense substitutions (R269H, R315W and R316C) affecting the intracellular N-terminal ankyrin domain of the TRPV4 ion channel in five families. Expression of mutant TRPV4 constructs in cells from the HeLa line revealed diminished surface localization of mutant proteins. In addition, TRPV4-regulated Ca2+ influx was substantially reduced even after stimulation with 4αPDD, a TRPV4 channel-specific agonist, and with hypo-osmotic solution. In summary, we describe a new hereditary channelopathy caused by mutations in TRPV4 and present evidence that the resulting substitutions in the N-terminal ankyrin domain affect channel maturation, leading to reduced surface expression of functional TRPV4 channels.
Congenital distal SMA (MIM%600175), scapuloperoneal SMA (SPSMA, MIM%181405) and hereditary motor and sensory neuropathy 2C (HMSN2C, MIM%606071) are distinct subtypes of spinal muscular atrophies and hereditary motor and sensory neuropathies characterized by distal and proximal muscle weakness and wasting and by autosomal dominant inheritance. Additional features such as vocal cord paralysis, scoliosis and/or arthrogryposis are likely to occur within families carrying these diseases1-4. Sensory abnormalities have also been described in HMSN2C. Risk loci for congenital distal SMA, SPSMA and HMSN2C have been linked to chromosome 12q23–24 (refs. 2-4). Although clinically distinct, these disorders show some phenotypic overlap, leading to the suggestion that they may result from mutations in the same gene4. Recently, non-neurological diseases have also been linked to the same chromosomal region5. We identified a large family (FAM_1; see anonymized pedigree, Supplementary Fig. 1) with ten living individuals showing a mild to severe congenital distal SMA, SPSMA or HMSN2C phenotype. We first carried out a genome-wide scan using 10K SNP Affymetrix arrays on 12 individuals of the family (left branch of the pedigree; Supplementary Fig. 1) and observed linkage to three chromosomal regions with log10 of odds (lod) scores >2 for several SNP markers, including the chromosome 12q23–24 region (data not shown). We constructed haplotypes by including additional distantly related family members (right branch of the pedigree; Supplementary Fig. 1). The genetic interval transmitted with the disease resides between SNPs rs2374688 and rs35426 (Chr. 12: 106,197,332–114,054,429 bp; Supplementary Table 1) and overlaps with the intervals reported for risk of congenital distal SMA, SPSMA and HMSN2C2-4.
In an affected individual from family FAM_1, we began sequencing all protein-coding exons and exon-intron boundaries of 19 genes but initially observed only known SNPs (Supplementary Table 2). However, sequencing of all protein-coding exons of TRPV4 (transient receptor potential vanilloid 4; chr. 12: 108,705,277–108,755,595; reverse strand) revealed a heterozygous C-to-T nucleotide change at position 943 in exon 6 (Supplementary Fig. 2a), which is predicted to cause the substitution of arginine with tryptophan at position 315 of TRPV4 (R315W). We then screened DNA samples from additional families showing one of the phenotypes described above, including two families previously reported1,3,4. All affected individuals from the chromosome 12q23–24-linked family (here called FAM_2) described by van der Vleuten et al. and diagnosed with congenital distal SMA and arthrogryposis1,3 had a G806A transition in exon 5 of TRPV4 leading to the amino acid substitution R269H (arginine to histidine) (Supplementary Fig. 2b). The family afflicted with HMSN2C named A5 in the report of McEntagart et al.4 carried the same R315W substitution as detected in FAM_1, whereas two other, unreported small families with HMSN2C (here called FAM_3 and FAM_4) carried the C946T transition changing arginine to cysteine at position 316 (R316C) (Supplementary Fig. 2a). The mutations causing these three substitutions were present in all affected individuals but were not found in 162 (looking at exon 5) and 304 (looking at exon 6) healthy European control individuals.
Apparent gain-of-function mutations in TRPV4 have recently been identified in a newly categorized family of autosomal dominant skeletal dysplasias characterized by varying degrees of short trunk and short limbs, ranging from the relatively mild type 3 brachyolmia, through Kozlowski spondylometaphyseal dysplasia, to the more severe metatropic dysplasia5,6. Although many of the affected individuals in our study showed mild to severe scoliosis (Supplementary Table 3), they did not have a short trunk, short stature, short limbs or brachydactyly. It seems likely that the scoliosis in these families is neuromuscular in etiology and caused by a different pathogenic mechanism. Additional support for a neuromuscular explanation for the scoliosis is provided by the finding that some infants were born with arthrogryposis, a clinical phenotype marked by multiple joint contractures likely resulting from reduced fetal movements in utero. Trpv4-knockout mice show increased bone mass due to reduced bone resorption7 and have impaired pressure sensation8 and abnormal osmotic regulation9. The sensory system has been tested extensively in Trpv4−/− mice8,9. In these animals, the threshold for response to noxious stimuli and the conduction velocity of myelinated nerves responding to stimuli were impaired8. The clinical phenotype of the affected individuals described here differs from the phenotypic abnormalities described for Trpv4−/− mice. Although some of the subjects we examined showed sensory abnormalities, mainly comprising reduced surface sensation, the human disorder much more resembles that of a predominantly motor neuron disease. More detailed studies addressing the function of the motor and sensory nerves in individuals with congenital distal SMA, SPSMA and HMSN2C and in Trpv4−/− mice might be able to elucidate common neurological abnormalities.
Based on structure and function, the mammalian TRPV family comprises four groups10: TRPV1/TRPV2, TRPV3, TRPV4 and TRPV5/TRPV6. The thermosensitive TRPV1, TRPV2, TRPV3 and TRPV4 channels are modestly permeable to Ca2+, with a permeability ratio PCa/PNa between ~1 and ~10 (ref. 11). The first description of TRPV4 and its functional characterization linked it to vertebrate mechanosensation12-15. Mechano- and osmosensory functions of TRPV4 in endothelia, in the alveolar unit of the lung and in intestinal sensory innervation have been subsequently described16-18.
All six TRPV family members share large C- and N-terminal cytosolic domains and a six-transmembrane-segment ion-transport domain (Fig. 1a) and are presumed to function as tetramers19. At their N terminus, the TRPV proteins harbor six ankyrin repeats (ANK) that have been reported to be critical determinants of channel maturation and function in multiple in vitro studies20-22. Crystal structures of the ankyrin repeat domains (ARD) of TRPV1, TRPV2 and TRPV6 have been shown to be highly similar23-25. Each ARD consists of six pairs of antiparallel inner and outer α-helices, followed by a ‘finger loop’ (Fig. 1b). The mutations we found resulted in a replacement of highly polar and positively charged arginines (R269H, R315W and R316C) with less polar or nonpolar residues at the outer helices of ANK4 and ANK5 (Fig. 1b). It is likely that these changes either affect the intrinsic ARD stability or directly abrogate critical protein-protein interactions required for proper TRPV4 channel maturation and function. Also, sequence comparisons of TRPV4 orthologs revealed conservation of each of the arginine residues across multiple vertebrate species, suggesting an important functional role for these residues and a potential pathogenic effect of substitutions (Supplementary Fig. 3).
Figure 1.

Schematic model of TRPV4. (a) The TRPV4 protein is composed of a cytosolic N-terminal region and six transmembrane domains (green), including the pore region (magenta) and an intracellular C-terminal tail. The N-terminal region contains the ankyrin repeat domain (ARD), which consists of six ankyrin repeats (ANK), highlighted here in different colors (ANK1–6). The substitutions reported in this study are located in the outer helices of ANK4 (R269H) and ANK5 (R315W and R316C). An asterisk marks position 620 (valine). The V620I substitution results in an activating TRPV4 mutant observed in brachyolmia. The first helix (from the N terminus) corresponds to the inner helix and the second to the outer helix. (b) Ribbon diagram of the cytosolic N-terminal ARD-containing domain of TRPV4 (amino acids 111–358). The ankyrin repeats (ANK1–6) are formed by antiparallel inner and outer helices; the respective fingers are color coded. The PACSIN binding site (amino acids 142–143) is indicated with an asterisk. Here 2pnn.pdb was chosen as a template; Lishko et al.24.
To evaluate the consequences of the TRPV4 mutations, we expressed differentially tagged (polyhistidine (His)- or Flag-tagged) mutant and wild-type human TRPV4 in HeLa cells. This cell line did not express any detectable levels of endogenous TRPV4, thereby contrasting it to several batches of HEK293 cells analyzed in our laboratory. Wild-type TRPV4 proteins were predominantly detected at the plasma membrane, whereas mutant TRPV4 proteins formed cytoplasmic aggregates without plasma membrane involvement, as shown by immunofluorescence (Fig. 2). When cotransfected at equimolar levels, wild-type TRPV4 protein could still be detected at the plasma membrane, but cytoplasmic aggregates were formed by both mutant and wild-type proteins (Fig. 3, upper). To exclude adverse effects of cytoplasmic mutant TRPV4 aggregates on HeLa cell viability, we determined membrane permeability as a measure of cell viability. No difference was observed for mutant and wild-type TRPV4 constructs (~15% of the cells were double-positive for DsRed and YO-PRO-1, indicating unchanged viability; Supplementary Fig. 4a–e).
Figure 2.

Immunolocalization of wild-type and mutant TRPV4 in transfected HeLa cells. Blue shows artificial coloring of nuclei by Hoechst; green shows anti–His tag staining. Cells were transfected with plasmid DNA (pIRES2-DsRed-Express vector) encoding His6-tagged wild-type TRPV4 (hTRPV4), three TRPV4 mutants (R269H, R315W and R316C) and two rescue mutants (R269K and R315K). Cells transfected with hTRPV4 showed localization of the protein to the plasma membrane. Mutants R269H, R315W and R316C were detected as patches distributed over the entire cytoplasm; no staining at the plasma membrane was observed. TRPV4 staining in the rescue mutants R269K and R315K was located predominantly at the plasma membrane.
Figure 3.

Coexpression of wild-type and mutant TRPV4 in transfected HeLa cells. Blue shows artificial coloring of nuclei by Hoechst staining. Green shows anti–His tag staining; red shows anti–Flag tag staining. Cells were cotransfected with plasmid DNA (backbones: pIRES2-AcGFP1 vector for the wild-type and pIRES2-DsRed-Express vector for the mutants) encoding Flag-tagged hTRPV4 and the His-tagged mutants (R269H and R315W; upper panels) or the His-tagged rescue mutants (R269K and R315K; lower panels). In cells cotransfected with hTRPV4 and the mutants R269H and R315W, the Flag-tagged wild-type protein (red) was predominantly seen at the plasma membrane, whereas the His-tagged mutant protein (green) was seen in the cytoplasm. Few mixed complexes (wild-type and mutant) (yellow) were seen. The His-tagged TRPV4 protein of the rescue mutants R269K and R315K (green) was detectable at the plasma membrane.
Next, we examined the functional properties of HeLa cells transfected with wild-type or mutant TRPV4 channels (Fig. 4). Calcium-imaging studies revealed no significant differences in basal calcium levels among the transfected cells (Fig. 4b and Supplementary Table 4a). However, cells transfected with wild-type TRPV4 responded with a rapid increase in intracellular calcium when challenged by hypo-osmotic solution, whereas the response of the different mutant TRPV4–transfected cells was significantly lower (P < 0.01) and heterogeneous between the different mutations (Fig. 4a,c and Supplementary Table 4b). The R269H and R316C mutants showed a significantly decreased (~30%) calcium peak compared to wild-type (Fig. 4c), whereas the calcium peak of the R315W mutant was decreased by ~75%. When HeLa cells coexpressed wild-type and any of the mutant TRPV4 proteins, we observed no significant change in basal calcium level (data not shown) or response to hypo-osmotic challenge (Fig. 4d and Supplementary Table 4c).
Figure 4.

Intracellular calcium changes of TRPV4-transfected HeLa cells. (a) Original traces show calcium response during hypo-osmotic challenge in HeLa cells transfected with wild-type or mutant TRPV4 channels (green, wild-type hTRPV4; light blue, R269H; red, R316C; violet, R315W). Control (dark blue) indicates a mock-transfected cell. Fl340/Fl380, ratio of fluorescence measures at 340 and 380 nm. (b) There was no difference in the basal calcium level of the transfected cells. (c) The hypo-osmotic solution–induced calcium response was significantly reduced in the mutants compared to hTRPV4 or to the rescue mutant (R269K). (d) Coexpression (+) of the mutant channels with the wild-type protein did not result in a significant difference in the Fura-2 ratio change evoked by the hypo-osmotic solution. **P < 0.01 and ***P < 0.001, indicating significant differences compared to cells expressing hTRPV4 only. hTRPV4#, double transfection with His- and Flag-tagged hTRPV4. Data are averaged values and s.d.
In patch-clamp studies, the mutant channels yielded significantly (90%, P < 0.001) smaller currents (Fig. 5a–c) when subjected to a hypo-osmotic challenge (reduction of extracellular osmolarity from 320 to 200 mOsm). Furthermore, activation with the TRPV4-specific agonist 4α-PDD resulted in significantly decreased (P < 0.001) currents in the mutant TRPV4-expressing cells, whereas, as in the Ca2+ study, cotransfected cells showed a wild-type phenotype (Fig. 5d,e). Mutant channels could not be activated when cells were shifted to 37 °C (Supplementary Fig. 5). The TRPV4 V620I gain-of-function mutant yielded significantly (P < 0.05) increased constitutively activated current and significantly (P < 0.05) elevated response after agonist application (Supplementary Fig. 6) in HeLa cells, as was previously demonstrated in HEK293 cells5. Based on our results using HeLa cells as a heterologous expression system, we suggest that the mutations described here lead to decreased stimulus-dependent TRPV4 channel activity.
Figure 5.

Effect of TRPV4 substitutions on TRPV4 activation by hypo-osmotic swelling or after 4α-PDD application. (a,b) Effect of hypo-osmotic challenge of the current density (pA/pF) on wild-type hTRPV4 or mutant R269H-, R315W- and R316C-transfected HeLa cells in response to a ramp protocol. (c) Pooled data from the same series as shown in a and b. Average basal inward and outward currents at −100 mV and +100 mV of HeLa cells expressing hTRPV4, mutants R269H, R315W and R316C, and rescue mutants R269K and R315K (n ≥ 4). *** indicates significant differences compared to cells expressing hTRPV4 (P < 0.001). Basal values are shown by white bars. The values of the hypo-osmotic challenge are indicated by gray bars. Mock-transfected cells were used as a control. (d) Averaged values before stimulation (white bars) and maximal values during stimulation by hypo-osmotic challenge (gray bars) of HeLa cells coexpressing mutant and wild-type channels. (e) Averaged values before stimulation (white bars) and maximal values after 4α-PDD application (gray bars) of HeLa cells expressing wild-type or mutant channels or coexpressing mutant and wild-type channels. Cotransfections are indicated in d and e with + (n ≥ 4). DsRed and GFP double-positive cells were selected for patch-clamp studies in the cotransfection studies. Numerical values are given as means ± s.e.m.
Cell surface biotinylation assays suggested that the functional alterations seen here might result from differences in the levels of surface-displayed wild-type and mutant TRPV4 proteins (Supplementary Fig. 7). Cell surface expression was markedly reduced for the TRPV4 mutants R269H and R315W and was completely abrogated for the R316C mutant.
TRPV4 protein localization and functionality were restored when the mutated amino acid residues were replaced by lysines (Figs. 2, 3,4c,d and 5c), suggesting that site-specific biochemical constraints at positions 269 and 315 are critical for TRPV4 channel localization and function. Previous studies have shown that TRP biogenesis involves protein folding, core glycosylation and oligomerization in the endoplasmic reticulum (ER), followed by transfer to the Golgi network and final transport of the tetramers to the plasma membrane14,16,26, and that substitutions or deletions in the ARDs result in defective trafficking19,20. Retention and ER-associated degradation have been described for a number of artificial ankyrin domain–deletion TRPV4 mutants or immature and unfolded TRPV4 monomers21. Mutations leading to the exposure of di-arginine motifs of type RXR, which are normally concealed within the tetrameric structure, may result in the retention of the misfolded or unfolded proteins within the ER, thereby preventing nonfunctional channel proteins from reaching the cell surface14,16,27. The TRPV4 protein contains four RXR motifs, two of which are located in the N-terminal (122RWR and 269RGR) and two in the C-terminal intracellular region of the protein. The arginine missense substitutions R269H, R315W and R316C disrupt one of these repeats, which allowed us to identify a new 315RR repeat that appears to affect proper TRPV4 channel maturation. The observed TRPV4 alterations are unlikely to interfere with recently published binding sites for ATP, Ca2+-calmodulin (corresponding to amino acids 189–222 in TRPV1)24 and PACSIN3 (amino acids 142–143 in TRPV4), which have been shown to affect channel sensitivity and localization22. Interaction with phosphatidylinositol phosphates (PIPs), and in particular phosphatidyl-4,5-bisphosphate (PI(4,5)P2), has been shown to regulate the function of various TRPs28,29. Candidate interaction motifs are pleckstrin homology–like domains, which have been identified as a cluster of positively charged residues. The proposed N-terminal pleckstrin homology–like motif of TRPV4 includes amino acids 114–125 (RHHSSDNKRWRKK) and is thus not directly affected by the mutations identified in this study.
Reduced surface expression accompanied by residual mutant TRPV4 channel activity suggests that the alterations do not directly affect the conductive properties of TRPV4 channels but do have a deleterious impact on channel assembly or trafficking. Based on current knowledge on TRPV channel maturation, we cannot reliably predict how tetramers comprising mutant TRPV4 subunits (that is, with mutant/wild-type ratios of 3:1, 2:2 and 1:3) will behave in surface trafficking and channel function, thus necessitating further studies, both in vitro and in vivo. Compared to the numerous nerve fibers in the healthy peripheral nerve showing intense TRPV4 staining and to the TRPV4 staining pattern of healthy skeletal muscle cells, the pattern seen for the diseased nerve of one subject with HMSN2C (and the R316C substitution) involved less positively stained nerve fibers and weaker cytoplasmic staining of the muscle cells (Fig. 6a–d). Haploinsufficiency can be considered the most likely underlying pathomechanism, although we cannot fully exclude adverse interaction of wild-type and mutant TRPV4 subunits.
Figure 6.
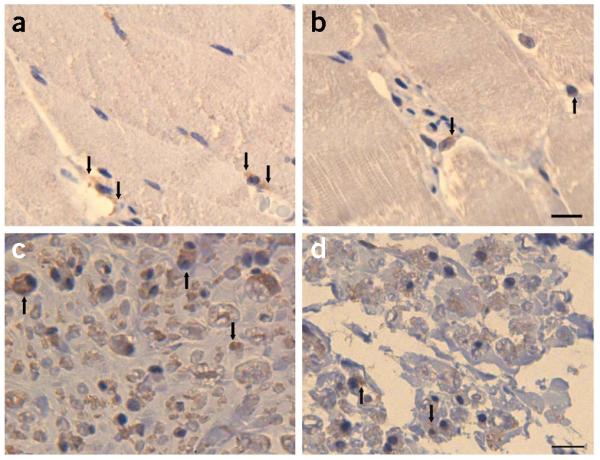
Immunohistochemistry (IHC). (a–d) Detection of TRPV4 (brown reaction product) in healthy skeletal muscle (a) and in a muscle biopsy (b) from an individual with HMSN2C (from FAM_4) harboring the TRPV4 R316C substitution. TRPV4 IHC on a nerve biopsy from a healthy control (c) and the HMSN2C patient (d). In normal tissue, TRPV4 protein is distributed in the cytoplasm (a, arrows). In the diseased tissue, reaction product is seen predominantly in the perinuclear region (b, arrows). In the healthy nerve tissue, more TRPV4-immunoreactive fibers (c, arrows) are seen than in the diseased nerve (d). Calibration bar, 10 μm.
In summary, this work identifies TRPV4 malfunction as the molecular basis for a new channelopathy underlying a heterogeneous group of neurodegenerative disorders. Our findings may lead to improved diagnosis of this diverse group of SMAs and HMSNs and may also contribute to the development of specific treatments.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the subjects and their families who participated in this study. This work was supported by the Austrian Science Fund (FWF, P19455-B05), the Oesterreichische Nationalbank (ÖNB, project 13010) and by the Interdisciplinary Centre for Clinical Research BIOMAT within the Faculty of Medicine, RWTH Aachen. J.S. is a Heisenberg fellow of the Deutsche Forschungsgemeinschaft (DFG). We thank M. Absenger, M. Hatz, A. Hof, H. Knausz, M. Schabhüttl, A. Gungl and T. Kueznik for expert technical assistance.
Appendix
ONLINE METHODS
Subjects
The study was approved by the local ethics committees of the participating universities. Informed consent was obtained from the study participants. Affected and unaffected family members and spouses underwent a detailed clinical, neurological and neurophysiological examination following standard methods, which was carried out by experienced neurologists, clinicians and human geneticists (M.A.-G., M.E.M., C.M.A.v.R.-A., R.S., H.J.S., C.L., S.U., J.S., N.E.A, B.S.-W. and H.L.). Index subjects were recruited during routine neurological examination in diverse neurological departments. Additional family members (including unaffected individuals and spouses) were then invited to participate in the study. A peripheral blood sample was taken from each participant for further genetic analysis. The affected individuals from families FAM_2 and A5 were not re-examined for this study. Paraffin-embedded material from biopsies of skeletal muscle and from the sural nerve of one HMSN2C-affected individual of FAM_4 harboring the TRPV4 R316C substitution, as well as normal control tissues, were obtained after informed consent.
Genome-wide scan
DNA was isolated from peripheral blood samples using a QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s protocol. Genetic analysis of FAM_1 included a genome-wide scan, which was carried out using the GeneChip Human Mapping 10K Array Xba 142 2.0. The genotypes were imported into the linkage analysis program and dChip 2006 (Build date: Dec 22 2005). Methods used for linkage analysis, which has also been carried out in the congenital distal SMA family from the Netherlands FAM_2 and in the New Zealand HMSN2C family A5, have been described elsewhere3,4.
Mutation detection
The TRPV4 coding region and splice sites were amplified using 15 primer pairs that were designed based on NCBI annotations for TRPV4 genomic reference (NC_000012) and mRNA (NM_021625.3) sequences and are available upon request. We amplified 20 ng of genomic DNA in a 10-μl reaction volume with 5 μl HotStar Master Mix (Qiagen) and 0.3 pM of each primer. The PCR conditions were as follows: initial denaturation at 95 °C for 12 min; 35 cycles of denaturation at 95 °C for 45 s, annealing at 57 °C for 30 s and extension at 72 °C for 30 s; final extension at 72 °C for 10 min. PCR products were purified using Nucleo Fast (Macherey-Nagel) clean-up PCR plates according to the manufacturer’s protocol. Sequences of amplified PCR products were determined using the ABI PRISM BigDye Terminator v3.1 Ready Reaction Cycle Sequencing Kit (Applied Biosystems) and analyzed on a 3730 DNA Analyzer ABI PRISM (Applied Biosystems). Chromatograms and sequences were analyzed and aligned against the reference sequence using the ABI SeqScape v2.5 software (Applied Biosystems).
Expression cloning
We cloned the wild-type TRPV4 transcript variant 1 coding sequence (corresponding to Ref.Seq. NM_021625) into pIRES2-DsRed-Express and pIRES2-AcGFP1 expression vectors (Clontech Laboratories Inc.) and generated N-terminally Flag- or His-tagged TRPV4 expression constructs. Mutants were generated using QuikChange II XL Site-Directed Mutagenesis (Stratagene) kit. The following mutant constructs were generated: TRPV4_R269H (G806A transition), TRPV4_R315W (C943T transition), TRPV4_R316C (C946T transition), TRPV4_R269K (codon CGT269AAA) and TRPV4_R315K (codon CGG315AAG). Oligonucleotide sequences used for site-directed mutagenesis are listed in Supplementary Table 5. All constructs were verified by DNA sequencing.
Cell culture
HeLa cells were grown in Dulbecco’s modified eagle medium (DMEM) and minimum essential medium (MEM) containing 10% fetal calf serum (FCS), 2 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin at 37 °C, 95% humidity and 5% CO2. One day before transfection, cells were split and seeded at 50% confluency directly into 24-well plates in medium without antibiotics. 1 μg plasmid DNA was used per transfection per well (24-well plate) using Lipofectamine 2000 (Invitrogen) reagent (1 μl Lipofectamine per μg DNA).
Immunofluorescence
24 h post-transfection, cells were fixed in 3.7% paraformaldehyde for 10 min, washed in PBS, permeabilized in 0.05% TritonX-100, washed in PBS, blocked in 1% normal mouse serum (DAKO) for 10 min and incubated with mouse HiLyte Fluor 647–labeled antibody to His tag (1 μg/ml, Tebu-bio) for 60 min. After washing in PBS, nuclei were counterstained with 1 μg/ml Hoechst 33342 in PBS for 15 min and rinsed again in PBS. All steps were performed at room temperature (22–24 °C). Cells were viewed with the confocal microscope LSM 510 Meta (Zeiss) with the following excitation/emission settings: 405/420–480 (Hoechst 33342); 543/560–615 (DsRed); 633/679–754 (HiLyte Fluor 647).
Cell surface biotinylation
HeLa cells were transfected with equal amounts of pIRES-DsRed vector (Clontech) expressing either Flag-tagged wild-type or mutant TRPV4 proteins, using Lipofectamine 2000 (Invitrogen). After 24 h incubation, the cells were harvested by trypsinization, washed twice with ice-cold PBS (pH 8.0) and incubated with 1 mM EZ-Link Sulfo-NHS-SS-Biotin (Pierce) for 30 min at room temperature. Subsequently, the cells were washed once with cold 50 mM Tris (pH 8.0) and once with cold PBS (pH 8.0) and lysed in PBS supplemented with 1.5% Triton X-100 and protease-inhibitor cocktail (Sigma). Biotinylated proteins were precipitated with NeutrAvidin agarose beads (Pierce) at 4 °C overnight, then eluted with sample loading buffer and analyzed by SDS-PAGE electrophoresis. Immunodetection on whole-cell lysates and biotinylation products was performed with antibodies to Flag tag (Sigma), pan-cadherin (Cell Signaling) and β-actin (Sigma).
Immunohistochemistry
Sections from paraffin-embedded material were cut, de-paraffinized and rehydrated. Antigen retrieval was performed for 10 min at 120 °C in DAKO Target Retrieval Solution 6.0 (DAKO Diagnostica) using a decloaking chamber (Biocare Medical). After cooling of the sections, three washes in PBS followed, and endogenous peroxidase activity was blocked by immersion of the sections in 1% H2O2 in methanol. Staining was performed according to the protocol of the UltraVision LP Large Volume Detection System (Thermo Scientific) starting with Ultra V Block for 5 min at room temperature, incubation with the antibody to TRPV4 (rabbit, 1:40, Sigma) for 30 min at room temperature, incubation with primary antibody enhancer for 10 min at room temperature and with horseradish peroxidase polymer for 30 min at room temperature, and detection with Liquid DAB+ Substrate Chromogen System for Autostainer (DAKO Diagnostica) for 5 min at room temperature. Sections were counterstained with hemalaun. Between all incubation steps, sections were rinsed three times in PBS. Sections were dehydrated and mounted in Tissue Tek Mounting Media 4494 Coverslipping Resin (Sanova).
Electrophysiology
24 h post-transfection, HeLa cells were prepared for patchclamp recordings as described previously30. Whole membrane currents were measured for at least five randomly selected Ds-Red–positive cells per TRPV4 expression construct or Ds-Red and GFP double-positive cells when coexpression studies were performed. The patch-clamp amplifier was an Axopatch 200B (Axon Instruments; sampling rate, 1 ms; 8-Pole Bessel filter 3kHz). Patch pipettes made of borosilicate glass tubes (GC 150; Clark Electromedical Instruments) had a resistance between 3 and 4 MΩ when back-filled with intracellular solution. A chloridized silver (Ag-AgCl) wire was used as a reference electrode. Cell capacitance and access resistance were continuously monitored. Between 50% and 70% of the series resistance was electronically compensated to minimize voltage errors. A ramp protocol was applied, consisting of a voltage step from the holding potential of 0 mV to −100 mV followed by a 400-ms linear ramp to +100 mV. Cell membrane capacitance values were used to calculate current densities. The flow-through chamber was perfused (2–3 ml/min) with a bath solution (see Supplementary Note for details about solutions used) using a gravity-driven perfusion system. All measurements were carried out at room temperature (see Supplementary Note for details on solutions used in electrophysiology).
Measurement of intracellular calcium concentration ([Ca2+]i)
The cells were cultured on glass cover slides until confluency and loaded in the dark for 45 min with 2 μM Fura-2 AM followed by washing with 1.8 mM Ca2+-containing Ringer solution. After 15 min, the single glass cover slide was mounted on the stage of a Zeiss 200M inverted epifluorescence microscope coupled to a PolyChrome V monochromator (Till Photonics) light source in a sealed, temperature-controlled RC-21B imaging chamber (Warner Instruments) and perfused with prewarmed solution (37 °C). Fluorescence images were obtained with alternate excitation at 340 and 380 nm. The emitted light was collected at 510 nm by an air-cooled Andor iXon camera. Measurements were made every 3 s. After 3 min of baseline recording, the cells were perfused with hypo-osmotic solution for 5 min, followed by 2 min wash-out with Ringer solution. Background fluorescence was recorded from each cover slide and subtracted before calculation. The acquired images were stored and subsequently processed offline with TILLvisION software (Till Photonics). [Ca2+]i was calculated as described previously30. Maximal and minimal ratio values were determined at the end of each experiment by first treating the cells with 1 μM ionomycin (maximal ratio) and then chelating all free Ca2+ with 10 mM EGTA (minimal ratio). Any cells not responding to ionomycin were discarded. The experiments were repeated at least three times. The average basal calcium level, as well as the hypo-osmotic solution–induced calcium peak of the individual transfected cells, was analyzed (n > 10 in each group) (see Supplementary Note for details on solutions used for Ca2+ imaging).
Statistical analysis of Ca2+ imaging and electrophysiology
Numerical values are given as means ± s.e.m. of n cells. Intergroup differences were assessed by a factorial analysis of variance with post hoc analysis with Fisher’s least-significant-difference test. P values < 0.05 were considered significant.
Fluorescent-activated cell sorting (FACS) analysis
To assess whether expression of TRPV4 mutants affects cell viability, HeLa or HEK293 cells were seeded in 12-well plates at 80–90% confluency and transfected the next day with 1.5 μg of pIRES2-DsRed Express (Clontech) vector DNA expressing wild-type TRPV4 or TRPV4 mutants, respectively, using Lipofectamine 2000 (Invitrogen). After 24 h of incubation, cells were harvested and resuspended in cold PBS (4 °C) at a density of 1 × 106 cells/ml. The cells were then stained with 1 μl of a 100 μM solution of the membrane-impermeating dye, YO-PRO-1 (Molecular Probes, Invitrogen) on ice for 30 min immediately followed by FACS analysis performed on an LSR II flow cytometer (BD Biosciences) using 488-nm excitation and FACSDiva software (BD Biosciences).
URL
Footnotes
METHODS: Methods and any associated references are available in the online version of the paper at http://www.nature.com/naturegenetics/.
Accession codes. GenBank: human TRPV4 cDNA, NM_021625; human TRPV4, NP_067638 IsoA. Pfam: ankyrin repeat, PF00023.
Note: Supplementary information is available on the Nature Genetics website.
AUTHOR CONTRIBUTIONS: M.A.-G., S.U., J.S., M.E.M., A.H.C., K.J.D., C.M.A.v.R.-A., N.E.A., H.L., B.S.-W., R.P., C.L., G.W.P., H.J.S., H.K. and T.R.P. recruited the study participants, acquired clinical data, conducted neurological and neurophysiological evaluations and performed linkage analysis. M.A.-G, C.G., L.P. and C.F. carried out the Affymetrix array linkage studies and identified the mutations. A.O., Z.B. and B.T. designed, carried out and analyzed the electrophysiological and Ca2+-imaging studies. E.F. conducted immunofluorescence and immunohistochemistry studies. H.S. conducted fluorescence-activated cell sorting (FACS) and biotinylation studies. A.K. performed structural biology and biocomputing analyses. A.H.C., M.E.M. and H.K. participated in the data analysis and reviewed the manuscript. M.A.-G. and C.G. analyzed the data, designed and supervised the study and wrote the manuscript.
References
- 1.Fleury P, Hageman GA. dominantly inherited lower motor neuron disorder presenting at birth with associated arthrogryposis. J. Neurol. Neurosurg. Psychiatry. 1985;48:1037–1048. doi: 10.1136/jnnp.48.10.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isozumi K, et al. Linkage of scapuloperoneal spinal muscular atrophy to chromosome 12q24.1-q24.31. Hum. Mol. Genet. 1996;5:1377–1382. doi: 10.1093/hmg/5.9.1377. [DOI] [PubMed] [Google Scholar]
- 3.van der Vleuten AJ, et al. Localisation of the gene for a dominant congenital spinal muscular atrophy predominantly affecting the lower limbs to chromosome 12q23-q24. Eur. J. Hum. Genet. 1998;6:376–382. doi: 10.1038/sj.ejhg.5200229. [DOI] [PubMed] [Google Scholar]
- 4.McEntagart ME, et al. Confirmation of a hereditary motor and sensory neuropathy IIC locus at chromosome 12q23-q24. Ann. Neurol. 2005;57:293–297. doi: 10.1002/ana.20375. erratum 57, 609 (2005) [DOI] [PubMed] [Google Scholar]
- 5.Rock MJ, et al. Gain-of-function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat. Genet. 2008;40:999–1003. doi: 10.1038/ng.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krakow D, et al. Mutations in the gene encoding the calcium-permeable ion channel TRPV4 produce spondylometaphyseal dysplasia, Kozlowski type and metatropic dysplasia. Am. J. Hum. Genet. 2009;84:307–315. doi: 10.1016/j.ajhg.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masuyama R, et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. 2008;8:257–265. doi: 10.1016/j.cmet.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki M, Mizuno A, Kodaira K, Imai M. Impaired pressure sensation in mice lacking TRPV4. J. Biol. Chem. 2003;278:22664–22668. doi: 10.1074/jbc.M302561200. [DOI] [PubMed] [Google Scholar]
- 9.Liedtke W, Friedman JM. Abnormal osmotic regulation in trpv4−/− mice. Proc. Natl. Acad. Sci. USA. 2003;100:13698–13703. doi: 10.1073/pnas.1735416100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pedersen SF, Owsianik G, Nilius B. TRP channels: an overview. Cell Calcium. 2005;38:233–252. doi: 10.1016/j.ceca.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 11.Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- 12.Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am. J. Physiol. Cell Physiol. 2004;286:C195–C205. doi: 10.1152/ajpcell.00365.2003. [DOI] [PubMed] [Google Scholar]
- 13.Liedtke W, Tobin DM, Bargmann CI, Friedman JM. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 2003;100(Suppl 2):14531–14536. doi: 10.1073/pnas.2235619100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nilius B, Watanabe H, Vriens J. The TRPV4 channel: structure-function relationship and promiscuous gating behaviour. Pflugers Arch. 2003;446:298–303. doi: 10.1007/s00424-003-1028-9. [DOI] [PubMed] [Google Scholar]
- 15.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD. OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. Cell Biol. 2000;2:695–702. doi: 10.1038/35036318. [DOI] [PubMed] [Google Scholar]
- 16.Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol. Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 17.Liedtke W. Molecular mechanisms of TRPV4-mediated neural signaling. Ann. NY Acad. Sci. 2008;1144:42–52. doi: 10.1196/annals.1418.012. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe H, Murakami M, Ohba T, Takahashi Y, Ito H. TRP channel and cardiovascular disease. Pharmacol. Ther. 2008;118:337–351. doi: 10.1016/j.pharmthera.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Erler I, Hirnet D, Wissenbach U, Flockerzi V, Niemeyer BA. Ca2+-selective transient receptor potential V channel architecture and function require a specific ankyrin repeat. J. Biol. Chem. 2004;279:34456–34463. doi: 10.1074/jbc.M404778200. [DOI] [PubMed] [Google Scholar]
- 20.Hellwig N, Albrecht N, Harteneck C, Schultz G, Schaefer M. Homo- and heteromeric assembly of TRPV channel subunits. J. Cell Sci. 2005;118:917–928. doi: 10.1242/jcs.01675. [DOI] [PubMed] [Google Scholar]
- 21.Arniges M, Fernández-Fernández JM, Albrecht N, Schaefer M, Valverde MA. Human TRPV4 channel splice variants revealed a key role of ankyrin domains in multimerization and trafficking. J. Biol. Chem. 2006;281:1580–1586. doi: 10.1074/jbc.M511456200. [DOI] [PubMed] [Google Scholar]
- 22.Cuajungco MP, et al. PACSINs bind to the TRPV4 cation channel. PACSIN 3 modulates the subcellular localization of TRPV4. J. Biol. Chem. 2006;281:18753–18762. doi: 10.1074/jbc.M602452200. [DOI] [PubMed] [Google Scholar]
- 23.McCleverty CJ, Koesema E, Patapoutian A, Lesley SA, Kreusch A. Crystal structure of the human TRPV2 channel ankyrin repeat domain. Protein Sci. 2006;15:2201–2206. doi: 10.1110/ps.062357206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lishko PV, Procko E, Jin X, Phelps CB, Gaudet R. The ankyrin repeats of TRPV1 bind multiple ligands and modulate channel sensitivity. Neuron. 2007;54:905–918. doi: 10.1016/j.neuron.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 25.Phelps CB, Huang RJ, Lishko PV, Wang RR, Gaudet R. Structural analyses of the ankyrin repeat domain of TRPV6 and related TRPV ion channels. Biochemistry. 2008;47:2476–2484. doi: 10.1021/bi702109w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erler I, et al. Trafficking and assembly of the cold-sensitive TRPM8 channel. J. Biol. Chem. 2006;281:38396–38404. doi: 10.1074/jbc.M607756200. [DOI] [PubMed] [Google Scholar]
- 27.Zerangue N, Schwappach B, Jan NY, Jan LY. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- 28.Nilius B, Owsianik G, Voets T. Transient receptor potential channels meet phosphoinositides. EMBO J. 2008;27:2809–2816. doi: 10.1038/emboj.2008.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Rossum DB, et al. Phospholipase Cγ1 controls surface expression of TRPC3 through an intermolecular PH domain. Nature. 2005;434:99–104. doi: 10.1038/nature03340. [DOI] [PubMed] [Google Scholar]
References
- 30.Weissmann N, et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl. Acad. Sci. USA. 2006;103:19093–19098. doi: 10.1073/pnas.0606728103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.