

Abstract
Constitutively activating internal tandem duplications (ITD) of FLT3 (FMS-like tyrosine kinase 3) are the most common mutations in acute myeloid leukemia (AML) and correlate with poor prognosis. Receptor tyrosine kinase inhibitors targeting FLT3 have developed as attractive treatment options. Because relapses occur after initial responses, identification of FLT3-ITD–mediated signaling events are important to facilitate novel therapeutic interventions. Here, we have determined the growth-inhibitory and proapototic mechanisms of 2 small molecule inhibitors of FLT3, AG1295 or PKC412, in hematopoietic progenitor cells, human leukemic cell lines, and primary AML cells expressing FLT3-ITD. Inactivation of the PI3-kinase pathway, but not of Ras–mitogen-activated protein (MAP) kinase signaling, was essential to elicit cytotoxic responses. Both compounds induced up-regulation of proapoptotic BH3-only proteins Bim and Puma, and subsequent cell death. However, only silencing of Bim, or its direct transcriptional activator FOXO3a, abrogated apoptosis efficiently. Similar findings were made in bone marrow cells from gene-targeted mice lacking Bim and/or Puma infected with FLT3-ITD and treated with inhibitor, where loss of Puma only provided transient protection from apoptosis, but loss of Bim preserved clonal survival upon FLT3-ITD inhibition.
Introduction
FLT3 (FMS-like tyrosine kinase 3) is a type III receptor tyrosine kinase (RTK) closely related to the platelet-derived growth factor (PDGF) receptor and c-Kit with important functions in the regulation of early hematopoietic cells. FLT3 is frequently mutated in patients with acute myeloid leukemia (AML), which correlates with poor prognosis and decreased patient survival.1-4 The most common mutations are internal tandem duplications (ITD) of the juxtamembrane domain, which cause ligand-independent dimerization and autophosphorylation. FLT3-ITD expression causes malignant transformation and factor-independent growth when expressed in factor-dependent cell lines.5,6
Development of RTK inhibitors selective for FLT3 has emerged as attractive drugs for treatment of AML patients. Several inhibitors have been described, such as AG1295, CEP701, PKC412, and SU-11 248, with cytotoxic effects to cell lines and primary AML cells in vitro expressing mutant FLT3. PKC412 is one of several FLT3 inhibitors that is currently evaluated in late-stage clinical trials in AML patients carrying FLT3 mutations.7 However, as single agents, these inhibitors are able to sustain only limited cytotoxic responses in AML patients, and relapse occurs after the initial response.8-11 Therefore combination therapy has emerged as a therapeutic strategy. Clarifying the downstream signaling components of FLT3-ITD could identify attractive targets for such intervention and enhance the long-term therapeutic benefits.
FLT3-ITD activates intracellular effector proteins mediating proliferation and survival. The Ras pathway and constitutive phosphorylation of mitogen-activated protein (MAP) kinase has been demonstrated in cells expressing FLT3-ITD.5,6 In primary AML blasts and in cell lines expressing FLT3-ITD, increased activation of the survival AKT kinase has been observed.12-14 AKT is transiently activated by normal FLT3 signaling, which leads to inhibition of apoptosis by phosphorylating FOXO3a, a Forkhead family member involved in apoptosis and cell-cycle control.15 Studies have confirmed that AKT inhibits FOXO3a in FLT3-ITD–expressing cells,13,14 suggesting that the pathway could be efficient for FLT3 intervention. Phosphorylated AKT is also found in AML samples expressing wild-type FLT3,16 indicating that overexpression of FLT3 is sufficient to trigger AKT activation.
Increasing evidence suggest that the mechanisms by which chemotherapeutic drugs and novel inhibitors eliminate malignant cells is mainly by apoptotic induction. The BH3-only proteins Bim (Bcl-2–interacting modulator of cell death) and Puma (p53 up-regulated modulator of apoptosis), proapoptotic members of the Bcl-2 family, can be activated in response to cytotoxic stimuli, including chemotherapeutic drugs.17 They have a prominent role among the BH3-only proteins, because they bind with high affinity to all antiapoptotic Bcl-2 family proteins,18 thereby promoting mitochondrial release of cytochrome c, which subsequently activates caspase 9 and caspase 3 and death effector molecules. Recent results suggest that Bim and Puma have overlapping effects as well as distinct roles in vivo. This was demonstrated in genetically modified mice in which glucocorticoid-induced apoptosis varied with cell type and was dependent on lack of Bim or Puma.19 Hence, exploring the regulation of Bim and Puma upon chemotherapeutic treatment may prompt new developments in treatment of AML. Studies indicate that BCR-ABL protein in chronic myelogenous leukemia (CML) and oncogenic FLT3-ITD support cell survival through down-regulation of Bim expression by a FOXO3a-dependent mechanism.13,14,20-22 Recently, it was demonstrated that FOXO3a increases Puma expression in response to growth factor deprivation in lymphoid cells and mouse embryonic fibroblasts, suggesting that Puma together with Bim may have overlapping functions as FOXO3a downstream targets,23 and that they may be involved in the effector mechanisms by which RTK inhibitors affect AML cells expressing FLT3-ITD.
Here we show that the FOXO3a pathway is essential in mediating apoptosis of FLT3-ITD–expressing cells upon treatment with AG1295 and PKC412. We found that FLT3-ITD activated AKT and ERK when introduced into the factor-dependent progenitor cell line FDC-P1. Inhibition of the PI3-kinase pathway and downstream FOXO3a was crucial for survival, and a phosphorylation-deficient, constitutively active variant of FOXO3a up-regulated both Bim and Puma during FLT3-ITD signaling and induced cell death. Importantly, Bim and Puma were up-regulated in human leukemic cell lines and in primary AML cells from patients harboring FLT3 mutations when treated with RTK inhibitor. Silencing of either Bim or FOXO3a, but not Puma, prevented apoptotic induction in FDC-P1 cells expressing FLT3-ITD. Consistently, FLT3-ITD–infected bone marrow cells from gene-targeted mice lacking Bim were insensitive to inhibition, whereas lack of Puma had only a transient and minor effect. These results indicate an essential contribution of FOXO3a repression by FLT3-ITD signaling to tumor cell survival by repression of Bim. Restoring or mimicking Bim activity by RTK inhibition and/or the application of BH3 mimetics may be a suitable strategy for pharmaceutic intervention in AML.
Methods
Reagents, cytokines, and antibodies
LY294002 and PD98059 were obtained from Calbiochem (San Diego, CA). AG1295 was purchased from Sigma-Aldrich (St Louis, MO), and PKC412 was kindly provided by Novartis (Basel, Switzerland). Recombinant murine interleukin-3 (IL-3) and murine and human FLT3 ligand (FL) were from PeproTech (London, United Kingdom). 4-Hydroxytamoxifen (4-OHT) was from Sigma-Aldrich. Antibodies for phospho-Ser473-Akt, phospho-ERK1/2 (Thr202/Tyr204), and cleaved caspase-3 (Asp175) were obtained from Cell Signaling Technology (Danvers, MA), and antibodies for phospho-Thr32-FOXO3a and phospho-Ser253-FOXO3a were from Upstate Biotechnology (Charlottesville, VA). FOXO3a antibody was obtained from Abcam (Cambridge, United Kingdom), Bim antibody was from Affinity BioReagents (Golden, CO), and Puma antibody was from ProSci (Poway, CA). Anti–human FLT3 and anti-pTyr (PY99) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was from Chemicon (Temecula, CA). Secondary antibodies were horseradish peroxidase–conjugated from The Jackson Laboratory (Bar Harbor, ME) or GE Healthcare Life Sciences (Piscataway, NJ).
Cells
MV4;11 was cultured in Iscove modified Dulbecco medium (IMDM; PAA Laboratories, Les Mureaux, France) with 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA), and 2 mM l-glutamine at 37°C in 5% CO2. FDC-P1 cells were cultured in the same medium supplemented with IL-3 containing supernatant. All other cell lines were cultured in RPMI 1640 (PAA Laboratories) with 10% FBS and 2 mM l-glutamine.
Establishment of cells expressing FLT3-ITD
For establishment of FDC-P1/FLT3-ITD, Phoenix-Eco cells were transfected with retroviral vector containing FLT3-ITD using standard CaPO4 precipitation, after which supernatants were filtered and used to infect FDC-P1 cells with 4 μg protamine sulfate/mL (Sigma-Aldrich) and IL-3. Transduced cells were isolated by selection with 800 μg G418/mL (PAA) for 1 week. Selected cells were maintained in the absence of cytokine for no longer than 2 months.
Immunoprecipitations and Western blot analysis
Cells (3 × 106−1 × 107) were washed in phosphate-buffered saline (PBS) and lysed in buffer containing 25 mM Tris, pH 7.5, 150 mM NaCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 10% glycerol, 1% Triton X-100, Complete Protease Inhibitor (Roche, Basel, Switzerland), and 1 mM Na3VO4. Lysates were precleared with protein A Sepharose (GE Healthcare Life Sciences) for 1 hour at 4°C followed by incubation overnight at 4°C with the indicated antibody. Immunocomplexes were captured with protein A Sepharose. After electrophoresis using NuPage precast 10% Bis-Tris gel (Invitrogen), and transfer to polyvinylidene fluoride (PVDF) membranes (Invitrogen), immunoblotting was performed with primary antibody overnight at 4°C and HRP-coupled secondary antibody for 1 hour. Blots were visualized by enhanced chemiluminescence (ECL; GE Healthcare Life Sciences).
Detection of apoptosis and cell-cycle analysis
Cells were labeled with annexin V–fluorescein isothiocyanate (FITC; BD Biosciences, San Jose, CA) and 5 μg/mL propidium iodide (PI; Sigma-Aldrich) and analyzed using a FACSCalibur and CellQuest software (BD Biosciences). For cell-cycle analysis, cells were fixed in 70% ethanol at −20°C for 20 minutes, washed in PBS, and resuspended in 3.5 mM Tris, 1 mM NaCl, 0,1% Nonidet P-40 (NP-40), 50 μg/mL PI, and 10 μg/mL RNaseA (Sigma-Aldrich). Cell-cycle status was analyzed by flow cytometry, and the amount of cells in G0/G1, S-phase, and G2/M was calculated.
Retroviral infections
FDC-P1/FLT3-ITD/FOXO3(A3):ER cells were generated by culturing FDC-P1/FLT3-ITD cells with viral supernatant from amphotropic packaging cells transfected with pBabe-Puro/FOXO3(A3):ER24 and 4 μg protamine sulfate/mL. Cells were selected for 1 week in 2 μg puromycin/mL (Sigma-Aldrich) and used immediately for experiments.
Infection of bone marrow progenitors and colony-forming assay
Retroviral vector MSCV-FLT3-ITD-IRES-EYFP25 was used to prepare high-titer viral supernatants by transient transfection of 293T cells using calcium phosphate. Isolation of lineage-depleted (Lin−) bone marrow cells from 6- to 12-week-old mice was performed with a lineage cell depletion kit on an AutoMACS device (Miltenyi Biotec, Bergisch Gladbach, Germany). The bim-/-,26 puma-/-,27 and bim-/- puma-/- 28 mice have previously been described. After overnight prestimulation with 25 ng/mL FL, 1 ng/mL IL-3, and 25 ng/mL kit ligand (KL), Lin− cells were incubated for 48 hours on retronectin-coated plates with retroviral supernatant from MSCV-FLT3-ITD-IRES-EYFP–transfected 293T cells. Enhanced yellow fluorescent protein (EYFP)+ cells were sorted on a FACSAria device (Becton Dickinson, Franklin Lakes, NJ) into 24-well plates containing 1 mL IMDM with 20% FBS, 0.65% bovine serum albumin (BSA) fraction V (Sigma-Aldrich), 0.05 mM 2-mercaptoethanol, human transferrin (iron-saturated; Pharmacia, Uppsala, Sweden), and 10 μg/mL bovine insulin (Sigma-Aldrich) without addition of cytokines. The same volume of 2% methylcellulose (Methocel; Fluka, Deisenhofen, Germany) was added, mixed by careful pipetting, and incubated in a humidified atmosphere with 5% CO2 at 37°C. Seven days later, colonies containing 50 or more cells were counted. All experiments with animals received institutional review board (IRB) approval and were performed according to the Austrian Tierversuchgesetz (BGB1. no. 501/1988 i.d.g.F) and the guidelines of the Animal Ethics Committee in Linköping, Sweden (Dnr 3-07).
Gene silencing
For silencing of murine Bim, FOXO3a, or Puma, SMARTpool small interfering RNAs (siRNAs) from Dharmacon Research (Lafayette, CO) were transfected into FDC-P1/ITD cells using Amaxa nucleofection (Cologne, Germany). Twenty-four hours before transfection, 5 × 106 cells were seeded in fresh complete medium. Cells were then collected by centrifugation, resuspended in Nucleofector solution T, and mixed with siRNA oligonucleotides to 0.7 or 1.4 μg Bim, 1.3 or 2.7 μg FOXO3a, or 0.5 or 2.0 μg Puma. An siRNA oligonucleotide to 1.5 μg green fluorescent protein (GFP; pmaxGFP) was used as a control (Amaxa). After transfection, cells were transferred into prewarmed (37°C) complete medium and analyzed for gene silencing and apoptosis after different time points.
Patient samples
The study was approved by the research ethical committee at the Faculty of Health Sciences, Linköping, Sweden. Informed consent was obtained from all patients in accordance with the Declaration of Helsinki. Bone marrow or peripheral blood samples from patients with de novo AML were subjected to Ficoll-Hypaque (GE Healthcare Life Sciences) density gradient centrifugation. Multiplex polymerase chain reaction (PCR) including digestion of amplicon with EcoRV for detection of FLT3-ITD was performed as described.2 After capillary electrophoresis separation, PCR fragments were analyzed, and their lengths and peak heights were estimated. Mutation amount as percent total DNA content was calculated from peak heights. All samples were morphologically confirmed to contain greater than 90% leukemic blasts by May-Grünwald-Giemsa staining on cytospins, and cells were cryopreserved in liquid nitrogen. After unthawing the samples, cells were allowed to rest for 4 hours in RPMI 1640 with 10% FBS before adding inhibitors. Effects of inhibitor on viability were studied after 72 hours by flow cytometry of PI-stained cells.
Real-time PCR
Total RNA was isolated using RNeasy mini kit (Qiagen, Hilden, Germany) and reversed transcribed to cDNA by SuperScript III Reverse Transcriptase (Invitrogen). Quantitative PCR was performed in 10 μL with 2× SYBR Green Mastermix (Applied Biosystems, Warrington, United Kingdom) containing DNA polymerase, dNTPs, buffer, 1 μM each forward and reverse primers, and 4-12 ng template using the 7500 FAST Real-Time System (Applied Biosystems). All samples were performed in triplicates, and human β-glucuronidase (GusB) was used as an endogenous control. Primer sequences designed with Primer express software (Applied Biosystems) were as follows: GusB forward primer: 5′-TGGTTGGAGAGCT-CATTTGGA-3′ and reverse primer: 5′-ACTCTCGTCGGTGACTGT-TCAG-3′; Bim forward primer: 5′-TGGCAAAGCAACCTTCTGATG-3′ and reverse primer: 5′-GCAGGCTGCAATTGTCTACCT-3′; and Puma forward primer: 5′-GAAGAGCAAATGAGCCAAACG-3′ and reverse primer: 5′-GGAGCAACCGGCAAACG-3′. Relative expression was calculated using the 2−ΔΔCt method.
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) was performed using a ChIP-Assay kit as recommended by the manufacturer (Upstate Biotechnology). Approximately 2 × 106 FDC-P1/FLT3-ITD cells, nontreated or treated with AG1295, were fixed in 1% formaldehyde at 37°C for 10 minutes and washed 2 times in PBS containing complete protease inhibitor. Lysates were sonicated at 4°C for 15 cycles of 30 seconds with a 15-second rest period in between cycles (Bioraptor; Diagenode, Liége, Belgium). Immunoprecipitations were done with normal rabbit immunoglobulin G (IgG) or with anti-FOXO3a antibody. The anti-FoxO3a antibody is a rabbit polyclonal antiserum raised against a C-terminal–specific peptide of human FOXO3a. After reversing cross-linking and incubation with proteinase K, DNA was recovered by phenol-chloroform extraction and ethanol precipitation. Immunoprecipitated DNA fragments were subjected to quantitative PCR in triplicate reactions using SYBR green on the 7500 FAST Real-Time System. Primers for detection of the Forkhead responsive element (FHRE) region in the murine Bim promotor were: 5′-GGGCGGGTACATTCT-GAGT-3′; 5′-CAGGCTGCGACAGGTAGTG-3′.23 The Δ cycle threshold (Ct) value of each sample was calculated by subtracting the Ct value for the input sample from the Ct value obtained for the immunoprecipitated sample. Fold increase of FOXO3a binding was then calculated using the 2−ΔΔCt method for each sample with nontreated cells as reference. PCRs with the same primers at 55°C for 32 cycles were also performed to visualize products on 1.5% agarose gels.
Statistical analysis
Statistical analysis was performed using the Student t test, and P values less than .05 were considered statistically significant.
Results
The PI-3 kinase/AKT pathway elicits critical growth stimulatory and prosurvival signals in FLT3-ITD–transduced FDC-P1 cells
We retrovirally infected the myeloid progenitor cell line FDC-P1 with a FLT3-ITD derived from an AML patient with a 7-amino acid duplication (EYEYDLK)29 spanning tyrosine residues 597 and 599 implied in the transduction of growth-promoting signals. Previously described cells expressing wild-type, nonmutated FLT3 receptor were used as controls.15 FLT3 expression was confirmed by immunoprecipitation followed by immunoblotting (Figure 1A). Upon cytokine deprivation, cells expressing wild-type FLT3 died rapidly, whereas cells expressing FLT3-ITD did not (Figure 1A). Because AKT and Ras signaling provide survival cues for FLT3-ITD–transformed cells, we investigated their phosphorylation status in FDC-P1/FLT3-ITD cells as well as the AKT downstream target FOXO3a. As expected, phosphorylation of AKT, ERK, and FOXO3a was apparent (Figures 1B,2D).
Figure 1. Growth stimulatory and antiapoptotic signals in FLT3-ITD–transduced FDC-P1 cells is mediated by the PI-3 kinase/AKT pathway.

FDC-P1 cells were retrovirally transduced with human FLT3-ITD. (A) Expression was confirmed by immunoprecipitation and immunoblotting for human FLT3. FDC-P1 cells expressing wild-type FLT3 or FLT3-ITD were cytokine-deprived and analyzed for survival during a 9-day incubation period by annexin V–FITC/PI staining and flow cytometry. One representative experiment of 3 performed yielding similar results is shown. (B) Whole-cell lysates were prepared from FDC-P1/FLT3 and FDC-P1/FLT3-ITD cells after 16 hours of cytokine deprivation followed by stimulation with 50 ng/mL FL, 10 ng/mL IL-3, or no cytokine addition. Western blot analysis was performed for p-AKT and p-FOXO3a. All blots were reprobed with a GAPDH antibody to demonstrate equal loading. (C) FDC-P1/FLT3-ITD cells treated with either 20 μM LY294002 or 50 μM PD98059 for 24 hours were stained for apoptotic cells with annexin V–FITC/PI and analyzed by flow cytometry. (D) Cell-cycle analysis was performed on FDC-P1/FLT3-ITD cells after 16 hours of treatment with either 20 μM LY294002 or 50 μM PD98059. Numbers of viable cells in S-phase were determined by flow cytometry. Bim expression was determined by Western blot analysis. Data are mean (± standard deviation [SD]) from 3 individual experiments performed in duplicates. **P < .01 (SD over control without inhibitor); ***P < .001 (SD).
Figure 2. Inhibition of FLT3-ITD signaling by AG1295 or PKC412 leads to induction of apoptosis, cell-cycle arrest, and inhibition of AKT and ERK phosphorylation.

FDC-P1/FLT3-ITD cells were treated for 24 hours with increasing concentrations of AG1295 (A) or PKC412 (B), then stained for FACS analysis with Annexin V–FITC/PI. (C) Cell-cycle analysis was performed on FDC-P1/FLT3-ITD cells after culturing with 10 μM AG1295 for 8 or 24 hours. Data from FACS analyses are mean (± SD) from 3 individual experiments. (D) Whole-cell lysates were prepared from FDC-P1/FLT3-ITD cells incubated for 24 hours in different concentrations of AG1295 and analyzed for p-AKT and p-ERK (1 representative blot is shown; n = 3).
Next, using specific pathway inhibitors, we demonstrated strong inhibition of survival by FLT3-ITD when applying LY294002, an efficient inhibitor of PI-3 kinase (Figure 1C). In contrast, addition of the MEK1/2 inhibitor PD98058 to block ERK activity did not affect survival induced by FLT3-ITD as much as LY294002. PI-3 kinase was also important for cell-cycle progression, because its inhibition led to G0/G1 arrest (Figure 1D). Only 6% of the cells were in S-phase after inhibiting PI-3 kinase for 24 hours compared with 29% without inhibitor and 21% with PD98059. This implies that the MAP kinase pathway is not as important for survival and proliferation as the PI-3 kinase pathway via FLT3-ITD in FDC-P1 cells. Because PI-3 kinase executes important survival functions by inhibiting the transcriptional activation of proapoptotic Bim, we examined the expression of Bim in FDC-P1/FLT3-ITD cells by Western blot analysis. Strong up-regulation of Bim was apparent after LY294002 treatment, whereas PD98059 only led to a small induction (Figure 1D).
RTK inhibitor–induced apoptosis involves AKT inactivation, down-regulation of phosphorylated FOXO3a, accumulation of proapoptotic Bim and Puma, and cleavage of caspase-3
To analyze the effects of RTK inhibitors on FLT3-ITD–bearing cells, we decided to use 2 previously described inhibitors with selective effects on FLT3. AG1295 has cytotoxic effects on FLT3-ITD–expressing cells, but is not clinically useful due to problems with solubility.30 PKC412 (N-benzoyl staurosporine), originally identified as an inhibitor of protein kinase C,31 is an efficient FLT3 inhibitor currently used in clinical trials. Its feasibility as targeted therapy in AML comes from studies showing low levels of toxicity on normal hematopoiesis32,33 and an ability to prolong survival in mice with activated FLT3-ITD–induced myeloproliferative disease.7,34
Treating FDC-P1/FLT3-ITD cells for 24 hours with increasing concentrations of AG1295 and PKC412 led to gradually increasing apoptosis in a dose-dependent manner (Figure 2A,B) and to cell-cycle arrest (Figure 2C). At later time points, viability declined even further, and after 72 hours, with high concentrations of AG1295 (30μM) or PKC412 (10 nM), all cells stained positive for annexin V and became apoptotic (not shown). While phosphorylation of AKT and ERK was attenuated (Figure 2D), the levels of BH3-only proteins Bim and Puma were increased, which correlated with the appearance of the cleaved active form of caspase-3 (Figure 3A). Treating cells with PKC412 led to similar results (Figure 3B). Because Bim and Puma are targets of FOXO3a,22-24 we examined the expression of phosphorylated FOXO3a in AG1295-treated cells. A time course study with increasing amounts of AG1295 showed that the phosphorylation of FOXO3a declined during treatment, and while phosphorylation of FOXO3a was reduced, levels of Bim, Puma, and cleaved caspase-3 increased (Figure 3C).
Figure 3. Inhibition of FLT3-ITD signaling by AG1295 or PKC412 leads to dephosphorylation of FOXO3a and up-regulation of proapopotic Bim and Puma.

FDC-P1/FLT3-ITD cells were treated for 24 hours with increasing concentrations of AG1295 (A) or PKC412 (B), then whole-cell lysates were prepared and analyzed for expression of Bim, Puma, and caspase-3. (C) FDC-P1/FLT3-ITD cells were treated with AG1295, and the amount of p-FOXO3a and expression of Bim, Puma, and caspase-3 was analyzed by Western blot analysis (n = 3). Anti-GAPDH antibody was used as a loading control. (D-F) FDC-P1/FLT3-ITD cells infected with pBabe:FOXO3(A3):ER were stimulated with 100 nM 4-OHT. After 8 and 24 hours, cells were analyzed for DNA content by PI staining and flow cytometry. Results are mean (± SD) from 2 experiments (D). Cells were also analyzed for apoptosis by flow cytometry after staining with annexin V–FITC/PI. Results are mean (± SD) from duplicates and repeated twice. **P < .01 (SD over control; E). Cell lysates were prepared at times indicated, and Western blot analysis was performed using Bim, Puma, or GAPDH antibodies (n = 2; F).
Apoptotic induction by FOXO3a in FLT3-ITD–expressing cells involves Bim and Puma
To address the importance of FOXO3a inactivation by FLT3-ITD signaling, we retrovirally overexpressed constitutively active, phosphorylation-deficient human FOXO3a fused to the estrogen receptor, FOXO3(A3):ER, in FDC-P1/FLT3-ITD cells. When 4-OHT was added, which leads to transcriptional activity of FOXO3a, fewer cells were maintained in S-phase and more cells accumulated G0 and in sub-G1 (Figure 3D), indicating apoptosis induction. Apoptosis was confirmed by annexin V staining (Figure 3E). Thus, suppression of FOXO3a by FLT3-ITD is important to inhibit apoptosis. Bim and Puma were strongly up-regulated during the first 24 hours (Figure 3F), suggesting a transcriptional regulation by FOXO3a.
Treatment with RTK inhibitors of human leukemic cell lines expressing mutated FLT3 leads to up-regulation of both Bim and Puma
If Bim and Puma are involved in apoptosis upon FLT3-ITD inhibition, they should be up-regulated in AML cells treated with RTK inhibitors. To investigate this, several human leukemic cell lines were treated for 96 hours with AG1295 or PKC412. Only the MV4;11 (expressing exclusively a mutated FLT3-ITD allele)35 and MonoMac-6 (carrying an activated FLT3-V592A mutation)25 cell lines were sensitive to treatment and showed a strong increase in numbers of apoptotic cells (Figure 4A). This was accompanied by up-regulation of Bim protein (Figure 4B). In contrast, leukemic cell lines expressing no FLT3 (THP-1) or wild-type FLT3 (NB4, RS4) showed no sensitivity, although NB4 showed a 2-fold increase of apoptotic cells after treatment with PKC412, despite no effect on Bim expression. Due to the lack of good quality antibodies, differences in the levels of Puma protein were difficult to determine. However, RNA levels of Puma and Bim were up-regulated after 24 hours of treatment with AG1295 or PKC412 (Figure 4C). These results mark a trend by RTK inhibitors to trigger a cytotoxic response mediated by Bim, and possibly Puma, in leukemic cells expressing mutated FLT3.
Figure 4. Treatment with AG1295 or PKC412 of human leukemic cell lines expressing mutated FLT3 leads to up-regulation of Bim and Puma and apoptosis induction.

Leukemic cell lines expressing no FLT3 (THP1), wild-type FLT3 (NB4, RS4), or mutated FLT3 (MV4;11, MonoMac-6) were seeded at a density of 105-106 cells/mL in the absence or presence of AG1295 or PKC412. (A) Seventy-two hours later, cells were analyzed for apoptosis by flow cytometry after staining with annexin V–FITC/PI. Data shown are mean (± SD) from 3 experiments. *P < .05 (SD over control without inhibitor); **P < .01 (SD); ***P < .001 (SD). (B) Expression of Bim and Puma protein was analyzed by Western blot analysis after 48 hours of treatment. (C) Real-time PCR using Bim and Puma primers from cells after 24 hours of treatment with 30 μM AG1295 (□) or 50 nM PKC412 ( ). The results are mean (± SD) from 2 experiments performed in triplicates and presented as relative expression compared with the control housekeeping gene GusB.
). The results are mean (± SD) from 2 experiments performed in triplicates and presented as relative expression compared with the control housekeeping gene GusB.
Bim and Puma are up-regulated in primary AML cells treated with PKC412
To assess the ability of RTK inhibitors to induce Bim or Puma, we exposed mononuclear cells from several AML patients with FLT3-ITD mutations to 2 concentrations of PKC412 (100 and 300 nM). AML cells from 4 patients were responsive to PKC412 and showed a decrease in numbers of viable cells after 72 hours (Figure 5A). Interestingly, the 2 patients with the highest amount of FLT3-ITD compared with total DNA content in the AML cells (25% and 37% for patients 1 and 4, compared with 19% and 18% for patients 2 and 3, respectively) were most responsive. Real-time PCR analysis demonstrated a significant up-regulation of Bim and Puma after 24 hours of PKC412 treatment, ranging from a 1.3- to 4.7-fold increase compared with no treatment (Figure 5B).
Figure 5. Cytotoxic effect of PKC412 on primary AML cells leads to up-regulation of Bim and Puma.
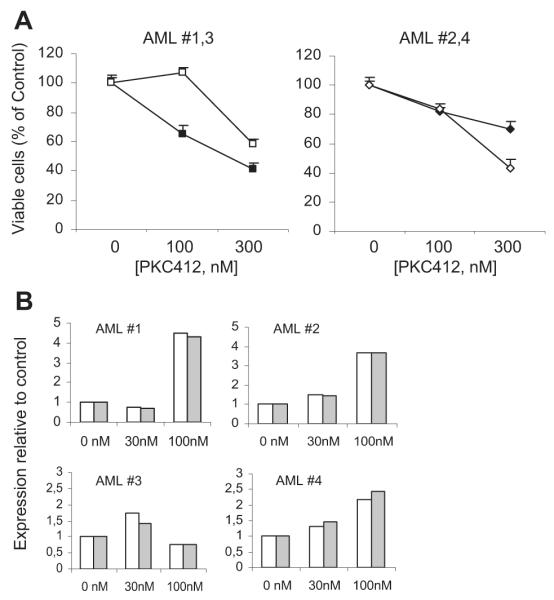
Mononuclear cells were prepared from cryopreserved bone marrow or peripheral blood samples of 4 AML patients. (A) Apoptosis assay was performed with PKC412 at 2 high concentrations (100 and 300 nM) on AML cells cultured for 72 hours, then stained with PI, and analyzed by flow cytometry. Data are mean (± SD) from duplicates. Symbols indicate patients as: ■, AML no. 1; □, AML no. 3; ◆, AML no. 2; ◇, AML no. 4. (B) The relative expression levels of Bim (□) and Puma ( ) mRNA from the same AML patients were measured by real-time PCR after 24 hours of treatment with 30 and 100 nM PKC412. Results are from triplicate reactions.
) mRNA from the same AML patients were measured by real-time PCR after 24 hours of treatment with 30 and 100 nM PKC412. Results are from triplicate reactions.
Silencing of FOXO3a or Bim suppresses apoptosis induced by AG1295 in FLT3-ITD cells
Apparently, suppression of FOXO3a-mediated transcriptional activation of Bim and Puma is an important step in FLT3-ITD transformation. Because inhibition of FLT3-ITD signaling via the receptor or via PI3-kinase induces apoptosis, it seems likely that AG1295 and PKC412 selectively lead to activation and killing via Bim and/or Puma. To assess the involvement of Bim and Puma in executing apoptosis, we performed gene silencing experiments. Amaxa nucleofection was used to transfect siRNA oligonucleotides to all known Bim transcripts (BimEL, BimL, and BimS) in FDC-P1/FLT3-ITD cells, which then were treated with AG1295 at 5 or 10 μM. Western blot analysis demonstrated that Bim expression increased in cells transfected with control siRNA and then treated with AG1295, whereas in cells transfected with Bim siRNA expression remained low (Figure 6A). We then analyzed the number of apoptotic cells after 24-hours of treatment with AG1295. In contrast to control siRNA, Bim siRNA was able to increase the number of viable cells significantly (Figure 6B).
Figure 6. Gene silencing of Bim or FOXO3a by siRNA prevents apoptosis induced by RTK inhibitor.

(A,B) FDC-P1/FLT3-ITD cells were transfected by nucleofection with siRNA specific for mouse Bim (0.7μg) or with a control siRNA (pmaxGFP; 1.5 μg). Four hours after transfection, AG1295 was added as indicated. Cells were harvested after 24 hours and analyzed for effects on Bim expression by Western blot analysis (A) or apoptosis after annexin V–FITC/PI staining and flow cytometry (B). Data shown are mean (+ SD) from 3 individual experiments. (C-F) FOXO3a siRNA oligonucleotides were introduced to FDC-P1/FLT3-ITD cells by nucleofection. Cell lysates were prepared after 24 hours for Western blot analysis (n = 2). In separate experiments, the expression of Bim was analyzed by Western blot analysis in cells transfected with pmaxGFP, 1.3 μg FOXO3a, or Bim siRNA (D). The numbers of viable cells were analyzed by annexin V–FITC/PI staining and flow cytometry after treatment with either AG1295 at 5 μM (E) or PKC412 at 5 nM (F) for 24 hours. The inhibitors were added to cells after 4, 24, or 48 hours after transfection. Data represent mean (± SD) from 2 experiments.
To confirm the role of FOXO3a upstream of Bim in FLT3-ITD inhibition, we transfected FDC-P1/FLT3-ITD cells with siRNAs to FOXO3a. As seen in Figure 6C, the expression of FOXO3a was significantly reduced after 24 hours of treatment. This was accompanied by reduction of Bim protein expression, indicating that silencing of FOXO3a affected Bim (Figure 6D). Next, we added RTK inhibitors at different time points after transfection (4, 24, and 48 hours, respectively), and the number of apoptotic cells was analyzed after 24 hours by annexin V–FITC/PI labeling. Flow cytometry revealed that treatment with AG1295 or PKC412 of cells transfected with control siRNA led to induction of apoptosis. This effect was reduced when transfecting FOXO3a siRNA and varied between 15% and 50% (Figure 6E,F). Together, these results suggest that Bim and FOXO3a significantly contribute to AG1295- and PKC412-induced apoptosis in cells expressing FLT3-ITD.
Bim is rate-limiting for RTK inhibitor–induced apopotosis of FLT3-ITD–expressing cells, but Puma can contribute
In addition to Bim, Puma can bind to all prosurvival Bcl-2 family members and may be involved in RTK inhibitor–mediated apoptotic induction of FLT3-ITD–bearing cells. To assess this possibility, we repeated the gene silencing experiments including siRNA to Puma. As seen in Figure 7A, silencing of Puma was successful in FDC-P1/FLT3-ITD cells. When comparing cells transfected with siRNA specific for Bim or Puma, and then treating the cells with 10 μM AG1295, Puma silencing increased the number of viable cells only marginally, whereas Bim silencing caused a profound survival benefit (Figure 7B). Interestingly, in cells transfected with siRNA to both Bim and Puma survival was enhanced compared with Bim alone, implying that Bim and Puma may synergize in RTK inhibitor–mediated apoptosis but the Puma knockdown efficiency achieved in our system may not have been sufficient to reveal a stronger biologic effect.
Figure 7. Bim is more critical than Puma in apoptotic induction of FLT3-ITD inhibition and is a direct transcriptional target of FOXO3a in FLT3-ITD cells treated with AG1295.

(A,B) FDC-P1/FLT3-ITD cells were transfected with siRNA control pmaxGFP (■) or siRNA to Bim (□), Bim + Puma ( ), or Puma (
), or Puma ( ). Cell lysates were prepared after 24 hours for Western blot analysis of Puma (A). In separate experiments, cells were transfected with siRNA as indicated, and 4 hours after transfection, AG1295 at 10 μM was added. Twenty-four hours later, cells were analyzed for apoptosis by flow cytometry after staining with annexin V–FITC and PI (B). Data shown are mean (± SD) from 1 representative experiment performed in duplicate and repeated twice. **P < .01 (SD over control siRNA); ***P < .001 (SD over control siRNA). (C,D) Wild-type (■), bim-/- (□), bim-/- puma-/- (
). Cell lysates were prepared after 24 hours for Western blot analysis of Puma (A). In separate experiments, cells were transfected with siRNA as indicated, and 4 hours after transfection, AG1295 at 10 μM was added. Twenty-four hours later, cells were analyzed for apoptosis by flow cytometry after staining with annexin V–FITC and PI (B). Data shown are mean (± SD) from 1 representative experiment performed in duplicate and repeated twice. **P < .01 (SD over control siRNA); ***P < .001 (SD over control siRNA). (C,D) Wild-type (■), bim-/- (□), bim-/- puma-/- ( ), or puma-/-(
), or puma-/-( ) bone marrow–derived Lin− progenitor cells infected with FLT3-ITD were FACS-sorted based on expression of EYFP. After treatment with 10 and 20 nM PKC412, the viability was assessed by flow cytometry after 72 hours in culture and compared with cells cultured without treatment (C). The bone marrow cells were also analyzed for colony formation in the absence of supportive cytokine but treated with 10 nM PKC412. Colony numbers were assessed after 7 days of culture (D). Data are mean (± SD) from 3 experiments. *P < .03 (SD compared with bone marrow–derived colonies from bim-/- mice). (E,F) ChIP-quantitative PCR analysis for FOXO3a binding to the Bim promotor. Sonicated DNA from FDC-P1/FLT3-ITD cells treated with AG1295 at 10 μM for 4 and 10 hours was immunoprecipitated with anti-FOXO3a or control rabbit IgG and amplified by quantitative PCR using primers specific for the Bim promotor. Relative expression of Bim was normalized to the input value and then compared with the corresponding untreated samples. Error bars represent SEM of triplicate reactions from 1 representative analysis of 2 separate experiments performed (E). PCRs were also visualized on 1.5% agarose gels stained with ethidium bromide (F).
) bone marrow–derived Lin− progenitor cells infected with FLT3-ITD were FACS-sorted based on expression of EYFP. After treatment with 10 and 20 nM PKC412, the viability was assessed by flow cytometry after 72 hours in culture and compared with cells cultured without treatment (C). The bone marrow cells were also analyzed for colony formation in the absence of supportive cytokine but treated with 10 nM PKC412. Colony numbers were assessed after 7 days of culture (D). Data are mean (± SD) from 3 experiments. *P < .03 (SD compared with bone marrow–derived colonies from bim-/- mice). (E,F) ChIP-quantitative PCR analysis for FOXO3a binding to the Bim promotor. Sonicated DNA from FDC-P1/FLT3-ITD cells treated with AG1295 at 10 μM for 4 and 10 hours was immunoprecipitated with anti-FOXO3a or control rabbit IgG and amplified by quantitative PCR using primers specific for the Bim promotor. Relative expression of Bim was normalized to the input value and then compared with the corresponding untreated samples. Error bars represent SEM of triplicate reactions from 1 representative analysis of 2 separate experiments performed (E). PCRs were also visualized on 1.5% agarose gels stained with ethidium bromide (F).
To further define the roles of Bim and Puma in apoptosis induced by FLT3 inhibition in a clean genetic system, we used cells from relevant BH3-only protein gene knockout mice. Lineage-depleted bone marrow cells from mice lacking Bim, Puma, or both, were transformed with a retrovirus containing the same FLT3-ITD that was used for FDC-P1 cells but now carrying EYFP as a marker to enable sorting of infected cells. We first assessed survival after 72 hours in suspension cultures of EYFP+ cells treated with 2 concentrations of PKC412 (10 and 20 nM) by staining with PI followed by flow cytometric analysis. This revealed that FLT3-ITD transformed Lin− bone marrow cells from bim-/- and bim-/- puma-/- mice were highly refractory to PKC412 treatment compared with wild-type cells (Figure 7C). Moreover, loss of puma appeared to delay cell death only marginally. To test whether loss of Bim or Puma were able to provide a long-term survival advantage, we next sorted EYFP+ cells into methylcellulose cultures lacking cytokines to study the clonal growth and transforming ability of FLT3-ITD in the absence of these BH3-only proteins. As demonstrated in Figure 7D, the majority of bim-/- and bim-/- puma-/- bone marrow cells that were able to form colony-forming units-cytokine (CFU-C) in the absence of cytokines formed also colonies in the presence of PKC412 (80.4% ± 7.7% and 79.1% ± 10.3%, respectively). In contrast, RTK-inhibition reduced the colony-formation potential of both wild-type and puma-/- mice to a similar extent (32.7% ± 8.5% and 46.6% ± 17.3%; P < .03 compared with Bim, respectively), confirming our results obtained in the short-term survival assays.
FOXO3a binds directly to the Bim promoter in FLT3-ITD cells treated with AG1295
Our studies indicate that FOXO3a-mediated up-regulation of Bim is essential for apoptotic induction of cells expressing FLT3-ITD and treated with FLT3-selective inhibitors. To determine whether this is due to direct binding of FOXO3a to the Bim promotor, we performed ChIP on the FHRE present in the murine and human Bim promotors. FOXO3a-DNA complexes were purified from FDCP1/FLT3-ITD cells treated with 10 μM AG1295 for 4 and 10 hours, respectively. Primers flanking the FHRE region were used for PCR assays. By quantitative PCR, the amount of DNA precipitated with anti-FOXO3a after treatment with AG1295 increased almost 60-fold compared with untreated cells, whereas control IgG failed to do so (Figure 7E). Specificity for the PCR was confirmed by visualizing PCR products on agarose gels stained with ethidium bromide (Figure 7F). Taken together, this confirms that FLT3-selective AG1295 induces direct binding of FOXO3a on to the Bim promotor and activates gene transcription.
Discussion
The mechanism of action of many anticancer drugs involves induction of apoptosis. While several genes are candidates to design strategies aiming at enhancing apoptosis, the BH3-only proteins are of special interest in that they mediate cytotoxic responses elicited by chemotherapeutic agents.36 Studies from gene knockout mice indicate that Bim and Puma have prominent roles in the response to diverse cytotoxic stimuli, probably because they bind with comparable affinity to all prosurvival Bcl-2 family proteins.37 Moreover, defective apoptosis promotes tumorigenesis by overexpression of prosurvival Bcl-2 family members, but also by loss or inactivation of proapoptotic relatives. Emerging evidence supports that elevated levels in particularly of Bim is a critical determinant in chemotherapy sensitivity,38 hence Bim may also confer resistance to cytotoxic drugs in malignant cells.
Deregulated activation of FLT3 is recognized as the most common genetic alteration in AML ranging from 23% to 35% in adult cases and 5% to 16% in pediatric studies.39 Multiple studies indicate that FLT3-ITD mutations confer a poor prognosis in AML patients.1,4,40,41 The most common aberrations are insertional mutations and/or insertions of amino acids within the juxtamembrane domain leading to duplications of key residues involved in negative feedback of receptor autophosphorylation. This confers permanent signaling via the Ras/MAP-kinase and the PI3-kinase/AKT pathways, which leads to functional inactivation of Bim, in part by repression via transcription factor FOXO3a. In CML, constitutive activation of the BCR-ABL oncoprotein exists in all patients, thereby providing the principle for the efficacy of therapy with the small-molecule inhibitor imatinib. Recent studies have demonstrated that BCR-ABL signaling leads to FOXO3a and Bim inactivation,20 which imatinib efficiently blocks.22 Imatinib activates several proapoptotic BH3-only proteins, but while combined loss of Bim and Bad abrogated apoptosis, loss of Bmf or Puma had no effect,38 indicating that different inhibitors can neutralize different BH3-only proteins.
We demonstrate that treatment with the well-documented RTK inhibitors AG1295 and PKC412 of autonomously growing progenitor cells expressing FLT3-ITD, or human leukemic cell lines carrying activating mutations of FLT3, leads to apoptosis induction via Bim due to inhibition of phosphorylation of AKT and FOXO3a. Previous studies have established that enforced expression of FLT3-ITD signals in progenitor cells leads to AKT-dependent inactivation of FOXO3a and repression of the Bim promoter.13,14 In FDC-P1/FLT3-ITD cells, the induction of FOXO3a activity by AG1295 or PKC412 treatment was accompanied by cell-cycle arrest followed by apoptosis. This is similar to reports proposing that imatinib targets FOXO3a to induce cell death in BCR-ABL–transformed cell lines.22,42 In line with this, AG1295-induced apoptosis was preceded by accumulation of the FOXO3a target Bim and to some extent Puma, and caspase-3 was converted to its active form. Importantly, silencing of either FOXO3a or Bim by siRNA prevented the induction of Bim and delayed apoptosis after AG1295 or PKC412 treatment. Although Puma was up-regulated by an inducible, transcriptionally active FOXO3a in FLT3-ITD–bearing cells upon RTK inhibitor treatment, silencing of Puma was not efficient in inhibiting apoptosis. However, the simultaneous treatment of FDC-P1/FLT3-ITD cells with siRNA to Bim and Puma led to additive effects, implying that they may have nonredundant effects. By using bone marrow cells from gene-targeted mice lacking either Bim or Puma, or both, we could show that bone marrow progenitors expressing FLT3-ITD and lacking both Bim and Puma were as resistant to PKC412 treatment as were cells from bim-/- mice. In contrast, lack of Puma did not significantly protect cells from FLT3 inhibition. Taken together, our observations suggest that transcriptional induction of Bim by FOXO3a upon blockade of FLT3 signaling triggers apoptosis in AML cells. By ChIP assays on AG1295-treated cells expressing FLT3-ITD, we provide evidence for direct binding of FOXO3a to the Bim promotor as a key mechanism for apoptotic induction via RTK inhibitors. Because Puma is essential for p53-mediated apoptosis in several cell types,19,43,44 treatment with p53-inducible chemotherapeutic drugs may augment the effects of RTK inhibition.
Whether mutant FLT3 receptors promote transformation and leukemogenesis by increasing quantitative signaling or inducing qualitative differences involving downstream targets is unclear. The tyrosine residue at position 599 within the ITD used in our study is important for binding of Src family kinases to FLT3 and contributes to FL-mediated ERK activation and proliferation.45 However, treatment of FDC-P1/FLT3-ITD cells with PD98059 had no immediate effect, and cell-cycle progression and survival were not much abrogated. In contrast, LY294002 blocking PI3-kinase showed strong inhibition and cells arrested in G0/G1 and subsequently died by apoptosis. AG1295 and PKC412 treatment led to AKT dephosphorylation, cell-cycle exit, induction of Bim and Puma expression, and the onset of apoptosis, suggesting the PI3-kinase pathway as a primary target for therapeutic intervention in AML.
Data from clinical trials in AML patients suggest that RTK inhibitors may effectively complement conventional chemotherapy.16,46-48 While clinical findings have identified mutations in the BCR-ABL gene that confer resistance to therapy, the similarity of most small-molecule inhibitors to inhibit ATP binding of mutated kinases predicts that AML will develop cellular resistance to multiple inhibitors. Acquired resistance to inhibitors after treatment of AML patients harboring mutated FLT3 receptors have been demonstrated.8,9 Thus, the treatment with drugs eliciting apoptotic responses via BH3-only proteins may have initial effects while gradually developing into more inefficient treatment, emphasizing the need for complementary treatment. Novel BH3 mimetics define a new class of anticancer agents that directly target prosurvival Bcl-2 proteins.49 Although effective as a single agent in killing tumor cells, ABT-737 should show synergy with other anticancer agents. Recently, it was shown that overexpression of prosurvival Bcl-2 family proteins leads to resistance against RTK inhibitors in a cell line with activating FLT3 mutations,50 indicating that BH3-only proteins are involved in apoptotic induction. Interestingly, the susceptibility to RTK inhibition was restored by treatment with ABT-737.
The majority of AML patients are likely to benefit from combined pharmaceutic strategies converging on FLT3 signaling and downstream signaling events, tilting the balance to proapoptotic Bcl-2 family members, in which Bim appears to play a significant role. It will now be of interest to focus on the role of Bim and other BH3-only proteins in the development of resistence to selective FLT3 drugs in AML.
Acknowledgments
We thank A. Strassser for the gift of knockout mice, and P. Coffer, J. Griffin, K. Lotfi, K. Spiekermann, and F. Öberg for reagents and cell lines. We are grateful to Dr G. Nilsson for critcial reading of the manuscript and to P. Druid, P. Hammar, and P. Tsapogas for technical help. PKC412 was a gift from Novartis Pharma AG (Basel, Switzerland).
This work was supported by grants from the Swedish Cancer Foundation (Stockholm, Sweden; 4249-B04-06XBB) and the Swedish Children’s Cancer Foundation (Stockholm, Sweden; PROJ07/047) to J.-I.J.; by grants from the Austrian Science Fund (FWF, Vienna, Austria; Y212-B13 START and SFB021) to A.V.; and grants from HKH Kronprinsessan Lovisas förening för barnasjukvård (Stockholm, Sweden), Axel Tielmans Minnesfond (Stockholm, Sweden), The County Council of Östergötland (Linköping, Sweden), the Cancer Foundation of Östergötland (Linköping, Sweden), and the Hans von Kantzows (Stockholm, Sweden), Magnus Bergvalls (Stockholm, Sweden), and the Ollie and Elof Ericssons (Åtvidaberg, Sweden) Foundations.
Footnotes
Authorship Contribution: A.N. and P.E. performed research and analyzed data; M.K. initiated the experimental part of the work, generated necessary cell lines, and performed research; V.L. performed experiments with gene-targeted bone marrow cells; E.W.-F.L. assisted with chromatin immunoprecipitation experiments; A.V. provided material from gene-targeted mice and analyzed data; J.-I.J. designed and performed research and drafted the manuscript; and all authors participated in the evaluation of artwork and actively participated in writing the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
References
- 1.Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 2.Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N-RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074–3080. [PubMed] [Google Scholar]
- 3.Stirewalt DL, Kopecky KJ, Meshinchi S, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97:3589–3595. doi: 10.1182/blood.v97.11.3589. [DOI] [PubMed] [Google Scholar]
- 4.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 5.Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96:3907–3914. [PubMed] [Google Scholar]
- 6.Hayakawa F, Towatari M, Kiyoi H, et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3–dependent cell lines. Oncogene. 2000;19:624–631. doi: 10.1038/sj.onc.1203354. [DOI] [PubMed] [Google Scholar]
- 7.Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433–443. doi: 10.1016/s1535-6108(02)00069-7. [DOI] [PubMed] [Google Scholar]
- 8.Clark JJ, Cools J, Curley DP, et al. Variable sensitivity of FLT3 activation loop mutations to the small molecule tyrosine kinase inhibitor MLN518. Blood. 2004;104:2867–2872. doi: 10.1182/blood-2003-12-4446. [DOI] [PubMed] [Google Scholar]
- 9.Grundler R, Thiede C, Miething C, Steudel C, Peschel C, Duyster J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood. 2003;102:646–651. doi: 10.1182/blood-2002-11-3441. [DOI] [PubMed] [Google Scholar]
- 10.Weisberg E, Roesel J, Bold G, et al. Anti-leukemic effects of the novel, mutant FLT3 inhibitor, NVP-AST487: effects on PKC412-sensitive and -resistant FLT3-expressing cells. Blood. 2008;112:5161–5170. doi: 10.1182/blood-2008-02-138065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heidel F, Solem FK, Breitenbuecher F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 12.Schwable J, Choudhary C, Thiede C, et al. RGS2 is an important target gene of Flt3-ITD mutations in AML and functions in myeloid differentiation and leukemic transformation. Blood. 2005;105:2107–2114. doi: 10.1182/blood-2004-03-0940. [DOI] [PubMed] [Google Scholar]
- 13.Scheijen B, Ngo HT, Kang H, Griffin JD. FLT3 receptors with internal tandem duplications promote cell viability and proliferation by signaling through Foxo proteins. Oncogene. 2004;23:3338–3349. doi: 10.1038/sj.onc.1207456. [DOI] [PubMed] [Google Scholar]
- 14.Brandts CH, Sargin B, Rode M, et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005;65:9643–9650. doi: 10.1158/0008-5472.CAN-05-0422. [DOI] [PubMed] [Google Scholar]
- 15.Jönsson M, Engström M, Jönsson JI. FLT3 ligand regulates apoptosis through AKT-dependent inactivation of transcription factor FoxO3. Biochem Biophys Res Commun. 2004;318:899–903. doi: 10.1016/j.bbrc.2004.04.110. [DOI] [PubMed] [Google Scholar]
- 16.Knapper S, Mills KI, Gilkes AF, Austin SJ, Walsh V, Burnett AK. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: the induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood. 2006;108:3494–3503. doi: 10.1182/blood-2006-04-015487. [DOI] [PubMed] [Google Scholar]
- 17.Puthalakath H, Strasser A. Keeping killers on a tight leash: transcriptional and post-translational control of the pro-apoptotic activity of BH3-only proteins. Cell Death Differ. 2002;9:505–512. doi: 10.1038/sj.cdd.4400998. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 19.Erlacher M, Michalak EM, Kelly PN, et al. BH3-only proteins Puma and Bim are rate-limiting for γ-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131–4138. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuribara R, Honda H, Matsui H, et al. Roles of Bim in apoptosis of normal and Bcr-Abl–expressing hematopoietic progenitors. Mol Cell Biol. 2004;24:6172–6183. doi: 10.1128/MCB.24.14.6172-6183.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skorski T, Bellacosa A, Nieborowska-Skorska M, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. EMBO J. 1997;16:6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Essafi A, Fernandez de Mattos S, Hassen YA, et al. Direct transcriptional regulation of Bim by FoxO3a mediates STI571-induced apoptosis in Bcr-Abl–expressing cells. Oncogene. 2005;24:2317–2329. doi: 10.1038/sj.onc.1208421. [DOI] [PubMed] [Google Scholar]
- 23.You H, Pellegrini M, Tsuchihara K, et al. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the proapoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 25.Spiekermann K, Dirschinger RJ, Schwab R, et al. The protein tyrosine kinase inhibitor SU5614 inhibits FLT3 and induces growth arrest and apoptosis in AML-derived cell lines expressing a constitutively activated FLT3. Blood. 2003;101:1494–1504. doi: 10.1182/blood-2002-04-1045. [DOI] [PubMed] [Google Scholar]
- 26.Bouillet P, Metcalf D, Huang DC, et al. Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity. Science. 1999;286:1735–1738. doi: 10.1126/science.286.5445.1735. [DOI] [PubMed] [Google Scholar]
- 27.Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 28.Erlacher M, Labi V, Manzl C, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006;203:2939–2951. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310–318. doi: 10.1182/blood.v99.1.310. [DOI] [PubMed] [Google Scholar]
- 30.Levis M, Allebach J, Tse KF, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–3891. doi: 10.1182/blood.v99.11.3885. [DOI] [PubMed] [Google Scholar]
- 31.Meyer T, Regenass U, Fabbro D, et al. A derivative of staurosporine (CGP 41 251) shows selectivity for protein kinase C inhibition and in vitro anti-proliferative as well as in vivo anti-tumor activity. Int J Cancer. 1989;43:851–856. doi: 10.1002/ijc.2910430519. [DOI] [PubMed] [Google Scholar]
- 32.Propper DJ, McDonald AC, Man A, et al. Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase C. J Clin Oncol. 2001;19:1485–1492. doi: 10.1200/JCO.2001.19.5.1485. [DOI] [PubMed] [Google Scholar]
- 33.Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60. doi: 10.1182/blood-2004-03-0891. [DOI] [PubMed] [Google Scholar]
- 34.Lee BH, Williams IR, Anastasiadou E, et al. FLT3 internal tandem duplication mutations induce myeloproliferative or lymphoid disease in a transgenic mouse model. Oncogene. 2005;24:7882–7892. doi: 10.1038/sj.onc.1208933. [DOI] [PubMed] [Google Scholar]
- 35.Quentmeier H, Reinhardt J, Zaborski M, Drexler HG. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia. 2003;17:120–124. doi: 10.1038/sj.leu.2402740. [DOI] [PubMed] [Google Scholar]
- 36.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen L, Willis SN, Wei A, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 38.Kuroda J, Puthalakath H, Cragg MS, et al. Bim and Bad mediate imatinib-induced killing of Bcr/ Abl+ leukemic cells, and resistance due to their loss is overcome by a BH3 mimetic. Proc Natl Acad Sci U S A. 2006;103:14907–14912. doi: 10.1073/pnas.0606176103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parcells BW, Ikeda AK, Simms-Waldrip T, Moore TB, Sakamoto KM. FMS-like tyrosine kinase 3 in normal hematopoiesis and acute myeloid leukemia. Stem Cells. 2006;24:1174–1184. doi: 10.1634/stemcells.2005-0519. [DOI] [PubMed] [Google Scholar]
- 40.Schnittger S, Schoch C, Dugas M, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 41.Frohling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–4380. doi: 10.1182/blood-2002-05-1440. [DOI] [PubMed] [Google Scholar]
- 42.Ghaffari S, Jagani Z, Kitidis C, Lodish HF, Khosravi-Far R. Cytokines and BCR-ABL mediate suppression of TRAIL-induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc Natl Acad Sci U S A. 2003;100:6523–6528. doi: 10.1073/pnas.0731871100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villunger A, Michalak EM, Coultas L, et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science. 2003;302:1036–1038. doi: 10.1126/science.1090072. [DOI] [PubMed] [Google Scholar]
- 44.Jeffers JR, Parganas E, Lee Y, et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell. 2003;4:321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 45.Heiss E, Masson K, Sundberg C, et al. Identification of Y589 and Y599 in the juxtamembrane domain of Flt3 as ligand-induced autophosphorylation sites involved in binding of Src family kinases and the protein tyrosine phosphatase SHP2. Blood. 2006;108:1542–1550. doi: 10.1182/blood-2005-07-008896. [DOI] [PubMed] [Google Scholar]
- 46.Fiedler W, Serve H, Dohner H, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105:986–993. doi: 10.1182/blood-2004-05-1846. [DOI] [PubMed] [Google Scholar]
- 47.Levis M, Pham R, Smith BD, Small D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004;104:1145–1150. doi: 10.1182/blood-2004-01-0388. [DOI] [PubMed] [Google Scholar]
- 48.Yee KW, Schittenhelm M, O’Farrell AM, et al. Synergistic effect of SU11248 with cytarabine or daunorubicin on FLT3 ITD-positive leukemic cells. Blood. 2004;104:4202–4209. doi: 10.1182/blood-2003-10-3381. [DOI] [PubMed] [Google Scholar]
- 49.Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 50.Kohl TM, Hellinger C, Ahmed F, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21:1763–1772. doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]