

Abstract
The WRN protein belongs to the RecQ family of DNA helicases and is implicated in replication fork restart, but how its function is regulated remains unknown. We show that WRN interacts with the 9.1.1 complex, one of the central factors of the replication checkpoint. This interaction is mediated by the binding of the RAD1 subunit to the N-terminal region of WRN and is instrumental for WRN relocalisation in nuclear foci and its phosphorylation in response to replication arrest. We also find that ATR-dependent WRN phosphorylation depends on TopBP1, which is recruited by the 9.1.1 complex in response to replication arrest. Finally, we provide evidence for a cooperation between WRN and 9.1.1 complex in preventing accumulation of DNA breakage and maintaining genome integrity at naturally-occurring replication fork stalling sites.
Taken together, our data unveil a novel functional interplay between WRN helicase and the replication checkpoint, contributing to shed light into the molecular mechanism underlying the response to replication fork arrest.
Keywords: RecQ helicases, Replication checkpoint, Genome Instability, Werner syndrome
INTRODUCTION
Werner syndrome protein (WRN) is a member of the RecQ family of DNA helicases. Mutations in the WRN gene lead to a human disorder the Werner syndrome (WS), characterised by a high incidence of cancer (Martin and Oshima 2000, Muftuoglu et al 2008b) and wide genomic instability manifested as chromosomal abnormalities (Muftuoglu et al 2008b). Mounting evidence strongly supports the idea that WRN plays a critical role in the maintenance of genome stability through faithful rescue of stalled replication forks. At the biochemical level, WRN shows a remarkable preference towards substrates that mimic structures associated with stalled replication forks (Brosh et al 2002, Machwe et al 2006) and WS cells exhibit enhanced instability at common fragile sites, chromosomal regions especially prone to replication fork stalling (Pirzio et al 2008). How WRN favours recovery of stalled forks and prevents DNA breakage upon replication perturbation is not fully understood. It has been suggested that WRN might facilitate replication restart either by promoting recombination or processing intermediates at stalled forks in a way that counteracts unscheduled recombination (Franchitto and Pichierri 2004, Pichierri 2007, Sidorova 2008). This hypothesis is supported by our recent findings indicating that loss of WRN results in excessive formation of double-stranded DNA breaks (DSBs) at stalled forks, which are subsequently repaired through recombination (Pirzio et al 2008).
Maintenance of genome stability during DNA synthesis requires the function of the replication checkpoint, which ensures proper handling of stalled forks and avoids DSB formation at replication intermediates. The replication checkpoint response is under the control of the ATR kinase that is recruited to stalled forks through ATRIP-mediated binding to RPA-coated stretches of ssDNA (Cimprich and Cortez 2008). Full activation of the checkpoint response requires the presence of the RAD9/RAD1/HUS1 (9.1.1) complex, which is loaded onto chromatin independently of ATR/ATRIP and stimulates the ATR activity through recruitment of the TopBP1 mediator protein (Cimprich and Cortez 2008). Indeed, TopBP1 associates directly with the 9.1.1 complex and, once recruited to stalled forks, facilitates the ATR/ATRIP-mediated phosphorylation of several other checkpoint proteins, in particular the downstream kinase CHK1 (Delacroix et al 2007, Furuya et al 2004, Zou et al 2002). In addition, the 9.1.1 complex acts as docking station for other proteins that are relocalised to replication forks or DNA damage sites, such as DNA polymerase beta, kappa and the DNA glycosylase NEIL1 (Guan et al 2007, Kai and Wang 2003, Toueille et al 2004). However, no functional interaction of the 9.1.1 complex with proteins that are directly correlated to replication fork processing and recovery has been identified so far.
Even though WRN is phosphorylated in an ATR-dependent manner during the response to replication fork arrest, loss of ATR activity does not abolish WRN recruitment to chromatin (Pichierri et al 2003), suggesting that other checkpoint factors may directly associate with WRN and affect its function in response to replication fork stalling. Interestingly, disruption of the 9.1.1 complex in mice results in the accumulation of DSBs at stalled forks and enhances chromosome fragility at genomic regions considered to be the equivalent of the human common fragile sites (Zhu and Weiss 2007).
Here we investigated the functional and physical interaction between WRN and the 9.1.1 complex and disclosed the essential role of 9.1.1 in WRN recruitment to stalled forks and in the regulation of its ATR-mediated phosphorylation.
RESULTS
The 9.1.1 complex is required for WRN relocalisation and phosphorylation following replication fork stalling
It is well recognized that in response to DNA damage or replication fork stalling WRN leaves the nucleolus and redistributes to nuclear foci (Constantinou et al 2000, Franchitto and Pichierri 2004). Since several proteins that participate in DNA repair and checkpoint response are loaded onto the chromatin by the 9.1.1 complex, we investigated whether WRN recruitment to stalled forks could be similarly regulated. To this aim, expression of the RAD9 subunit was down-regulated by RNAi in HeLa cells leading to the disruption of the entire complex (Figure 1A). RNAi-treated cells were challenged with HU or CPT and examined for the ability of WRN to form foci. The majority of WRN was detected in nucleoli in untreated cells, whereas, after treatments, it relocalised to nuclear foci in cells transfected either with empty vector (mock) or control siRNA (siCtrl), but not in RAD9-depleted cells (Figure 1B).
Figure 1. Abrogation of RAD9 function impairs WRN subnuclear relocalisation and phosphorylation after HU-treatment.


(A) Depletion of RAD9 by RNAi. HeLa cells were transfected with siRNAs directed against GFP (siCtrl) or RAD9 (siRAD9) and cell lysates were prepared at the indicated time prior to immunoblotting with anti-RAD9 and anti-RAD1 antibodies. Anti-WRN antibody was used as loading control. (B) WRN relocalisation in HeLa cells transfected with Ctrl or RAD9 siRNAs and 48h later exposed for 6h to HU or CPT prior to immunofluorescence with anti-WRN antibody. In the panel, representative images from the untreated and the HU-treated cells are shown. Graph shows quantification of the nuclei presenting WRN focal staining under different experimental conditions. (C) WRN relocalisation in HeLa cells transfected with Ctrl or RAD9 siRNAs and 48h later exposed for 3h to etoposide (Etop) or bleomycin (Bleo) prior to immunofluorescence with anti-WRN antibody. In the panel, representative images from the untreated and the etoposide-treated cells are shown. Quantification of the nuclei presenting WRN focal staining after etoposide or bleomycin exposure is shown in the graph. (D) Depletion of WRN by RNAi. HeLa cells were transfected with siRNAs directed against GFP (siCtrl) or WRN and cell lysates prepared at the indicated time prior to immunoblotting with anti-WRN antibody. Anti-RAD9 antibody was used to verify that siWRN did not produce disruption of the 9.1.1 complex. Anti-PCNA antibody was used as loading control. (E) RAD9 relocalisation in nuclear foci in cells depleted of WRN. HeLa cells were transfected with Ctrl or WRN siRNAs and 48h thereafter treated for 6h with HU or CPT prior to immunofluorescence with anti-RAD9 antibody. In the panel, representative images from untreated and CPT-treated cells are shown. The percentage of nuclei showing RAD9 focal staining for each experimental condition is reported in the graph. (F) HeLa cells were transfected with siRNAs directed against GFP (siCtrl) or RAD9 (siRAD9) and treated with 10μM CPT for 6h, then cell lysates were prepared to immunoblotting with anti-RAD9 antibody. Anti-tubulin (Tub.) antibody was used as loading control. (G) WRN phosphorylation in RAD9-depleted cells. Mock and RAD9 RNAi-transfected HeLa cells were treated with 2mM HU or 10μM CPT for 6h, then cell extracts were immunoprecipitated (IP) using anti-WRN antibody. WRN phosphorylation was evaluated for the presence of a phospho-reactive band (IB) using anti-pST/Q antibodies. The total amount of WRN immunoprecipitated was determined by anti-WRN antibody (IB).
Data are presented as means of three independent experiments. Error bars represent standard error.
Although both WRN and RAD9 (i.e. the 9.1.1 complex altogether) are implicated in DSB and base-excision repair (BER) (Cheng et al 2004, Harrigan et al 2006, Lan et al 2005, Roos-Mattjus et al 2003, Toueille et al 2004), treatment of RAD9-depleted cells with agents producing cell cycle-independent breaks (etoposide) or base-damage to DNA (bleomycin) did not affect WRN relocalisation in nuclear foci (Figure 1C). Similarly, transfection of HeLa cells using a single siRAD9 oligonucleotide, designed to target a region different from those targeted by the pool of four independent siRAD9 sequences (see also “Materials and Methods”), efficiently down-regulated RAD9 protein and prevented formation of WRN foci specifically after replication arrest induced by HU (Supplementary Figure 1). Consistently with a specific requirement of the 9.1.1 complex in the formation of WRN foci, introduction of an RNAi-resistant RAD9 protein in cells transfected with RAD9 siRNA restored the ability of WRN to localize in nuclear foci after replication arrest (Supplementary Figure 2).
Since RAD9 is necessary for WRN recruitment, we next examined whether WRN is required for RAD9 foci formation following replication stress. We down-regulated WRN expression in HeLa cells (Figure 1D) and analysed formation of RAD9 foci by immunofluorescence. Down-regulation of WRN in untreated cells resulted in a small fraction of nuclei showing RAD9 localisation in foci (Figure 1E). The percentage and brightness of nuclei with RAD9 foci enhanced following HU or CPT exposure irrespective of the presence of WRN and appeared even increased in siWRN-treated cells (Figure 1E).
We have previously reported that in response to replication arrest, WRN is phosphorylated by an ATR-dependent mechanism and assembles into nuclear foci even in cells over-expressing the kinase dead form of ATR (Pichierri et al 2003). Consistently, time-course analysis showed that WRN phosphorylation occurs early after replication arrest when WRN begins to form nuclear foci (Supplementary Figure 3A and B). We then investigated the dependence of the ATR-dependent WRN phosphorylation on the 9.1.1 complex. To test this hypothesis, HeLa cells, in which RAD9 had been depleted (Figure 1F), were treated with HU or CPT and cell lysates were subjected to WRN immunoprecipitation (Figure 1G). Using an antibody that specifically recognises phospho S/TQ motifs (pS/TQ), phosphorylation of WRN was barely detectable in untreated conditions but strongly increased after treatments (Figure 1G). Depletion of RAD9 resulted however in reduced WRN immunoreactivity to the pS/TQ antibody, suggesting that most, if not all, of the phosphorylation of WRN is RAD9-dependent (Figure 1G). In agreement with the observed RAD9-independent WRN relocalisation following bleomycin treatment (Figure 1C), RAD9 depletion did not affect S/TQ phosphorylation of WRN after DNA damage induced by this agent (data not shown).
Altogether, these results suggest that the presence of a functional 9.1.1 complex is specifically required to regulate WRN relocalisation into nuclear foci upon replication fork stalling and is also necessary for the ATR-dependent phosphorylation of WRN.
WRN phosphorylation but not its relocalisation depends on the replication checkpoint mediator protein TopBP1
In human cells and model organisms, phosphorylation of several ATR targets (Kumagai et al 2006, Liu et al 2006, Mordes et al 2008) is stimulated by TopBP1, which requires RAD9 for its chromatin loading (Delacroix et al 2007). Since WRN phosphorylation was defective in the absence of RAD9, we examined the possibility that this impairment might depend on the loss of TopBP1 function. HeLa cells were depleted of TopBP1 by RNAi (Figure 2A) and treated with HU to analyse the WRN phosphorylation status (Figure 2B). Western blotting analysis showed that loss of TopBP1 prevented the HU-induced phosphorylation at S/TQ sites of WRN (Figure 2B). Consistently with the observed RAD9-independent WRN foci formation after bleomycin treatment (Figure 1C), TopBP1 down-regulation by RNAi did not affect phosphorylation of WRN at S/TQ sites following this kind of DNA damage (Supplementary Figure 4). Altogether, these findings establish a role for TopBP1 in the in vivo regulation of WRN phosphorylation in response to perturbed replication.
Figure 2. Analysis of the requirement of TopBP1 for WRN relocalisation and phosphorylation upon perturbed replication.

(A) Depletion of TopBP1 by RNAi. HeLa cells were transfected with siRNAs directed against GFP (siCtrl) or TopBP1 (siTopBP1) and cell lysates immunoblotting with anti-TopBP1 antibody. Anti-PCNA antibody was used as loading control. (B) Analysis of WRN phosphorylation at S/TQ sites in TopBP1-depleted cells after HU treatment. Ctrl and TopBP1 RNAi-transfected HeLa cells were treated with 2mM HU for 8h prior to lysis and immunoprecipitation using an anti-WRN antibody. WRN phosphorylation was evaluated by immunoblotting in the WRN immunoprecipitates with an anti-pST/Q antibody (IB: pS/TQ). The total amount of the immunoprecipitated WRN protein was determined by anti-WRN immunobloting (IB: WRN). Immunoprecipitation using normal rabbit IgG (IgG) was used as a negative control. (C) WRN relocalisation in nuclear foci in cells with abrogated TopBP1 function. HeLa cells were transfected with TopBP1 siRNAs and 48h later were exposed for 6h to HU or CPT prior to immunofluorescence with anti-WRN antibody. In the panel, representative images from the untreated and the HU- or CPT-treated cells are shown. Graph shows quantification of the nuclei presenting WRN focal staining after HU or CPT treatment. Data are presented as means of three independent experiments. Error bars represent standard error.
To further address the importance of TopBP1 for WRN function, the ability of TopBP1-depleted cells to recruit WRN after replication arrest was investigated. No differences in the level of WRN foci formation was observed however in cells treated with either HU or CPT (Figure 2C).
Thus, although TopBP1 is not needed for WRN recruitment in response to replication arrest, it is essential for subsequent WRN phosphorylation by ATR.
Co-localisation and increased association between WRN and 9.1.1 complex after replication arrest
Since the 9.1.1 complex regulates WRN recruitment and phosphorylation, we next wanted to test whether WRN could associate with the 9.1.1 complex in response to replication arrest. Cell lysates were immunoprecipitated with an anti-WRN antibody and analysed for the presence of the subunits of the 9.1.1 complex. The results show that WRN associated with RAD9 and RAD1 in untreated cells and this association appeared to be stimulated by HU and CPT (Figure 3A). Interestingly, reciprocal co-immunoprecipitation experiments confirmed the WRN/RAD9 interaction and its enhancement by replication arrest (Figure 3B).
Figure 3. The 9.1.1 complex and WRN co-immunoprecipitate and co-localise upon replication arrest.

(A) WRN immunoprecipitates the 9.1.1 complex. HeLa cells were treated with 2mM HU or 20μM CPT for 6h, then cell lysates were immunoprecipitated (IP) using anti-WRN antibody and normal IgG as a negative control. The presence of RAD9 and RAD1 was assessed by immunoblotting (IB) using the indicated antibodies. Inputs contained 20% of the total lysates used for immunoprecipitation. (B) RAD9 immunoprecipitates WRN. HeLa cells were treated with 2mM HU or 20μM CPT for 6h. Cell extracts were immunoprecipitated (IP) with anti-RAD9 antibody and normal IgG as a negative control. The presence of WRN was evaluated by immunoblotting (IB) using the indicated antibody. (C) Analysis of WRN and RAD9 co-localisation after replication arrest. HeLa cells were treated with 2mM HU for 8h and subjected to immunofluorescence using mouse anti-WRN and rabbit anti-RAD9 antibodies. Representative images from HeLa cells untreated or treated with HU for 8h are presented. Insets show an enlarged portion of the nuclei for a better evaluation of the co-localisation status of WRN with RAD9 foci.
Given that both WRN and RAD9 form nuclear foci in response to replication perturbation, we tested whether WRN co-localized with the 9.1.1 complex by immunofluorescence studies. Prior to treatment, the majority of WRN was in the nucleolus, whereas RAD9 was distributed in the nucleoplasm in a faint scattered form (Figure 3C). However, in response to HU, WRN formed distinct nuclear foci that largely co-localized with those of RAD9 (Figure 3C).
Thus, WRN and the 9.1.1 complex interact in vivo and replication fork arrest stimulates their association and co-localization at nuclear foci.
Direct interaction of the N-terminal region of WRN with the 9.1.1 complex via RAD1
To confirm a direct interaction between WRN and the 9.1.1 complex, we performed in vitro binding assay using recombinant proteins. Full-length WRN was split into three fragments: N-WRN (aa 1-550), H-WRN (aa 506-949) and C-WRN (aa 940-1432), which were expressed as GST-fusion proteins in bacteria (Figure 4A). Purified GST-fusion proteins were used as bait in GST pull-down experiments with HeLa nuclear extracts. Since each subunit of the complex does not exist in isolation in the cell, we used the presence of RAD9 and RAD1 as readout of the entire complex in the pulled down material. Western blotting analysis revealed that only the GST-N-WRN fragment was able to pull-down the 9.1.1 complex from nuclear extracts and multiple bands corresponding to RAD9 were detected (Figure 4A). Given that RAD9 exists in unphosphorylated and phosphorylated isoforms, it is possible that both were pulled down in our experiments.
Figure 4. Interaction between the N-terminal region of WRN and the 9.1.1 complex via RAD1.

(A) The N-terminal region of WRN interacts with RAD1. GST-tagged peptides corresponding to the N-, H-, and C- regions of the WRN protein were purified from E. coli and incubated with 5μg of HeLa nuclear extracts (NE). After separation on SDS-PAGE, the presence of RAD9 and RAD1 in the pull-down material was assessed by immunoblotting using the corresponding antibodies. The 5% of total NE was loaded as input. Coomassie Blue (CB) staining was used to show the equal input of the GST-tagged WRN fragments. (B) Far western analysis of the WRN interaction with the 9.1.1 complex. Recombinant 9.1.1 complex was separated using SDS-PAGE and blotted onto nitrocellulose membrane. Ponceau staining shows equal loading and transfer between the lanes (left panel). Single lanes were incubated with: no probe (lane 1), purified Flag-14-3-3 (lane 2) or purified Flag-WRN (lane 3) and subjected to immunoblotting using anti-Flag antibody to detect association of WRN to 9.1.1 complex (right panel). (C) Schematic representation of the N-terminal sub-fragments of WRN used to map the 9.1.1 interaction site. (D) Association between the different N-terminal sub-fragments of WRN and RAD1. GST-tagged peptides corresponding to the five different N-terminal sub-fragments were purified from E. coli and incubated with in-vitro-translated (IVT) 35S-labelled RAD1. After separation on SDS-PAGE and blotting, the presence of RAD1 in the pull-down material was assessed by autoradiography. Ponceau red staining of the blot shows the amount of N-terminal sub-fragments used in the pull-down analysis. Incubation of IVT 35S-labelled RAD1 with GST or beads alone was used as a control. Boxes indicate the identity and position of each fragment. (E) GST-tagged peptides corresponding to the five different N-terminal sub-fragments were purified from E. coli and incubated with 2μg of HeLa nuclear extracts. After separation on SDS-PAGE, the presence of RAD1 in the pull-down material was assessed by immunoblotting using an anti-RAD1 antibody. The 1/5 of total NE was loaded as input. Ponceau red staining of the blot shows the amount of N-terminal sub-fragments used in the pull-down analysis. Boxes indicate the identity and position of each fragment.
To define which subunit of the 9.1.1 complex interacted with WRN, Far Western blotting assay was employed. Recombinant 9.1.1 complex was separated by SDS-PAGE and transferred to PVDF prior to incubation with purified recombinant full-length WRN containing an N-terminal Flag-tag. Far Western analysis revealed that full-length WRN interacted with RAD1 subunit of the 9.1.1 complex and that interaction was specific, as RAD1 did not interact with the irrelevant Flag-tagged protein, Flag-14-3-3 (Figure 4B). Next, we generated four GST-fusion peptides containing various portions of the N-terminal region of WRN (Figure 4C) to identify the region involved in binding RAD1 by GST pull-down experiments. As Figure 4D shows, binding of in vitro-translated radiolabelled RAD1 was observed in the lane corresponding to fragment aa 80-120, suggesting that an amino-acid sequence located in this region may mediate interaction of the N-terminal portion of WRN with RAD1. To further confirm this observation, we incubated HeLa nuclear extracts with the GST-fused N-terminal fragments of WRN and assessed the binding with the 9.1.1 complex by anti-RAD1 Western blotting. Consistently with results shown in Figure 4D, Western blotting analysis revealed that RAD1 was pulled down by fragment 80-120 and the full-length N-WRN region (aa 1-550) (Figure 4E).
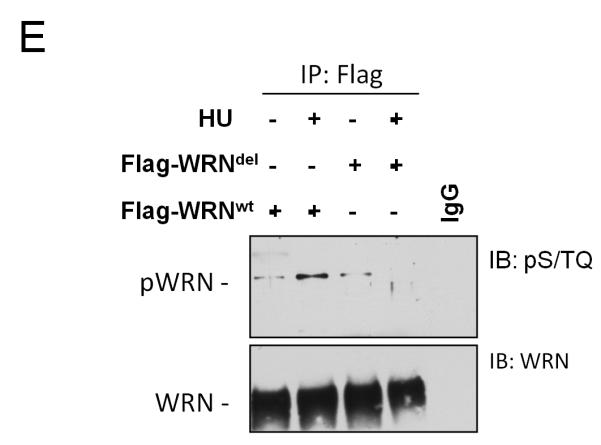
We next investigated the requirement of an intact aa 80-120 sequence for the in vivo interaction of WRN with the 9.1.1 complex and for WRN foci formation. Taking into account the structural features of the N-terminal region of WRN (Perry et al 2006), we decided to remove only a smaller region encompassing residues 112-121. Indeed, deletion of the entire aa 80-120 sequence eliminates three contiguous β strands most likely affecting folding of the protein. In contrast, removal of the eight amino acids does not affect the β strands, but eliminates a connecting loop that protrudes out of the exonuclease domain and that could be easily implicated in protein-protein interaction (Perry et al 2006). As shown in Figure 5A, the WRNdel allele was expressed as a full-length protein in WS cells at a level comparable to that of the WRNwt protein. GST pull-down experiments showed that expression of the deletion mutant form of WRN almost completely abolished interaction with RAD1 (Figure 5B) and, although it properly retained in nucleoli, failed to relocalise in foci after replication arrest (Figure 5C). Consistently, the WRNdel protein failed to co-immunoprecipitate the 9.1.1 complex (Figure 5D) and get phosphorylated at S/TQ sites after replication arrest (Figure 5E). Our previous studies reported that localization of WRN in nuclear foci after HU treatment depends on the MRE11 complex, at least in a subset of S-phase nuclei and very early after replication arrest (Franchitto and Pichierri 2004). To analyse whether the WRN/9.1.1 association was required for WRN association with MRE11 and to have insight into the cross-talk between these two events, we transfected 293T cells with the Flag-tagged wild-type WRN or WRNdel construct and analysed the association of WRN with MRE11 by CoIP. As shown in Supplementary Figure 4A, association with MRE11 is greatly reduced in the WRNdel mutant even though MRE11 foci formed normally in WS cells expressing the WRNdel protein (Supplementary Figure 5B).
Figure 5. Impaired WRN relocalisation in nuclear foci and association with 9.1.1 complex in cells expressing a WRN mutant bearing a deletion in the RAD1-bindign region.

(A) Western blotting on extracts from WS cells stably expressing the Flag-tagged wild-type WRN (WRNwt) or the 112-121 deletion mutant (WRNdel) showing levels of WRN using an anti-WRN antibody. WS cells were used as a negative control and tubulin as loading control.
(B) Analysis of the association with 9.1.1 of the WRN mutant protein with deletion in the 9.1.1-binding region. Five μg of wild-type or 112-121 deletion mutant GST-tagged N-terminal fragment of WRN (1-550) was purified from E. coli and incubated with 2μg of HeLa nuclear extracts. After release in sample buffer and separation on SDS-PAGE, the presence of RAD1 in the pull-down material was assessed by immunoblotting using an anti-RAD1 antibody. One-tenth of the released material was subjected to immunoblotting using an anti-GST antibody to visualize the amount of GST-NWRN fragments. Pull-down using uncoupled GST-binding beads was used as negative control.
(C) Analysis of WRN relocalisation to nuclear foci after replication arrest. Images show WRN nuclear distribution with or without an 8h HU treatment. The inset shows the percentage of WRN positive nuclei. Data are presented as means of three independent experiments+/− standard errors.
(D) Analysis of WRN-9.1.1 association in cells expressing WRNdel. 293T cells transiently expressing the Flag-tagged WRNwt or the Flag-tagged WRNdel protein were treated with 2mM HU for 8h prior to lysis and immunoprecipitation using anti-Flag antibody. The presence of RAD9 and RAD1 were assessed by immunoblotting (IB) using the indicated antibodies. Immunoprecipitation using normal IgG was used as a negative control. Inputs contained 15% of the total lysates used for immunoprecipitation. A fraction of the lysate (1/50) was also analysed by immunoblotting to evaluate the amount of the wild-type and mutant form of WRN expressed in 293T cells. RAD9 immunoblotting was used to confirm the presence of RAD9 in the lysates and as loading control.
(E) Analysis of WRN phosphorylation at S/TQ sites in the 112-121 deletion WRN mutant after HU treatment. Cells expressing the wild-type and the WRNdel mutant were treated with 2mM HU for 6h prior to lysis and immunoprecipitation using an anti-Flag antibody. WRN phosphorylation was evaluated by immunoblotting in the WRN immunoprecipitates with an anti-pST/Q antibody (IB: pS/TQ). The total amount of the immunoprecipitated WRN protein was determined by anti-WRN immunobloting (IB: WRN). Immunoprecipitation using normal mouse IgG (IgG) was used as a negative control.
We concluded that N-terminal region of WRN binds the RAD1 subunit of the 9.1.1 complex and that this interaction is functionally related to the ability of WRN to relocalise in nuclear foci upon replication arrest.
WRN and the 9.1.1 complex act in a common pathway to avoid DNA damage and ensure viability after replication fork stalling
After replication arrest, WRN-deficient cells accumulate DSBs (Franchitto and Pichierri 2004) and similarly to RAD9- and HUS1-deficient cells show enhanced chromosome fragmentation (Hopkins et al 2004, Weiss et al 2000), thus we asked whether WRN and the 9.1.1 complex interaction could be involved in preventing DSB formation at stalled replication forks. We used γ-H2AX-immunofluorescence to evaluate the level of DSBs induced by HU treatment in WS cells stably-transfected or not with the wild-type WRN (Pirzio et al 2008), and in which RAD9 was down-regulated by RNAi. Our analysis showed that the percentage of γ-H2AX-positive nuclei observed in WS cells after HU was comparable to that seen in RAD9-depleted cells (Figure 6A). In the absence of both WRN and RAD9, we were not able to detect any further increase of γ-H2AX-positive nuclei, even though the number of nuclei with high fluorescence enhanced at 6h after treatment in WS cells transfected with RAD9 siRNAs (Figure 6A and B). Consistent with these results, analysis of DSBs by neutral comet assay indicated that the level of DSBs formed following HU treatment in WSWRN or WS cells transfected with RAD9 siRNAs is similar (Supplementary Figure 6). To verify the existence of a functional cross-talk between WRN and the 9.1.1 complex in response to replication fork arrest, cell viability was assessed in WRN-proficient or deficient cells either transfected with Ctrl or RAD9 siRNAs. We used a short-term assay to overcome the use of transient gene knock-down by RNAi. As shown in Figure 6C, the percentage of dead cells in WRN- or RAD9-deficient cells was similar, whereas cells in which both WRN and RAD9 were absent resulted slightly more sensitive to HU treatment, even though no additive effect was observed. A distinct, yet important cell viability pathway under replication perturbation, depends on the FANC proteins (Pichierri et al 2004). In striking contrast to what observed in the WRN/RAD9 co-depleted cells, WRN RNAi in FANCD2-deficient cells (FA-D2) almost doubled the number of dead cells compared to what observed in their wild-type counterpart (FA-D2+FANCD2) after WRN RNAi (Figure 6D).
Figure 6. Enhanced levels of DNA damage in WS cells in which RAD9 function was abrogated.


(A) Analysis of γ-H2AX foci in WSWRN (wild-type) or WS in which RAD9 function was depleted. Cells were transfected with siRNAs directed against GFP (siCtrl) or RAD9 (siRAD9) and 48h afterwards were exposed for 3h or 6h to HU prior to immunofluorescence with anti- γ-H2AX antibody. In the panel, representative images from the untreated or HU-treated cells are shown. Graphs show quantification of the nuclei presenting γ-H2AX focal staining after HU treatment in WSWRN cells (left graph) or WS cells (right graph). (B) The graph shows the γ-H2AX fluorescence intensity. The experiment was carried out as described in (A). (C) Evaluation of cell viability by LIVE/DEAD assay. WS and WSWRN cells were transfected with siRNAs directed against GFP (siCtrl) or RAD9 (siRAD9) and treated with HU for 24h at the indicated doses. Cell viability was evaluated as described in “Materials and Methods”. Data are presented as percent of dead cells. (D) FA-D2 and FA-D2 cells complemented with the FANCD2 protein (FA-D2+FANCD2) were transfected with siRNAs directed against GFP (siCtrl) or WRN (siWRN) and treated with HU for 24h at the indicated doses. Cell viability was evaluated as described in “Materials and Methods”. Data are presented as percent of dead cells.
Data are presented as means of three independent experiments. Error bars represent standard errors.
These results suggest that WRN and the 9.1.1 complex work in a common pathway deputed to prevent the occurrence of DSBs and cell death after replication fork arrest.
WRN and the 9.1.1 complex act in a common pathway to regulate fragile site stability
Our data indicate that WRN and the 9.1.1 complex operate in a common pathway to prevent DSB formation following replication fork arrest. Since in human cells there are genomic loci where fork stalling occurs frequently and spontaneously because of the propensity of these regions to adopt secondary structures (Glover et al 2005, Pirzio et al 2008, Zhu and Weiss 2007), we examined the role of WRN and RAD9 in maintaining stability of common fragile sites. The sensitivity to aphidicolin was initially evaluated in wild-type cells in which WRN, RAD9 or both were depleted by RNAi. Cells were treated or not with aphidicolin and 24 h later metaphase chromosomes were collected and scored for total gaps and breaks. The analysis showed that cells lacking WRN or RAD9 resulted extremely sensitive to aphidicolin treatment and that the knock-down of WRN or RAD9 was responsible for chromosome instability (Figure 7A). However, compound WRN and RAD9 deficiency did not give rise to an additive chromosomal breakage phenotype (Figure 7A). Of note, in the absence of aphidicolin the double WRN/RAD9 knock-down displayed an induction of chromosomal breakage similar to that observed in each single deficiency (Figure 7A). To assess whether chromosomal damage detected in WRN- or RAD9-depleted cells took place at specific DNA regions, the expression of the most frequent human common fragile sites FRA3B, FRA7H and FRA16D was examined by FISH. In comparison to wild-type cells, knock-down of WRN or RAD9 led to a similar increase in gaps and breaks occurring at fragile sites, irrespective of the presence of aphidicolin (Table 1). Moreover, FISH analysis revealed that the level of fragile site induction observed in double WRN/RAD9 knock-down cells was not significantly higher than that observed in each single deficiency (Table 1). To demonstrate that interaction of WRN with the 9.1.1 complex is indeed essential to the WRN-dependent function in the maintenance of genome stability, we analysed formation of gaps and breaks in chromosomes of WS cells transfected with the WRNdel mutant that cannot efficiently interact with the 9.1.1 complex (see Figure 5). As shown in Figure 7B, transfection of the wild-type WRN protein reduced the number of gaps and breaks detectable after aphidicolin treatment in WS cells, whereas the WRNdel mutant did not revert the chromosome instability associated to WRN deficiency.
Figure 7. Evaluation of chromosomal damage in response to aphidicolin in WRN- and RAD9-depleted cells.

Average overall chromosome gaps and breaks per cell in WSWRN (wild-type) cells in which WRN, RAD9 or WRN/RAD9 were down-regulated by RNAi. Cells were treated with different doses of aphidicolin (Aph) and harvested 24h later. Representative Giemsa-stained metaphases of cells treated or not with 0.2μM aphidicolin are reported. Insets show an enlarged portion of the metaphases for a better evaluation of chromosomal gaps or breaks.
(B) Average overall chromosome gaps and breaks per cell in WS cells stably transfected with the wild-type form of WRN (WRNwt) or the WRNdel mutant (WRNdel). Cells were treated with different doses of aphidicolin (Aph) and harvested 24h later.
Data are presented as means of three independent experiments. Error bars represent standard error.
| Cell line | Treatment1 | Mean gaps and breaks per Cell |
FRA3B loci with a break (%) |
Total breaks attributable to FRA3B (%) |
FRA16D loci with a break (%) |
Total breaks attributable to FRA16D (%) |
FRA7H loci with a break (%) |
Total breaks attributable to FRA7H (%) |
|---|---|---|---|---|---|---|---|---|
| Wild-type | − APH | 0.5 | 0 | 0 | 0 | 0 | 0 | 0 |
| + APH | 0.8 | 2 | 5 | 1 | 2.5 | 1 | 2.5 | |
| siWRN | − APH | 1.8 | 15 | 18 | 6 | 6.7 | 9 | 10 |
| + APH | 2.4 | 35* | 27 | 16* | 8.2 | 17* | 14 | |
| siRAD9 | − APH | 2.1 | 13 | 16 | 9 | 7.6 | 10 | 9.5 |
| + APH | 2.8 | 32* | 22 | 19* | 7.8 | 21* | 15 | |
| siWRNRAD9 | − APH | 2.4 | 22 | 19 | 10 | 8.8 | 11 | 9.2 |
| + APH | 3 | 31* | 28 | 21* | 10 | 22* | 14 |
Notes: 24h 0,05μM aphidicolin;
p<0.1 (Student’s t-test)
These results confirm that WRN and the 9.1.1 complex act in a common pathway for maintaining the integrity of genomic regions where replication forks naturally stall.
DISCUSSION
The WRN protein is one of the most important factors contributing to the maintenance of genome stability during the S-phase. Although several pieces of evidence indicate that WRN may function facilitating the recovery of stalled or collapsed replication forks (Sidorova 2008), little is known about how WRN is regulated by the main genome surveillance pathway acting during DNA replication: the ATR-dependent checkpoint (Cimprich and Cortez 2008). In this study, we describe a novel functional correlation between WRN and the 9.1.1 complex, which is an essential component for ATR-dependent checkpoint signalling activation, specifically stimulated in response to replication perturbation and correlated with maintenance of chromosome integrity at stalled forks.
It is well known that WRN relocalises to sites of stalled replication forks (Constantinou et al 2000, Franchitto and Pichierri 2004) and that contains three different DNA binding regions conferring ability to bind to various replication intermediates in vitro (Shen et al 1998, von Kobbe et al 2003). However, how WRN delocalises from nucleoli and is recruited to stalled forks have not been fully elucidated yet. Our results indicate that recruitment of WRN to nuclear foci following replication fork arrest completely depends on the presence of a functional 9.1.1 complex, suggesting that in vivo association of WRN with its substrates is mediated by the replication checkpoint. Although the 9.1.1 complex participates in the response to various types of DNA damage (Bao et al 2004, Roos-Mattjus et al 2003), RAD9 is dispensable for WRN assembly in nuclear foci after DSBs or base damage, but it is critical for WRN relocalisation following replication arrest. However, we cannot rule out that 9.1.1-dependent WRN recruitment in nuclear foci after replication inhibition reflects the action of WRN at sites where replication forks collapse and DSBs are formed.
We previously reported that a functional MRE11 complex is required for correct formation of WRN foci (Franchitto and Pichierri 2004). Our previous and present data are not, however, contradictory. Indeed, loss of a functional MRE11 complex prevents formation of WRN foci only in a subset of S-phase cells (Franchitto and Pichierri 2004), whereas RAD9 down-regulation completely prevents localization of WRN in nuclear foci. Thus, WRN-MRE11 association probably takes place once WRN is recruited to damaged forks by the 9.1.1-dependent mechanism, as evidenced by the reduced ability of the WRNdel mutant to immunoprecipitate MRE11 and by its proficiency to form MRE11 foci after replication arrest. From this point of view, WRN and MRE11 could co-operate only at a subset of forks, as suggested by the requirement of MRE11 at spontaneously-collapsed forks (Schlacher et al 2011), and MRE11-dependent WRN foci formation (Franchitto and Pichierri 2004) may reflect the action on substrates processed by MRE11. Similarly to MRE11 deficiency, also ATR depletion reduces, without completely abolishing, the number of nuclei with WRN foci (Ammazzalorso et al 2010). Overall, our previous and current observations are consistent with the 9.1.1 complex being upstream in the events that regulate the formation of WRN foci, while the interaction with MRE11 and ATR-dependent phosphorylation modulate their assembly. Since WRN has been implicated in several genome maintenance pathways (Cheng et al 2007), the specific requirement of 9.1.1 complex for the response to replication arrest represents the first proof that different WRN functions may be regulated by defined protein-protein interactions. For instance, DNA damage-induced relocalisation of WRN could be specifically regulated by acetylation, c-Abl-dependent phosphorylation (Blander et al 2002, Cheng et al 2003, Muftuoglu et al 2008a) or by a yet-to-be identified interaction. It is worth noting that DNA damage and viability in the double knock-down cells are slightly enhanced compared to that observed in each single deficiency. This is not surprising since the 9.1.1 complex may be involved in regulating other functions of the replication checkpoint and not just in the WRN-dependent prevention of DSBs accumulation at stalled forks, a situation similar to that recently reported in the cross-talk between WRN and ATR (Ammazzalorso et al 2010). The replication checkpoint coordinates the cellular response to replication arrest and its complete activation involves two independent events: loading of the ATR/ATRIP heterodimer and of the 9.1.1 complex onto chromatin (Cimprich and Cortez 2008). WRN is phosphorylated in an ATR-dependent manner after HU or CPT treatment (Ammazzalorso et al 2010, Pichierri et al 2003) and down-regulation of the 9.1.1 complex impairs phosphorylation of WRN at S/TQ sites. It has been reported that one of the most important functions of the 9.1.1 complex is the recruitment of TopBP1 through its association with RAD9 (Delacroix et al 2007, Lee et al 2007, Mordes et al 2008). Indeed, TopBP1-9.1.1 interaction is required for proper phosphorylation by ATR of downstream checkpoint factors (Delacroix et al 2007, Kumagai et al 2006, Liu et al 2006, Mordes et al 2008). Our results indicate that depletion of TopBP1 by RNAi abrogates WRN phosphorylation but does not affect relocalisation to foci, suggesting that defective phosphorylation of WRN observed in RAD9-depleted cells could correlate with loss of TopBP1 recruitment and mediator function. Thus, association of WRN with 9.1.1 is essential for its recruitment to stalled forks, but TopBP1 is needed for subsequent phosphorylation by ATR. Interestingly, the interaction of the 9.1.1 complex with WRN is mediated by the RAD1 subunit, while RAD9 interacts directly with TopBP1 (Lee et al 2007), suggesting that WRN and TopBP1 may actually associate with the 9.1.1 complex at the same time. Our data also indicate that relocalisation to nuclear foci of WRN precedes modifications by ATR. This is consistent with previous data suggesting that WRN foci formation is not abrogated by loss of ATR function (Pichierri et al 2003) and with our observations showing that ATR regulates the strength of the WRN association with chromatin (Ammazzalorso et al 2010). To the best of our knowledge, WRN represents the first example of human RecQ helicase whose subnuclear dynamics is finely tuned by the replication checkpoint. Consistently with a functional role of 9.1.1 complex in bringing up WRN to stalled forks, WRN and the 9.1.1 complex co-immunoprecipitate and such association is stimulated under replication stress conditions. It is worth noting that the sequences of the N-terminal region of WRN involved in the interaction with the RAD1 subunit of 9.1.1 are located in proximity to the reported PCNA binding site (Rodriguez-Lopez et al 2003). The 9.1.1 complex is structurally similar to the PCNA homotrimer (Parrilla-Castellar et al 2004) and RAD1 is the 9.1.1 subunit that most resembles PCNA from the structural point of view (Dore et al 2009, Xu et al 2009). Thus, it is tempting to speculate that PCNA or 9.1.1 complex may associate with WRN in a mutually-exclusive manner. Further experiments will be required to directly address this point and analyse if PCNA may be required to support WRN foci formation in other settings. The physical and functional interaction between WRN and the 9.1.1 complex suggests that they act in a common pathway. Indeed, both WRN-deficient cells and the 9.1.1 complex mutants show similar phenotypes under replication stress conditions, including enhanced chromosome breakage and expression of common fragile sites (Hopkins et al 2004, Pirzio et al 2008, Scappaticci et al 1982, Weiss et al 2000, Zhu and Weiss 2007). We show that concomitant depletion of WRN and RAD9 does not cause any additive accumulation of DSBs or cell death after replication arrest. Similarly, WRN- and RAD9-depleted cells, exhibit a comparable instability at common fragile sites, which is not increased abrogating 9.1.1 complex function in a WRN-deficient background. This latter evidence is noteworthy because bridges together ATR, 9.1.1 complex and WRN in a common pathway involved in preventing breakage at genomic regions representing natural replication fork stalling sites and considered as hotspots of instability in precancerous lesions (Glover et al 2005, Halazonetis et al 2008). It is conceivable that, even though the 9.1.1 complex is required for several of the replication checkpoint activities, the WS-like phenotype observed in the 9.1.1 mutants under conditions of replication stress could derive from the inability of the cells to recruit WRN, thus resulting in mishandling of stalled forks.
Altogether, our results suggest a novel functional link between WRN and the key checkpoint factor 9.1.1 complex in response to replication arrest. They also demonstrate that WRN and 9.1.1 collaborate in a common pathway to prevent accumulation of DSBs and genome stability specifically at naturally-occurring hotspots of replication fork stalling, thus contributing to improve our knowledge of how the replication stress response is regulated in human cells.
MATERIALS AND METHODS
Cell cultures
HeLa and 293T cells were from American Type Culture Collection (ATCC) and the SV40-transformed WS fibroblast cell line (AG11395) was obtained from Coriell Cell Repositories (Camden, NJ, USA). AG11395 fibroblasts retrovirally-transduced with full length cDNA encoding wild-type WRN (WSWRN) were described previously (Pirzio et al 2008). FANCD2-mutant (PD20) SV40-transformed fibroblast and their FACND2-complemented counterpart (PD20+FANCD2) are from the Oregon Health and Science University (OHSU) Fanconi Anemia Cell Repository.
All the cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) containing 10% FBS (Boehringer Mannheim) and incubated at 37 °C in a humidified 5% CO2 atmosphere.
RNA interference and transfection
RAD9, TopBP1 and WRN expression was knocked down by transfection with SMARTpool siRNAs (Dharmacon) directed against proteins of interest at the final concentration of 10nM. Transfection was performed using Interferin (Polyplus) according to the manufacturer’s instructions. As a control, a siRNA duplex directed against GFP was used.
Immunoprecipitation and Western blotting
For immunoprecipitation cells were lysed as described (Franchitto and Pichierri 2004), in the presence of 10U of benzonase, and 1mg of total cell extract incubated ON at + 4 °C with either anti-WRN (Santa Cruz biotechnologies) or anti-RAD9 (Calbiochem) antibody coupled to M280 tosylactivated Dynabeads (Invitrogen). For the analysis of the WRNdel mutant, 20×107 293T cells transiently transfected with the WRNwt or WRNdel expression plasmids were lysed as described above and incubated ON at + 4 °C with anti-Flag (Sigma-Aldrich) antibody coupled to M280 tosylactivated Dynabeads (Invitrogen). For the analysis of whole protein content, cells were washed with PBS and lysed in standard RIPA buffer. Twenty micrograms of total protein were resolved by SDS-PAGE and transferred to nitrocellulose (PROTRAN, Schleicher & Schuell). For Western blotting analysis the following antibodies were used: anti-RAD9 (Calbiochem), anti-RAD1 (Santa Cruz Biotechnology), anti-TopBP1 (Bethyl Laboratories), anti-WRN (Santa Cruz Biotechnology), anti-pST/Q (Cell Signaling), anti-PCNA (Santa Cruz Biotechnology), anti-Flag and anti-β-tubulin (Sigma-Aldrich). Incubation with antibodies was performed for 2h at RT. Proteins were visualised using ECL plus according to the manufacturer’s instructions (Amersham) and normalised to PCNA or tubulin level in each extract.
Generation of WRN fragments and GST pull-down
DNA sequences corresponding to amino acids 1 to 550 (N-WRN), 506 to 940 (H-WRN), and 949 to 1432 (C-WRN) of WRN protein were amplified by PCR from the pFLAGWRN2 plasmid. Similarly, DNA sequences corresponding to the different N-WRN sub-fragments were generated by PCR using specific primer pairs. The PCR products were subsequently purified and sub-cloned into pGEX4T-1 for subsequent expression in bacteria as GST fusion. The resulting vectors were subjected to sequencing to ensure that no mutations were introduced into the WRN sequence and were used for transforming BL21 cells (Stratagene). Expression of GST and the three GST fusion proteins were induced upon addition of 1mM IPTG for 2h at 37 °C. GST, GST-N-WRN, GST-H-WRN, GST-C-WRN and the different GST-N-WRN sub-fragments were affinity-purified using GSH-magnetic beads (Promega). Pull-down was carried out incubating 2μg of each fragment with 5μg or 2μg of HeLa NE for 16h at +4 °C in Tris/HCl supplemented with 100mM NaCl, 0,4% Triton X-100 and protease inhibitors. As negative controls, NE were incubated also with GST and beads alone. Analysis of RAD1 binding to the GST-N-WRN sub-fragments was carried out using 35S-labelled RAD1 prepared by TnT reaction (Promega). Two single 50μl TnT reactions were pooled and then diluted into incubation buffer supplemented with 1mM DTT and 1mM MgCl2. Three hundred microliter of the diluted probe was incubated with the GST-N-WRN fragments, GST or empty beads for 4h at +4 °C. After release, samples were separated by SDS-PAGE prior to Coomassie staining and Phosphorimaging.
Far Western
Recombinant 9.1.1 complex (1μg/μl, Alexis-Biochemicals) was subjected to SDS-PAGE and transferred onto a PVDF membrane. Membrane was stained with Ponceau Red solution (Sigma) and subsequently washed in denaturation buffer (6M Guanidine/HCl) at +4 °C and then in denaturation buffer diluted serially 1:2 with PBS, 1mM DTT, 0.2% NFDM at +4 °C. Each lane was then incubated with recombinant purified Flag-WRN (kindly provided by Dr. Lucio Comai, University of Southern California, Los Angeles, USA), recombinant Flag-14-3-3 protein (kindly provided by Dr. Marco Lalle, Istituto Superiore di Sanità, Rome, Italy), or without probe. PVDF strips were then incubated with mouse anti-FLAG (Sigma) and secondary horseradish peroxidase-conjugated (HRP)-antibodies. Detection was performed with ECL (Amersham).
Immunofluorescence
After appropriate treatment cells grown on 22×22mm glass coverslips were harvested at the indicated time and processed for immunofluorescence after in situ fractionation as described in (Franchitto and Pichierri 2004, Franchitto et al 2008). For each time point, at least 200 nuclei were examined and foci were scored at 60× magnification. Only nuclei showing more than five bright foci were considered as a positive.
LIVE/DEAD staining
HeLa cells were transfected with siRNAs directed against GFP (control), WRN, RAD9 or WRN/RAD9 and treated for 18h with 2mM HU. We used a short-term assay to overcome the use of transient gene knock-down by RNAi. Viability was evaluated by the LIVE/DEAD assay (Sigma-Aldrich) according to the manufacturer’s instructions. Cell number was counted in randomly chosen fields and expressed as percent of dead cells (number of red nuclear stained cells/total cell number). For each time point, at least 200 cells were counted.
Fragile site induction and Fluorescence in situ hybridization
Fragile sites were induced treating WSWRN (wild-type) or WS cells, in which RAD9 function was abrogated by RNAi, with aphidicolin (0.2μM) for 24h. Metaphase cells were collected and prepared as previously reported (Pirzio et al 2008). For each condition of treatment the number of breaks and gaps was observed on Giemsa-stained metaphases.
FISH analyses were performed as described after treatment with 0.05μM aphidicolin (Pirzio et al 2008, Wilke et al 1996) using BACs (kindly provided by Dr. Daniela Toniolo Dibit-HSR, Milano, Italy and Dr. Mariano Rocchi University of Bari, Italy), mapping to fragile site regions as probes.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Drs L. Comai and M. Lalle for providing recombinant proteins and Drs D. Toniolo and M. Rocchi for providing BACs for FISH analysis. This work was supported by Fondazione Telethon to P.P. (grant n. GGP04094) and, in part, by grants from Associazione Italiana per la Ricerca sul Cancro (AIRC) to P.P. (grant n. IG9294) and A.F. (grant n. IG4400)
Footnotes
CONFLICT OF INTEREST SECTION The authors declare no conflict of interest.
REFERENCES
- Ammazzalorso F, Pirzio LM, Bignami M, Franchitto A, Pichierri P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010;29:3156–3169. doi: 10.1038/emboj.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao S, Lu T, Wang X, Zheng H, Wang LE, Wei Q, et al. Disruption of the Rad9/Rad1/Hus1 (9-1-1) complex leads to checkpoint signaling and replication defects. Oncogene. 2004;23:5586–5593. doi: 10.1038/sj.onc.1207753. [DOI] [PubMed] [Google Scholar]
- Blander G, Zalle N, Daniely Y, Taplick J, Gray MD, Oren M. DNA damage-induced translocation of the Werner helicase is regulated by acetylation. J Biol Chem. 2002;277:50934–50940. doi: 10.1074/jbc.M210479200. [DOI] [PubMed] [Google Scholar]
- Brosh RM, Jr., Waheed J, Sommers JA. Biochemical characterization of the DNA substrate specificity of Werner syndrome helicase. J Biol Chem. 2002;277:23236–23245. doi: 10.1074/jbc.M111446200. [DOI] [PubMed] [Google Scholar]
- Cheng WH, von Kobbe C, Opresko PL, Fields KM, Ren J, Kufe D, et al. Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Mol Cell Biol. 2003;23:6385–6395. doi: 10.1128/MCB.23.18.6385-6395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng WH, von Kobbe C, Opresko PL, Arthur LM, Komatsu K, Seidman MM, et al. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J Biol Chem. 2004;279:21169–21176. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- Cheng WH, Muftuoglu M, Bohr VA. Werner syndrome protein: functions in the response to DNA damage and replication stress in S-phase. Exp Gerontol. 2007;42:871–878. doi: 10.1016/j.exger.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, Hickson ID, et al. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delacroix S, Wagner JM, Kobayashi M, Yamamoto K, Karnitz LM. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007;21:1472–1477. doi: 10.1101/gad.1547007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore AS, Kilkenny ML, Rzechorzek NJ, Pearl LH. Crystal structure of the rad9-rad1-hus1 DNA damage checkpoint complex--implications for clamp loading and regulation. Mol Cell. 2009;34:735–745. doi: 10.1016/j.molcel.2009.04.027. [DOI] [PubMed] [Google Scholar]
- Franchitto A, Pichierri P. Werner syndrome protein and the MRE11 complex are involved in a common pathway of replication fork recovery. Cell Cycle. 2004;3:1331–1339. doi: 10.4161/cc.3.10.1185. [DOI] [PubMed] [Google Scholar]
- Franchitto A, Pirzio LM, Prosperi E, Sapora O, Bignami M, Pichierri P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J Cell Biol. 2008;183:241–252. doi: 10.1083/jcb.200803173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya K, Poitelea M, Guo L, Caspari T, Carr AM. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 2004;18:1154–1164. doi: 10.1101/gad.291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover TW, Arlt MF, Casper AM, Durkin SG. Mechanisms of common fragile site instability. Hum Mol Genet. 2005;14 Spec No. 2:R197–205. doi: 10.1093/hmg/ddi265. [DOI] [PubMed] [Google Scholar]
- Guan X, Bai H, Shi G, Theriot CA, Hazra TK, Mitra S, et al. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007;35:2463–2472. doi: 10.1093/nar/gkm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Harrigan JA, Wilson DM, 3rd, Prasad R, Opresko PL, Beck G, May A, et al. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006;34:745–754. doi: 10.1093/nar/gkj475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins KM, Auerbach W, Wang XY, Hande MP, Hang H, Wolgemuth DJ, et al. Deletion of mouse rad9 causes abnormal cellular responses to DNA damage, genomic instability, and embryonic lethality. Mol Cell Biol. 2004;24:7235–7248. doi: 10.1128/MCB.24.16.7235-7248.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai M, Wang TS. Checkpoint activation regulates mutagenic translesion synthesis. Genes Dev. 2003;17:64–76. doi: 10.1101/gad.1043203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell. 2006;124:943–955. doi: 10.1016/j.cell.2005.12.041. [DOI] [PubMed] [Google Scholar]
- Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, Oshima J, et al. Accumulation of Werner protein at DNA double-strand breaks in human cells. J Cell Sci. 2005;118:4153–4162. doi: 10.1242/jcs.02544. [DOI] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J Biol Chem. 2007;282:28036–28044. doi: 10.1074/jbc.M704635200. [DOI] [PubMed] [Google Scholar]
- Liu S, Bekker-Jensen S, Mailand N, Lukas C, Bartek J, Lukas J. Claspin operates downstream of TopBP1 to direct ATR signaling towards Chk1 activation. Mol Cell Biol. 2006;26:6056–6064. doi: 10.1128/MCB.00492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machwe A, Xiao L, Groden J, Orren DK. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–13946. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- Martin GM, Oshima J. Lessons from human progeroid syndromes. Nature. 2000;408:263–266. doi: 10.1038/35041705. [DOI] [PubMed] [Google Scholar]
- Mordes DA, Glick GG, Zhao R, Cortez D. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 2008;22:1478–1489. doi: 10.1101/gad.1666208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M, Kusumoto R, Speina E, Beck G, Cheng WH, Bohr VA. Acetylation regulates WRN catalytic activities and affects base excision DNA repair. PLoS ONE. 2008a;3:e1918. doi: 10.1371/journal.pone.0001918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet. 2008b;124:369–377. doi: 10.1007/s00439-008-0562-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrilla-Castellar ER, Arlander SJ, Karnitz L. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amst) 2004;3:1009–1014. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, Han S, et al. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol. 2006;13:414–422. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- Pichierri P, Rosselli F, Franchitto A. Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene. 2003;22:1491–1500. doi: 10.1038/sj.onc.1206169. [DOI] [PubMed] [Google Scholar]
- Pichierri P, Franchitto A, Rosselli F. BLM and the FANC proteins collaborate in a common pathway in response to stalled replication forks. EMBO J. 2004;23:3154–3163. doi: 10.1038/sj.emboj.7600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichierri P. Interplay between wrn and the checkpoint in s-phase. Ital J Biochem. 2007;56:130–140. [PubMed] [Google Scholar]
- Pirzio LM, Pichierri P, Bignami M, Franchitto A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol. 2008;180:305–314. doi: 10.1083/jcb.200705126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Lopez AM, Jackson DA, Nehlin JO, Iborra F, Warren AV, Cox LS. Characterisation of the interaction between WRN, the helicase/exonuclease defective in progeroid Werner’s syndrome, and an essential replication factor, PCNA. Mech Ageing Dev. 2003;124:167–174. doi: 10.1016/s0047-6374(02)00131-8. [DOI] [PubMed] [Google Scholar]
- Roos-Mattjus P, Hopkins KM, Oestreich AJ, Vroman BT, Johnson KL, Naylor S, et al. Phosphorylation of human Rad9 is required for genotoxin-activated checkpoint signaling. J Biol Chem. 2003;278:24428–24437. doi: 10.1074/jbc.M301544200. [DOI] [PubMed] [Google Scholar]
- Scappaticci S, Cerimele D, Fraccaro M. Clonal structural chromosomal rearrangements in primary fibroblast cultures and in lymphocytes of patients with Werner’s Syndrome. Hum Genet. 1982;62:16–24. doi: 10.1007/BF00295599. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-Strand Break Repair-Independent Role for BRCA2 in Blocking Stalled Replication Fork Degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JC, Gray MD, Oshima J, Loeb LA. Characterization of Werner syndrome protein DNA helicase activity: directionality, substrate dependence and stimulation by replication protein A. Nucleic Acids Res. 1998;26:2879–2885. doi: 10.1093/nar/26.12.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova JM. Roles of the Werner syndrome RecQ helicase in DNA replication. DNA Repair (Amst) 2008;7:1776–1786. doi: 10.1016/j.dnarep.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toueille M, El-Andaloussi N, Frouin I, Freire R, Funk D, Shevelev I, et al. The human Rad9/Rad1/Hus1 damage sensor clamp interacts with DNA polymerase beta and increases its DNA substrate utilisation efficiency: implications for DNA repair. Nucleic Acids Res. 2004;32:3316–3324. doi: 10.1093/nar/gkh652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C, Thoma NH, Czyzewski BK, Pavletich NP, Bohr VA. Werner syndrome protein contains three structure-specific DNA binding domains. J Biol Chem. 2003;278:52997–53006. doi: 10.1074/jbc.M308338200. [DOI] [PubMed] [Google Scholar]
- Weiss RS, Enoch T, Leder P. Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes Dev. 2000;14:1886–1898. [PMC free article] [PubMed] [Google Scholar]
- Wilke CM, Hall BK, Hoge A, Paradee W, Smith DI, Glover TW. FRA3B extends over a broad region and contains a spontaneous HPV16 integration site: direct evidence for the coincidence of viral integration sites and fragile sites. Hum Mol Genet. 1996;5:187–195. doi: 10.1093/hmg/5.2.187. [DOI] [PubMed] [Google Scholar]
- Xu M, Bai L, Gong Y, Xie W, Hang H, Jiang T. Structure and functional implications of the human rad9-hus1-rad1 cell cycle checkpoint complex. J Biol Chem. 2009;284:20457–20461. doi: 10.1074/jbc.C109.022384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Weiss RS. Increased common fragile site expression, cell proliferation defects, and apoptosis following conditional inactivation of mouse Hus1 in primary cultured cells. Mol Biol Cell. 2007;18:1044–1055. doi: 10.1091/mbc.E06-10-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Cortez D, Elledge SJ. Regulation of ATR substrate selection by Rad17-dependent loading of Rad9 complexes onto chromatin. Genes Dev. 2002;16:198–208. doi: 10.1101/gad.950302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.