

Abstract
A series of novel indole-based analogues were prepared and their affinities for sigma receptors were determined using in vitro radioligand binding assays. The results of this study identified several compounds with nanomolar sigma-2 affinity and significant selectivity over sigma-1 receptors. In particular, 2-(4-(3-(4-fluorophenyl)indol-1-yl)butyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (9f) was found to display high affinity at sigma-2 receptors with good selectivity (σ-1/σ-2 = 395). The pharmacological binding profile for this compound was established with other relevant nonsigma sites.
Keywords: Sigma-2 receptors, indole
1. Introduction
Sigma receptors are classified into two subtypes denoted sigma-1 and sigma-2. [1] The sigma-1 receptor has been purified and cloned in several species and is well characterized at the functional and structural level [2,3]. It is expressed in the central nervous system and is also widely distributed in peripheral organs and tissues such as heart and spleen [4]. The sigma-2 receptor suffers from a lower degree of knowledge, in part because this protein has not yet been cloned. These receptors are associated with functions and disorders such as inflammation [5], depression [6,7], anxiety [8], Alzheimer’s disease [9], epilepsy and drug abuse [10–12]. Cancer diagnosis and treatment is also an area of great interest in current sigma receptor research. Indeed sigma receptor overexpression in malignant tissues suggest that tumors may be visualized by SPECT or PET imaging using radiolabeled sigma ligands [13–16]. The sigma-2 subtype is upregulated in proliferative cells where the density of sigma-2 receptors was found to be 10-fold higher than in quiescent cells [17]. Additional studies have shown that sigma ligands, especially sigma-2 agonists, can inhibit proliferation and induce apoptosis in tumor cells which give sigma ligands possible application as agents for the treatment of cancer [18,19]. Therefore, the finding of selective sigma-2 receptor ligands may aid in the isolation and characterization of this receptor, but also could lead to the development of new therapeutics, particularly in oncology.
Several classes of structurally diverse compounds have been shown to possess a high affinity for sigma receptors (for a review see Narayanan et al. [20]). While several selective, high-affinity sigma-1 ligands are available [21–25], potent sigma-2 receptor selective ligands are less common. Siramesine (Lu28-179, σ-1 = 17 nM, σ-2 = 0.12 nM) and 5-bromo-N-(4-(6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)butyl)-2,3-dimethoxybenzamide (σ-1 = 12,900 nM, σ-2 = 8.2 nM) are two of the most highly selective sigma-2 ligands identified to date [26,27].
In our previous work we reported the synthesis and biological evaluation of a series of benzoazoles with high, mixed affinity for sigma-1 and sigma-2 receptors [28]. Among the compounds described in this early study, 3-(4-(4-cyclohexylpiperazin-1-yl)butyl)benzo[d]thiazol-2(3H)-one (1) exhibited a subnanomolar sigma-2 affinity and a modest preference for sigma-2 versus sigma-1 with a selectivity ratio of 11 (Figure 1). The goal of the present study was to prepare novel sigma ligands with selectivity for sigma-2 and we decided to use this compound as our lead structure.
Figure 1.

Structures of 1 and its indole analogue 4a
2. Results and discussion
2.1. Chemistry
The strategy we chose involved the replacement of the benzothiazole heterocycle of 1 by the more stable and versatile indole ring (Figure 1). Several parts of the new molecule 4a were then modified to develop new indole-based sigma receptor ligands. These modifications were made at three different positions: an acetyl group was introduced at the 5 position of the indole ring, the 1-cyclohexylpiperazine (A-ring, Figure 2) was replaced by other cyclic amines (B- and C-ring) and finally different aryl groups (D-, E- and F-rings) were introduced at the 3 position of the heterocycle.
Figure 2.

R2- and R3-rings
The introduction of an acetyl group on the 5 position of the indole was motivated by previous work in our laboratory in the benzoazole series (data not shown) which hinted at this group giving better sigma-2 selectivity. Similarly, we decided to replace the piperazine group of 4a by two cyclic amines that were described in the literature as potentially sigma-2 preferring and gave good results selectivity-wise in the benzoazole series: 1-(4-fluorophenyl)piperazine (Bring) and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (C-ring) [27]. Furthermore, the decision to introduce an aryl group in the C-3 indole position was inspired by the work of Perregaard who successfully converted a 3-(aminobutyl)indole with mixed affinity for sigma receptors to the sigma-2 selective ligand siramesine by adding a 4-fluorophenyl ring in the NH position of the indole [26]. The aryl groups we chose were the unsubstituted phenyl group (D-ring), the metabolically more stable 4-fluorophenyl ring (E-ring), and the H-bond acceptor and phenyl-bioisostere furan ring (F-ring).
The preparation of compounds 4a–4f is outlined in Scheme 1. Treatment of indole (2a) or 5-acetyl-indole (2b) with 1,4-dibromobutane in the presence of potassium hydroxide in DMF gave the bromo derivatives 3a–3b. These were then coupled with 1-cyclohexylpiperazine (A-ring, Figure 2), 1-(4-fluorophenyl)piperazine (B-ring) or 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline (C-ring) in the presence of potassium carbonate in DMF to afford target compounds 4a–4f.
Scheme 1.

Synthesis of indole derivatives4a–4f. Reagents and conditions: (a) 1,4-dibromobutane, KOH, TBAI, DMF, 0°C→rt, 2 h; (b) Cyclic amine, K2CO3, DMF, 60°C, 2 h.
The synthesis of final compounds 9a–9i is described in Scheme 2. Intermediates 6a–6c were prepared with excellent yields by reaction of commercially available 3-bromo-1-(phenylsulfonyl)indole (5) with the appropriate boronic acid under Suzuki conditions [29]. Indoles 6a–6c underwent an easy and clean deprotection with magnesium and ammonium chloride in methanol to give the corresponding derivatives 7a–7c. N-alkylation of the heterocyles with 1,4-dibromobutane gave the bromo intermediates 8a–8c which were then reacted with A-, B- and C-ring to give 3-substituted-indole derivatives 9a–9i.
Scheme 2.

Synthesis of 3-substituted-indole derivatives. Reagents and conditions: (a) Arylboronic acid, K2CO3, Pd(Pφ3)4, benzene/EtOH, reflux, 16 h; (b) Mg, NH4Cl, MeOH/THF, rt, 2 h; (c) 1,4-dibromobutane, KOH, TBAI, DMF, 0°C→rt, 2 h; (d) Cyclic amine, K2CO3, DMF, 60°C, 2 h.
2.2. Pharmacology
All final compounds were tested for in vitro affinity at sigma-1 and sigma-2 receptors using well-established assay conditions [30–34]. The sigma-1 receptors were labeled with 5 nM [3H](+)-pentazocine and the sigma-2 receptors were labeled with 3 nM [3H]di-o-tolylguanidine (DTG) in the presence of 300 nM (+)-pentazocine to block sigma-1 receptors. Non-specific binding was determined in the presence of 10 μM haloperidol. Ten concentrations of each sigma compound (0.1–1000 nM) were used in the assays. The chemical structures, sigma binding affinities and selectivity ratios of the new compounds are summarized in Table 1. In addition, the affinity of 9f for monoamine transporters and several serotonin and dopamine receptors was determined. A binding profile of the compound was also prepared by NovaScreen/Caliper Life Sciences (Hanover, MD). The results of these experiments are summarized in Tables 2 and 3.
Table 1.
Sigma receptor binding affinities and selectivity ratios of 1, 4a–4f, 9a–9i and haloperidola
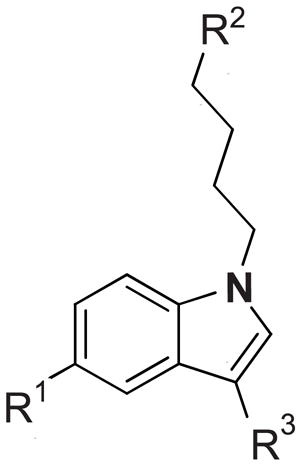 | ||||||
|---|---|---|---|---|---|---|
| Compd | R1 | R2 | R3 | σ-1 (Ki, nM) | σ-2 (Ki, nM) | σ-1/σ-2 |
| 1 | - | - | - | 4.17 ± 0.62 | 0.39 ± 0.06 | 10.7 |
| 4a | H | A-ring | H | 3.28±0.32 | 1.90±0.16 | 1.7 |
| 4b | H | B-ring | H | 71.50±1.53 | 13.32±0.61 | 5.4 |
| 4c | H | C-ring | H | 140.7±5.7 | 3.66±0.83 | 38.4 |
| 4d | CH3CO | A-ring | H | 5.33±2.12 | 2.31±0.02 | 2.3 |
| 4e | CH3CO | B-ring | H | 2.13±0.31 | 3.30±0.30 | 0.6 |
| 4f | CH3CO | C-ring | H | 14.55±1.42 | 1.42±0.10 | 10.2 |
| 9a | H | A-ring | D-ring | 92.43±1.90 | 12.30±2.34 | 7.5 |
| 9b | H | B-ring | D-ring | 222.33±22.41 | 9.96±1.19 | 22.3 |
| 9c | H | C-ring | D-ring | 713.5±163.5 | 46.29±7.18 | 15.4 |
| 9d | H | A-ring | E-ring | 90.87±12.30 | 22.55±1.10 | 4.0 |
| 9e | H | B-ring | E-ring | 1202±73.89 | 83.33±3.90 | 14.4 |
| 9f | H | C-ring | E-ring | 2948.4±374.4 | 7.45±0.71 | 395.8 |
| 9g | H | A-ring | F-ring | 34.50±4.84 | 10.07±2.01 | 3.4 |
| 9h | H | B-ring | F-ring | 279.0±40.5 | 157.0±1.6 | 1.8 |
| 9i | H | C-ring | F-ring | 613.66±38.29 | 12.02±1.15 | 51.1 |
| haloperidol | 3.35±0.80 | 80.60±14.10 | 0.042 | |||
Affinities (Ki in nM) were determined in rat brain homogenates. Sigma-1 receptors were labeled with [3H](+)-pentazocine. Sigma-2 receptors were labeled with [3H]DTG in the presence of (+)-pentazocine to block sigma-1 receptors. Nonspecific binding was determined in the presence of haloperidol. The values in this table represent the mean ± SEM from replicate assays.
Table 2.
Nonsigma protein binding affinities of compound 9fa
| Radioligand | Nonspecific binding | Ki (nM) | |
|---|---|---|---|
| Monoamine Transporters | |||
| Dopamineb | 40–80 pM [125I]RTI-55 | 5 μM mazindol | 5000 ± 1000 |
| Serotoninb | 40–80 pM [125I]RTI-55 | 5 μM imipramine | 295 ± 82 |
| Norepinephrineb | 40–80 pM [125I]RTI-55 | 5 μM mazindol | 1350 ± 110 |
| Other Receptors | |||
| Dopamine D1c | 0.18 nM [3H]SCH-23390 | 1 μM SCH-23390 | > 10000 |
| Dopamine D2d | 0.2–0.5 nM [3H]YM-09151-2 | 1 μM chlorpromazine | 870 ± 260 |
| Dopamine D3d | 0.2–0.5 nM [3H]YM-09151-2 | 1 μM chlorpromazine | 544 ± 90 |
| Serotonin 5-HT1Ae | 0.5 nM [3H]8-OH-DPAT | 1 μM dihydroerotamine | 1510 ± 410 |
| Serotonin 5-HT2Ae | 0.1 nM [125I]DOI | 10 μM 5-HT | 1270 ± 310 |
Affinities (Ki in nM) were determined using standard assays conditions. The values in this table represent the mean ± SEM from replicate assays. Values of >10000 nM signify that there was less than 30% displacement of the radioligand at that concentration.
HEK293 cells expressing hBAT, hSERT or hNERT.
LhD1 cells.
CHOp-D2 or CHOp-D3 cells.
HEK-h5-HT1A or HEK-h5-HT2A cells.
Table 3.
Summary of binding profile of 9f in 64 radioligand/enzyme assays at 10−5M and 10−7Ma
| Assay name | 50% inhibition (10−7M) | 50% inhibition (10−5M) |
|---|---|---|
| Adrenergic, α1 | No | Yes |
| Adrenergic, α2 | No | Yes |
| Adrenergic, β1 | No | Yes |
| Cannabinoid, CB1 | No | Yes |
| Cannabinoid, CB2 | No | Yes |
| Dopamine D4.2 | No | Yes |
| Glycine, Strychnine | Yes | Yes |
| Histamine, H1 | No | Yes |
| Histamine, H2 | No | Yes |
| Muscarinic, M1 | No | Yes |
| Muscarinic, M2 | No | Yes |
| Muscarinic, central | No | Yes |
| Opioid, μ | No | Yes |
| Calcium channel, type L (BZT) | No | Yes |
| Sodium, Site 2 | No | Yes |
| Neurokinin, NK2 (NKA0 | No | Yes |
Details of each assay condition can be accessed through Caliper’s web site at www.caliperls.com.
As shown in Table 1, the lead compound 1 had a high affinity for sigma-1 and sigma-2 receptors (4.17 and 0.39 nM, respectively) and a small selectivity for sigma-2 versus sigma-1 receptors (σ-1/σ-2 = 10.7). The replacement of the benzothiazolone core by an indole core resulted in no change in affinity for sigma-1 receptors and a slight decrease in affinity for sigma-2. As a result, 4a does not exhibit a preference for sigma-2 receptors, but this decrease in selectivity is not significant, while the change of the central template gives us access to a whole new class of potent sigma ligands. The next step was the replacement of the piperazine moiety with two different putative sigma-2 preferring elements (B- and C-ring). The 1-(4-fluorophenyl)piperazine compound 4b showed only a slight selectivity (σ-1/σ-2 ratio of 5) while the 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline derivative 4c exhibited an interesting selectivity of 38 along with a high affinity for the sigma-2 receptor (Ki = 3.66 nM). Introduction of an acetyl group in the C-5 position of the indole provided compounds (4d–4f) with high affinities for both sigma receptors, but no meaningful selectivity. Compounds 9a–9i were subsequently prepared to examine the influence of various aryl cycles attached in position 3 of the indole and, as depicted in Table 1, these pharmacomodulations gave mixed results. Generally most of the compounds displayed decreased affinities for both sigma receptors and a small selectivity. However 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolines 9f and 9i proved to be potent and selective sigma-2 selective ligands. In particular, compound 9f with a 4-fluorophenyl ring in the C-3 position of its core ring presented a high affinity for the sigma-2 receptor (7.45 nM) and a weak affinity of 2948 nM for the sigma-1 receptor, giving this indole one of the best sigma-1/sigma-2 selectivity ratios (σ-1/σ-2 = 395) described so far. Because many ligands for sigma receptors also bind to non-sigma sites, we decided to submit 9f to an extensive selectivity profile characterization. As can be seen from Table 2, compound 9f had essentially no affinity for dopamine and norepinephrine transporters and a low affinity for serotonin transporters. It also showed no measurable affinity for the D1 receptor and very low affinities for dopaminergic receptors D2 and D3 and serotoninergic receptors 5-HT1A and 5-HT2A. The compound was then tested by NovaScreen in 64 radioligand/enzyme assays (neurotransmitter related, steroids, ion channels, second messengers, prostaglandins, growth factors/hormones, brain/gut peptides, enzymes) at two concentrations of 10−5 M and 10−7 M. The summary of this “profiling” is presented in Table 3. A radioligand displacement of more than 50% was observed in 16 assays at the concentration of 10−5 M and in only the glycine, strychnine-sensitive assay at the concentration of 10−7 M. This compound seems to be a reasonably clean sigma-2 selective ligand and therefore could be of great interest as a lead for further development or as a pharmacological tool.
3. Conclusion
We synthesized a series of 4-(indol-1-yl)butan-1-amines and evaluated their binding affinity for sigma receptors. Several high-affinity sigma-2 receptor ligands with significant selectivity for sigma-2 versus sigma-1 were identified. The best result was obtained with the ligand bearing a 4-fluorophenyl ring in the C-3 position of its indole core and a 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline as the distal cyclic amine. This compound, which displayed a high affinity for the sigma-2 receptor (7.45 nM) and a weak affinity for the sigma-1 receptor (2948 nM), was also submitted to an extensive selectivity profile characterization and only exhibited moderate affinity for the glycine, strychnine-sensitive assay at the 10−7 M concentration.
4. Experimental protocols
4.1. Chemistry
Reagents and starting materials were obtained from commercial suppliers and were used without purification. Precoated silica gel GF Uniplates from Analtech were used for thin-layer chromatography (TLC). Column chromatography was performed on silica gel 60 (Sorbent Technologies). Melting points were determined on a Electrothermal 9100 apparatus and are uncorrected. 1H and 13C NMR spectra were obtained on a Bruker 500MHz, 400MHz, or Bruker 400MHz Ultra Shield. The high resolution mass spectra (HRMS) were recorded on a Waters Micromass Q-Tof Micro mass spectrometer with a lock spray source. The mass spectra (MS) were recorded on a WATERS ACQUITY Ultra Performance LC with ZQ detector in ESI or APCI mode. Elemental analysis (C, H, N) were recorded on an elemental analyzer, Perkin-Elmer CHN/SO Series II Analyzer. Chemical names were generated using ChemDraw Ultra (CambridgeSoft, version 10.0). Except where otherwise noted, 1H and 13C NMR data for final compounds are given for materials in their salt form.
4.1.1. General procedure for the synthesis of 1-(4-bromobutyl)indole and derivatives (3a, 3b and 8a–8c)
The method adopted for the synthesis of 1-(4-bromobutyl)indole (3a) is described. Potassium hydroxyde (3.83 g, 68.3 mmol) and tetrabutylammonium iodide (0.2 g, 0.54 mmol) were added, under mechanical stirring, to a solution of indole (2a) (2 g, 17.1 mmol) in anhydrous DMF (25 mL). The reaction mixture was stirred at room temperature for 45 min. and cooled to 0°C. 1,4-Dibromobutane was then added and the mixture was stirred for 15 min. at 0°C and for 1 h at room temperature. The mixture was poured into 70 mL of water, extracted with methylene chloride (3 × 50 mL), and the combined organic layers were washed with brine and dried. The solvent was removed in vacuo, and the residue was chromatographed on a silica gel column using a gradient of hexanes/ethyl acetate (10:0 to 9:1) as the eluent to give 2.92 g (68%) of 1-(4-bromobutyl)indole as a pale yellow oil. 1H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 8.2 Hz, 1H), 7.29 (t, J = 7.2 Hz, 1H), 7.19 (t, J = 7.7 Hz, 1H), 7.12 (d, J = 3.0 Hz, 1H), 6.58 (d, J = 2.9 Hz, 1H), 4.15 (t, J = 6.8 Hz, 2H), 3.38 (t, J = 6.5 Hz, 2H), 2.02 (quint, J = 7.3 Hz, 2H), 1.86 (quint, J = 7.0 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 127.75, 127.69, 121.63, 121.15, 119.47, 109.39, 101.43, 101.37, 45.54, 33.20, 30.06, 28.91. MS (ESI) m/z 252 [M+H]+ for 79Br, 254 [M+H]+ for 81Br.
4.1.2. 1-[1-(4-Bromobutyl)indol-5-yl]-ethanone (3b)
This compound was prepared from 5-acetylindole (2b) as described for 3a. 67% yield, white solid. mp 67–68 °C. 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 7.90 (d, J = 8.7, 1H), 7.35 (d, J = 8.7, 1H), 7.16 (d, J = 3.0, 1H), 6.63 (d, J = 3.1, 1H), 4.18 (t, J = 5.3, 2H), 3.38 (t, J = 6.4, 2H), 2.67 (s, 3H), 2.05 – 2.00 (m, 2H), 1.87 – 1.80 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 198.21, 138.44, 129.59, 129.20, 128.06, 123.39, 121.89, 109.13, 103.41, 45.72, 32.79, 29.84, 28.83, 26.61. MS (ESI) m/z 316 [M+Na]+ for 79Br, 318 [M+Na]+ for 81Br.
4.1.3. General procedure for the synthesis of 1-(4-(4-cyclohexylpiperazin-1-yl)butyl)indole and other final products (4a–4f, 9a–9i)
The method adopted for the synthesis of 1-(4-(4-cyclohexylpiperazin-1-yl)butyl)indole dioxalate (4a) is described. K2CO3 (0.2 g, 1.43 mmol) and 1-cyclohexylpiperazine (0.081 g, 0.47 mmol) were added, under mechanical stirring, to a solution of 3a (0.12 g, 0.47 mmol) in anhydrous DMF (4 mL). The reaction mixture was heated at 60°C for 2 h. After cooling, the mixture was poured into 20 mL of water, extracted with ethyl acetate (3 × 30 mL), and the combined organic layers were washed with saturated aqueous NaCl and dried. The solvent was removed in vacuo, and the residue was chromatographed on a silica gel column using methylene chloride/methanol (97:3) as the eluent. 1-(4-(4-cyclohexylpiperazin-1-yl)butyl)indole was isolated as a dioxalate salt (white solid, 0.073 g, 30%). mp 230–233 °C. 1H NMR (400 MHz, DMSO-d6, 60°C): δ 7.55 (br s, 4H), 7.53 (d, J = 7.9 Hz, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.33 (d, J = 3.0 Hz, 1H), 7.12 (t, J = 7.4 Hz, 1H), 7.00 (t, J = 7.4 Hz, 1H), 6.42 (d, J = 2.8 Hz, 1H), 4.18 (t, J = 6.9 Hz, 2H), 3.00 (s, 4H), 2.83-2.77 (m, 5H), 2.57 (t, J = 7.3 Hz, 2H), 1.95-1.93 (m, 2H), 1.83-1.78 (m, 4H), 1.61-1.58 (m, 1H), 1.52-1.47 (m, 2H), 1.35-1.19 (m, 4H), 1.13-1.07 (m, 1H). 13C NMR (100 MHz, DMSO-d6, 60°C): δ 162.58, 135.49, 128.16, 127.95, 120.64, 120.14, 118.54, 109.42, 100.22, 63.20, 55.54, 49.78, 46.85, 44.91, 27.00, 26.65, 24.2, 24.38, 22.23. Anal. calcd for C26H37N3O8: C, 60.10; H, 7.18; N, 8.09. Found: C, 60.58; H, 6.80; N, 8.09. HRMS (TOF ES+) calcd for C22H34N3 [M+H]+ 340.2753, found 340.2759.
4.1.4. 1-(4-(4-(4-Fluorophenyl)piperazin-1-yl)butyl)indole oxalate (4b)
This compound was prepared from 3a and 1-(4-fluorophenyl)piperazine as described for 4a. 25% yield, white solid. mp 160–162 °C. 1H NMR (400 MHz, DMSO-d6): δ 7.54 (d, J = 7.8 Hz, 1H), 7.48 (d, J = 8.1 Hz, 1H), 7.13-6.98 (m, 6H), 6.42 (s, 1H), 4.21-4.18 (m, 2H), 3.27-2.95 (m, 10H), 1.78-1.63 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 164.38, 156.47 (J = 236.0 Hz), 146.73, 135.58, 128.59, 128.15, 118.91, 117.67 (J = 7.1 Hz), 115.43 (J = 22.0 Hz), 109.75, 100.54, 55.29, 50.97, 46.67, 44.94, 27.09, 21.16. Anal. calcd for C24H28FN3O4·¼ H2O: C, 64.63; H, 6.44; N, 9.42. Found: C, 64.60; H, 6.05; N, 9.32. HRMS (TOF ES+) calcd for C22H27N3F [M+H]+ 352.2189, found 352.2206.
4.1.5. 2-(4-(Indol-1-yl)butyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline oxalate (4c)
This compound was prepared from 3a and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline as described for 4a. 51% yield, white solid. mp 186-187 °C. 1H NMR (400 MHz, DMSO-d6): δ 11.57 (br s, 2H), 7.55 (d, J = 7.8 Hz, 1H), 7.49 (d, J = 8.2 Hz, 1H), 7.38 (d, J = 3.0 Hz, 1H), 7.13 (t, J = 7.3 Hz, 1H), 7.02 (t, J = 7.4 Hz, 1H), 6.76 (s, 1H), 6.73 (s, 1H), 6.43 (d, J = 2.8 Hz, 1H), 4.20 (t, J = 6.6 Hz, 2H), 4.13 (s, 2H), 3.72 (s, 3H), 3.71 (s, 3H), 3.31-3.29 (m, 2H), 3.10-3.06 (m, 2H), 2.93-2.91 (m, 2H), 1.83-1.71 (m, 4H). 13C NMR (100 MHz, DMSO-d6): δ 164.62, 148.16, 147.57, 135.57, 128.60, 128.15, 123.55, 120.99, 120.66, 120.44, 118.91, 111.49, 109.77, 109.76, 100.53, 55.55, 55.48, 54.48, 51.61, 48.83, 44.93, 27.03, 24.65, 21.21. HRMS (TOF ES+) calcd for C23H29N2O2 [M+H]+ 365.2229, found 365.2242.
4.1.6. 1-{1-[4-(4-Cyclohexyl-piperazin-1-yl)-butyl]indol-5-yl}-ethanone dioxalate (4d)
This compound was prepared from 3b and 1-cyclohexylpiperazine as described for 4a. 31% yield, white solid. mp 224–227 °C. 1H-NMR (free amine, 400 MHz, CDCl3): δ 8.16 (d, J = Hz, 1H), 7.74 (t, J = 8.1 Hz, 1H), 7.23 (t, J = 8.5 Hz, 1H), 7.06-7.03 (m, 1H), 6.48-6.45 (m, 1H), 4.04-3.99 (m, 2H), 2.63 (s, 4H), 2.53-2.51 (m, 3H), 2.45 (s, 4H), 2.45 (s, 1H), 2.26-2.21 (m, 2H), 1.85 (s, 2H), 1.73-1.70 (m, 4H), 1.52 (d, J = 10.0 Hz, 1H), 1.37-1.35 (m, 2H), 1.14-1.13 (m, 4H), 1.03-0.99 (s, 1H). 13C NMR (100 MHz, free amine, CDCl3) δ 198.09, 138.33, 129.46, 129.27, 127.94, 123.23, 121.53, 109.25, 103.00, 64.08, 57.19, 51.83, 48.37, 46.29, 27.96, 27.79, 26.56, 25.69, 25.43, 23.74. Anal. calc. for C28H39N3O9: C, 59.88; H, 7.00; N, 7.48. Found: C, 59.54; H, 6.83; N, 7.60. HRMS (TOF ES+) calcd for C24H36N3O [M+H]+ 382.2858, found 382.2875.
4.1.7. 1-(1-{4-[4-(4-Fluorophenyl)-piperazin-1-yl]-butyl}indol-5-yl)-ethanone dioxalate (4e)
This compound was prepared from 3b and 1-(4-fluorophenyl)piperazine as described for 4a. 37% yield, white solid. mp 140–142 °C. 1H NMR (free amine, 400 MHz, DMSO-d6) δ 8.28 (s, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.56 (d, J = 8.7 Hz, 1H), 7.49 (d, J = 3.0 Hz, 1H), 7.00 (t, J = 8.9 Hz, 2H), 6.87 (dd, J = 8.3, 5.4 Hz, 2H), 6.61 (d, J = 2.5 Hz, 1H), 4.21 (t, J = 6.9 Hz, 2H), 2.98 (s, 4H), 2.58 (s, 3H), 2.39 (s, 4H), 2.26 (t, J = 7.1 Hz, 2H), 1.87-1.64 (m, 2H), 1.39 (d, J = 7.0 Hz, 2H). 13C NMR (free amine, 100 MHz, DMSO-d6) δ 197.82, 156.39 (J = 234.0 Hz), 148.37, 138.57, 130.92, 129.29, 128.01, 123.30, 121.38, 117.41 (d, J = 7.6 Hz), 115.64 (d, J = 21.8 Hz), 110.22, 102.98, 57.46, 53.05, 49.40, 46.03, 40.42, 40.21, 40.00, 39.59, 28.13, 27.02, 23.84. Anal. calc. for C28H32FN3O9·5/4H2O: C, 56.42; H, 5.79; N, 7.10. Found: C, 56.41; H, 5.83; N, 7.05. HRMS (TOF ES+) calcd for C24H29N3OF [M+H]+ 394.2295, found 394.2290.
4.1.8. 1-{1-[4-(6,7-Dimethoxy-3,4-dihydro-1H-isoquinolin-2-yl)-butyl]indol-5-yl}-ethanone oxalate (4f)
This compound was prepared from 3b and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline as described for 4a. 41% yield, white solid. mp 170–171 °C. 1H NMR (free amine, 400 MHz, CDCl3) δ 8.29 (s, 1H), 7.86 (d, J = 8.7 Hz, 1H), 7.35 (d, J = 8.7 Hz, 1H), 7.17 (s, 1H), 6.59 (d, J = 10.8 Hz, 2H), 6.48 (s, 1H), 4.19-4.15 (m, 3H), 3.82-3.81 (m, 6H), 3.48 (s, 2H), 2.79-2.76 (m, 2H), 2.64 (s, 3H), 2.50-2.47 (m, 2H), 1.94-1.90 (m, 2H), 1.61-1.59 (m, 3H), 1.28-1.25 (m, 2H). 13C NMR (free amine, 100 MHz, CDCl3) δ 198.20, 162.50, 147.42, 138.47, 129.40, 128.00, 126.00, 123.28, 121.66, 111.37, 109.47, 109.26, 103.07, 57.26, 55.90, 55.44, 50.79, 46.40, 36.42, 31.37, 28.09, 26.56, 24.28. Anal. calc. for C27H32N2O7·H2O: C, 63.02; H, 6.66; N, 5.44. Found: C, 63.26; H, 6.15; N, 5.46. HRMS (TOF ES+) calcd for C25H31N2O3 [M+H]+ 407.2335, found 407.2343.
4.1.9. General procedure for the synthesis of 3-aryl-1-(phenylsulfonyl)indoles 6a-6c
The method adopted for the synthesis of 3-phenyl-1-(phenylsulfonyl)indole (6a) is described. To a solution of phenylboronic acid (0.49 g, 4.1 mmol) in benzene (21 mL) and ethanol (10 mL) were added potassium carbonate (0.680 g, 4.4 mmol) and 3-bromo-1-(phenylsulfonyl)indole (5) (1.2 g, 3.4 mmol). The reaction mixture was stirred at room temperature and deoxygenated by passing through it a steam of argon for 15 min. Palladium triphenylphosphine tetrakis (0.039 mg, 0.034 mmol) was added and the solution was refluxed for 16 h. After cooling, the mixture was poured into 25 mL of water, extracted with ethyl acetate (3 × 30 mL), washed with saturated aqueous NaCl and dried. The solvent was removed in vacuo, and the residue was chromatographed on a silica gel column using hexane/ethyl acetate (9:1) to give 0.955 g (84%) of 3-phenyl-1-(phenylsulfonyl)indole as a white solid. mp 144–146 °C. 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.2 Hz, 1H), 7.95 (d, J = 7.3 Hz, 2H), 7.81 (d, J = 7.9 Hz, 1H), 7.73 (s, 1H), 7.63 (d, J = 7.4 Hz, 2H), 7.57-7.27 (m, 8H). 13C NMR (100 MHz, DMSO-d6) δ 137.07, 135.34, 134.00, 130.37, 129.52, 127.30, 126.59, 126.08, 124.79, 120.13, 113.85, 99.44. MS (APCI) m/z 334.9 [M+H]+.
4.1.10. 3-(4-Fluorophenyl)-1-(phenylsulfonyl)indole (6b)
This compound was prepared from 3-bromo-1-(phenylsulfonyl)indole (5) and 4-fluorophenylboronic acid as described for 6a. 91% yield, white solid. 1H NMR (400 MHz, CDCl3): δ 8.07 (d, J = 8.3 Hz, 1H), 7.92 (d, J = 7.5 Hz, 2H), 7.70 (d, J = 7.9 Hz, 1H), 7.65 (s, 1H), 7.55-7.50 (m, 3H), 7.43 (t, J = 7.9 Hz, 2H), 7.36 (t, J = 7.4 Hz, 1H), 7.28 (t, J = 7.4 Hz, 1H), 7.14 (t, J = 8.6 Hz, 2H), 13C NMR (100 MHz, CDCl3): δ 162.56 (d, J = 245.5 Hz), 138.38, 135.67, 134.11, 129.73 (d, J = 7.9 Hz), 129.54, 129.45, 129.20 (d, J = 3.3 Hz), 127.03, 125.29, 123.92, 123.40, 122.99, 120.43, 116.09 (d, J = 21.4 Hz), 114.07. MS (EI) m/z 374 [M+Na]+.
4.1.11. 3-(Furan-3-yl)-1-(phenylsulfonyl)-1H-indole (6c)
This compound was prepared from 3-bromo-1-(phenylsulfonyl)indole (5) and furan-3-boronic acid as described for 6a. 97% yield, yellow solid. mp 88–91 °C. 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 8.3 Hz, 1H), 8.03 – 7.88 (m, 2H), 7.83 (s, 1H), 7.78 – 7.63 (m, 2H), 7.56 – 7.49 (m, 2H), 7.47 – 7.35 (m, 3H), 7.33 – 7.27 (m, 1H), 6.71 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 143.45, 139.07, 138.10, 135.46, 133.90, 129.32, 129.14, 126.79, 125.13, 123.70, 122.44, 120.48, 117.67, 115.41, 113.85, 109.66. MS (APCI) m/z 323.9 [M]+.
4.1.12. General procedure for the synthesis of 3-arylindoles 7a–7c
The method adopted for the synthesis of 3-phenylindole (7a) is described. Mg (2.12 g, 875 mmol) and ammonium chloride (0.04 g, 0.8 mmol) were added to a stirred solution of 6a (0.85 g, 2.5 mmol) in methanol (80 mL) and THF (20 mL). The exothermic mixture was stirred for 2 h at room temperature and was concentrated under reduced pressure. A saturated solution of ammonium chloride (30 mL) was then added and the mixture was extracted with ethyl acetate (3*100 mL). The organic layer was washed with water and brine, dried and evaporated. The residue was chromatographed on a silica gel column using hexane/ethyl acetate (8:2) to give 0.28 g (60%) of 3-phenyl-1H-indole as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 7.4 Hz, 1H), 7.96 (s, 1H), 7.83 (d, J = 7.1 Hz, 2H), 7.61 (t, J = 7.7 Hz, 2H), 7.55 – 7.34 (m, 3H), 7.31 (d, J = 2.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 136.77, 135.74, 129.02, 127.62, 126.19, 125.80, 122.55, 122.12, 120.51, 119.94, 118.20, 111.69. MS (APCI) m/z 193.1 [M]+.
4.1.13. 3-(4-Fluorophenyl)indole (7b)
This compound was prepared from 6b as described for 7a. 80% yield, white solid. mp 105–107 °C. 1H NMR (400 MHz, DMSO-d6): δ 11.40 (s, 1H), 7.85 (d, J = 7.6 Hz, 1H), 7.73-7.67 (m, 3H), 7.51 (d, J = 8.0 Hz, 1H), 7.25 (t, J = 8.8 Hz, 2H), 7.19 (t, J = 7.2 Hz, 2H), 7.12 (t, J = 7.2 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 161.47, 159.07, 136.78, 132.21, 128.09, 128.01, 124.84, 123.22, 121.37, 119.54, 118.70, 115.47, 115.26, 114.67, 111.88. MS (ESI) m/z 210 [M-H]+.
4.1.14. 3-(Furan-3-yl)indole (7c)
This compound was prepared from 6c as described for 7a. 89% yield, yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.06 (s, 1H), 7.98-7.81 (m, 2H), 7.60 (s, 1H), 7.40 (d, J = 7.7 Hz, 1H), 7.37-7.21 (m, 2H), 6.78 (s, 1H). 13C NMR (100 MHz, CDCl3) δ 143.06, 137.74, 136.57, 125.74, 122.55, 121.57, 120.30, 119.88, 119.72, 111.52, 109.92, 109.06. MS (APCI) m/z 184.0 [M]+.
4.1.15. 1-(4-Bromobutyl)-3-phenylindole (8a)
This compound was prepared from 3-phenylindole (7a) as described for 3a. 77% yield, colorless oil. 1H NMR (400 MHz, CDCl3) δ 8.05-8.03 (m, 1H), 7.74 (d, J = 7.0 Hz, 2H), 7.52 (t, J = 7.7 Hz, 2H), 7.43 (d, J = 8.2 Hz, 1H), 7.40-7.22 (m, 4H), 4.32-4.11 (m, 2H), 3.48-3.33 (m, 2H), 2.18-2.00 (m, 2H), 1.95-1.88 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 136.80, 135.62, 128.85, 127.40, 126.38, 125.88, 125.39, 122.11, 120.18, 120.08, 117.09, 109.68, 45.57, 33.05, 30.0, 28.85. MS (APCI) m/z 327 [M]+ for 79Br, 329 [M]+ for 81Br.
4.1.16. 1-(4-Bromobutyl)-3-(4-fluorophenyl)indole (8b)
This compound was prepared from 7b as described for 3a. 83% yield, colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.86 (d, J = 7.9 Hz, 1H), 7.60-7.56 (m, 2H), 7.37 (d, J = 8.2 Hz, 1H), 7.26 (t, J = 7.7 Hz, 1H), 7.20-7.16 (m, 2H), 7.12 (t, J = 8.7 Hz, 2H), 4.18 (t, J = 6.8 Hz, 2H), 3.38 (t, J = 6.5 Hz, 2H), 2.08-2.01 (m, 2H), 1.92-1.85 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 161.60 (d, J = 243.0 Hz), 136.87, 131.75 (d, J = 3.3 Hz), 128.99 (d, J = 7.6 Hz), 126.50, 125.28, 122.35, 120.27, 120.03, 116.41, 115.79 (d, J = 21.1 Hz), 109.82, 45.77, 33.08, 30.19, 29.03. MS (ESI) m/z 346 [M+H]+ for 79Br, 348 [M+H]+ for 81Br.
4.1.17. 1-(4-Bromobutyl)-3-furan-3-ylindole (8c)
This compound was prepared from 7c as described for 3a. 75% yield, yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.89-7.76 (m, 2H), 7.54 (t, J = 1.6 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.34-7.26 (m, 1H), 7.25-7.15 (m, 2H), 6.80-6.64 (m, 1H), 4.15-4.09 (m, 2H), 3.45 (t, J = 6.5 Hz, 2H), 2.06-2.00 (m, 2H), 1.91-1.78 (m, 3H). 13C NMR (100 MHz, CDCl3) δ 142.96, 137.49, 136.62, 126.30, 124.88, 122.11, 120.13, 119.85, 119.62, 109.79, 109.59, 107.87, 45.47, 33.03, 29.96, 28.84. MS (APCI) m/z 327 [M]+ for 79Br, 329 [M]+ for 81Br.
4.1.18. 1-[4-(4-Cyclohexyl-piperazin-1-yl)-butyl]-3-phenylindole dioxalate (9a)
This compound was prepared from 8a and 1-cyclohexylpiperazine as described for 4a. 21% yield, white solid. mp 242–244 °C. 1H NMR (free amine, 400 MHz, CDCl3) δ 7.96 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 7.3 Hz, 2H), 7.45 (t, J = 7.7 Hz, 2H), 7.39 (d, J = 8.2 Hz, 1H), 7.32-7.23 (m, 3H), 7.19 (t, J = 7.4 Hz, 1H), 4.18 (t, J = 7.0 Hz, 2H), 2.70 (s, 4H), 2.55 (s, 4H), 2.47-2.30 (m, 2H), 2.03-1.86 (m, 4H), 1.81 (s, 2H), 1.75-1.63 (m, 1H), 1.60-1.55 (m, 2H), 1.37-1.17 (m, 5H), 1.12 (s, 1H). 13C NMR (free amine, 100 MHz, CDCl3) δ 136.74, 135.66, 128.76, 127.29, 126.26, 125.71, 125.51, 121.86, 120.04, 119.86, 116.74, 109.73, 63.99, 57.62, 52.52, 48.60, 46.26, 28.35, 28.01, 25.97, 25.66, 24.13. HRMS (TOF ES+) calcd for C28H38N3 [M+H]+ 416.3066, found 416.3081.
4.1.19. 1-{4-[4-(4-Fluorophenyl)-piperazin-1-yl]-butyl}-3-phenylindole oxalate (9b)
This compound was prepared from 8a and 1-(4-fluorophenyl)piperazine as described for 4a. 28% yield, white solid. mp 195–197 °C. 1H NMR (free amine, 400 MHz, CDCl3) δ 8.00 (d, J = 7.9 Hz, 1H), 7.71 (d, J = 7.5 Hz, 2H), 7.55-7.38 (m, 3H), 7.31 (d, J = 5.6 Hz, 3H), 7.23 (d, J = 7.4 Hz, 1H), 6.98 (t, J = 8.5 Hz, 2H), 6.88 (s, 2H), 4.21 (t, J = 6.9 Hz, 2H), 3.12 (s, 4H), 2.58 (s, 4H), 2.52-2.36 (m, 2H), 1.97 (m, 2H), 1.62 (m, 2H). 13C NMR (free amine, 100 MHz, CDCl3) δ 158.39, 156.02, 147.94, 136.82, 135.71, 128.78, 127.33, 126.33, 125.62 (d, J = 23.6 Hz), 121.99, 120.00 (d, J = 17.7 Hz), 117.83 (d, J = 7.6 Hz), 115.50 (d, J = 22.0 Hz), 109.73, 77.40, 77.08, 76.76, 57.82, 53.16, 50.03, 46.29, 28.07, 24.18. Anal. calc. for C30H32FN3O4·⅓H2O: C, 68.82; H, 6.29; N, 8.03. Found: C, 68.95; H, 6.03; N, 8.01. HRMS (TOF ES+) calcd for C28H31N3F [M+H]+ 428.2502, found 428.2516.
4.1.20. 6,7-Dimethoxy-2-(4-(3-phenylindol-1-yl)butyl)-1,2,3,4-tetrahydroisoquinoline oxalate (9c)
This compound was prepared from 8a and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline as described for 4a. 30% yield, white solid. mp 189–190 °C. 1H NMR (free amine, 400 MHz, CDCl3) δ 7.98 – 7.96 (m, 2H), 7.68 (d, J = 7.2 Hz, 2H), 7.53-7.36 (m, 3H), 7.31 (s, 1H), 7.29-7.24 (m, 2H), 7.19 (t, J = 7.3 Hz, 1H), 6.59 (s, 1H), 6.49 (s, 1H), 4.22-4.19 (m, 2H), 3.83 (m, 6H), 3.51 (s, 2H), 2.81-2.79 (m, 2H), 2.70-2.68 (m, 2H), 2.53-2.50 (m, 2H), 1.97-1.95 (m, 2H), 1.66-1.64 (m, 2H). 13C NMR (free amine, 100 MHz, CDCl3) δ 162.56, 147.59, 147.28, 136.83, 136.83, 135.73, 135.73, 128.74, 127.28, 126.32 (d, J = 6.9 Hz), 126.09, 125.67, 125.56, 121.86, 119.93 (d, J = 14.3 Hz), 116.71, 111.46, 109.68 (d, J = 19.9 Hz), 57.51, 55.93, 55.57, 50.94, 46.27, 36.42, 28.48, 28.10, 24.49. Anal. calc. for C31H34N2O6·½H2O: C, 69.00; H, 6.54; N, 5.19. Found: C, 68.60; H, 6.11; N, 5.12. HRMS (TOF ES+) calcd for C29H33N2O2 [M+H]+ 441.2542, found 441.2521.
4.1.21. 1-(4-(4-Cyclohexylpiperazin-1-yl)butyl)-3-(4-fluorophenyl)indole dioxalate (9d)
This compound was prepared from 8b and 1-cyclohexylpiperazine as described for 4a. 28% yield, white solid. mp 248–249 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.98 (br s, 4H), 7.81 (d, J = 7.6 Hz, 1H), 7.70-7.65 (m, 3H), 7.53 (d, J = 7.8 Hz, 1H), 7.26-7.09 (m, 4H), 4.20 (s, 2H), 2.98-2.47 (m, 11H), 1.88-1.75 (m, 6H), 1.56-1.47 (m, 3H), 1.23-1.04 (m, 5H). 13C NMR (125 MHz, DMSO-d6): δ 164.73, 163.36, (d, J = 241.0 Hz), 136.53, 131.91, 128.18 (d, J = 7.6 Hz), 126.72, 125.28, 121.61, 119.86, 119.18, 115.64 (d, J = 20.8 Hz), 114.01, 110.44, 63.11, 56.27, 51.13, 47.54, 45.33, 27.33, 26.32, 25.30, 24.89, 22.86. Anal. calcd for C32H40FN3O8: C, 62.63; H, 6.57; N, 6.85. Found: C, 62.48; H, 6.25; N, 6.76. HRMS (TOF ES+) calcd for C28H37N3F [M+H]+ 434.2972, found 434.2987.
4.1.22. 3-(4-Fluorophenyl)-1-(4-(4-(4-fluorophenyl)piperazin-1-yl)butyl)indole oxalate (9e)
This compound was prepared from 8b and 1-(4-fluorophenyl)piperazine as described for 4a. 54% yield, white solid. mp 214–216 °C. 1H NMR (500 MHz, DMSO-d6): δ 11.40 (br s, 2H), 7.84 (d, J = 7.9 Hz, 1H), 7.76 (s, 1H), 7.70-7.67 (m, 2H), 7.58 (d, J = 8.2 Hz, 1H), 7.26 (t, J = 8.6 Hz, 2H), 7.22 (t, J = 7.6 Hz, 1H), 7.13 (t, J = 7.5 Hz, 1H), 7.07 (t, J = 8.7 Hz, 2H), 6.99-6.96 (m, 2H), 4.25 (t, J = 6.7 Hz, 2H), 3.26 (br s, 4H), 3.10 (br s, 4H), 2.97 (br s, 2H), 1.85-1.83 (m, 2H), 1.66 (br s, 2H). 13C NMR (125, MHz, DMSO-d6): δ 164.05, 160.38 (d, J = 240.4 Hz), 156.43 (d, J = 235.1 Hz), 146.76, 136.44, 131.86 (d, J = 3.0 Hz), 128.18 (d, J = 7.7 Hz), 126.70, 125.29, 121.63, 119.88, 119.16, 117.62 (d, J = 7.7 Hz), 115.61 (d, J = 21.0 Hz), 115.43 (d, J = 21.8 Hz), 114.08, 110.37, 55.31, 51.01, 46.72, 45.07, 27.04, 21.29. Anal. calcd for C30H31F2N3O4: C, 67.28; H, 5.83; N, 7.85. Found: C, 67.08; H, 5.80; N, 7.73. HRMS (TOF ES+) calcd for C28H30N3F2 [M+H]+ 446.2408, found 446.2391.
4.1.23. 2-(4-(3-(4-Fluorophenyl)indol-1-yl)butyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline oxalate (9f)
This compound was prepared from 8b and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline as described for 4a. 53% yield, white solid. mp 182–184 °C. 1H NMR (500 MHz, DMSO-d6): δ 10.52 (br s, 2H), 7.84 (d, J = 8.0 Hz, 1H), 7.75 (s, 1H), 7.70-7.67 (m, 2H), 7.58 (d, J = 8.2 Hz, 1H), 7.26 (t, J = 8.8 Hz, 2H), 7.22 (t, J = 7.6 Hz, 1H), 7.13 (t, J = 7.4 Hz, 1H), 6.77 (s, 1H), 6.72 (s, 1H), 4.26 (t, J = 6.8 Hz, 2H), 4.15 (br s, 2H), 3.72 (s, 3H), 3.70 (s, 3H), 3.32 (br s, 2H), 3.13-3.09 (m, 2H), 2.92 (br s, 2H), 1.90-1.85 (m, 2H), 1.75 (br s, 2H). 13C NMR (125 MHz, DMSO-d6): δ 164.34, 160.39 (d, J = 240.4 Hz), 148.15, 147.55, 136.45, 131.85, 128.20 (d, J = 7.7 Hz), 126.69, 125.32, 123.50, 121.64, 120.61, 119.90, 119.17, 115.61 (d, J = 21.0 Hz), 114.11, 111.46, 110.40, 109.67, 55.53, 55.48, 54.51, 51.70, 48.90, 45.05, 26.92, 24.68, 21.29. HRMS (TOF ES+) calcd for C29H32N2O2F [M+H]+ 459.2448, found 459.2456.
4.1.24. 1-[4-(4-Cyclohexyl-piperazin-1-yl)-butyl]-3-furan-3-ylindole dioxalate (9g)
This compound was prepared from 8c and 1-cyclohexylpiperazine as described for 4a. 21% yield, white solid. mp 240–243 °C. 1H NMR (free amine, 400 MHz, CDCl3) δ 7.85-7.69 (m, 2H), 7.49 (s, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.27-7.20 (m, 2H), 7.16 (t, J = 7.4 Hz, 1H), 6.69 (s, 1H), 4.11 (t, J = 6.8 Hz, 2H), 2.72 (s, 4H), 2.54 (s, 4H), 2.45 (s, 1H), 2.40-2.27 (m, 2H), 1.94 (s, 2H), 1.87-1.81 (m, 4H), 1.61 (m, 2H), 1.55-1.48 (m, 2H), 1.28-1.22 (m, 3H), 1.15-1.08 (s, 1H). 13C NMR (free amine, 100 MHz, CDCl3) δ 142.90, 137.34, 136.57, 126.20, 125.09, 121.91, 120.03, 119.69, 119.67, 109.74, 109.69, 107.53, 64.17, 57.43, 52.06, 48.47, 46.14, 28.10, 27.92, 25.81, 25.54, 23.98. Anal. calc. for C30H39N3O9·½H2O: C, 60.59; H, 6.79; N, 7.07. Found: C, 60.20; H, 6.39; N, 7.01. HRMS (TOF ES+) calcd for C26H36N3O [M+H]+ 406.2858, found 406.2874.
4.1.25. 1-{4-[4-(4-Fluorophenyl)-piperazin-1-yl]-butyl}-3-furan-3-ylindole dioxalate (9h)
This compound was prepared from 8c and 1-(4-fluorophenyl)piperazine as described for 4a. 46% yield, white solid. mp 172–174 °C. 1H NMR (free amine, 400 MHz, CDCl3) δ 7.93-7.75 (m, 2H), 7.55-7.54 (m, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.31-7.20 (m, 3H), 7.01-6.96 (m, 2H), 6.89-6.86 (m, 2H), 6.73 (s, 1H), 4.18 (t, J = 7.0 Hz, 2H), 3.11 (t, J = 4 Hz, 4H), 2.56 (t, J = 4 Hz, 4H), 2.44-2.40 (m, 2H), 2.03-1.85 (m, 2H), 1.63-1.59 (m, 2H). 13C NMR (free amine, 100 MHz, CDCl3) δ 158.35, 154.92 (d, J = 212.2 Hz), 147.98 (d, J = 2.2 Hz), 142.94, 137.45, 136.64, 126.29, 125.06, 121.96, 119.92 (d, J = 36.5 Hz), 117.78 (d, J = 7.6 Hz), 115.62, 115.40, 109.81, 109.70, 107.62, 57.90, 53.22, 50.11, 46.26, 28.14, 24.28. Anal. calc. for C30H32FN3O9·½H2O: C, 59.40; H, 5.48; N, 6.93. Found: C, 59.47; H, 5.15; N, 6.92. HRMS (TOF ES+) calcd for C26H29N3OF [M+H]+ 418.2295, found 418.2298.
4.1.26. 2-(4-(3-(Furan-3-yl)indol-1-yl)butyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline oxalate (9i)
This compound was prepared from 8c and 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline as described for 4a. 42% yield, white solid. mp 156 °C. 1H NMR (free amine, 400 MHz, CDCl3) δ 7.90-7.78 (m, 2H), 7.52 (s, 1H), 7.40 (d, J = 8.1 Hz, 1H), 7.30-7.24 (m, 2H), 7.21 (t, J = 7.4 Hz, 1H), 6.72 (s, 1H), 6.61 (s, 1H), 6.51 (s, 1H), 4.18 (t, J = 6.9 Hz, 3H), 3.86-6.84 (m, 6H), 3.52 (s, 2H), 2.84-2.80 (m, 2H), 2.69 (t, J = 5.7 Hz, 2H), 2.52 (t, J = 7.2 Hz, 2H), 2.05-1.84 (m, 2H), 1.72-1.60 (m, 2H). 13C NMR (free amine, 100 MHz, CDCl3) δ 147.56, 147.24, 142.89, 137.40, 136.68, 126.48, 126.27, 126.14, 125.10, 121.93, 120.05, 119.76, 119.70, 111.39, 109.80, 109.75, 109.51, 107.55, 57.65, 55.94, 55.91, 55.72, 51.06, 46.23, 28.64, 28.14, 24.58. HRMS (TOF ES+) calcd for C27H31N2O3 [M+H]+ 431.2335, found 431.2356.
4.2. Pharmacology
In vitro competition binding assays were performed as follows. Preparation of rat brain membrane and binding assays for the σ1 and σ2 receptor were performed using methods published previously in detail [30-34]. In brief, homogenates of whole rat brain excluding the cerebellum (400–500 μg) were taken in test tubes and incubated with 5 nM [3H](+)-pentazocine to label σ-1 receptors, or 3 nM [3H]-DTG in the presence of 300 nM (+)-pentazocine to label σ-2 receptors. Non-specific binding was determined in the presence of 10 μM haloperidol. Ten concentrations of each sigma compound ranging from 0.1-1000 nM were incubated for 120 min at 25° C in 50 mM Tris-HCl, pH 8.0 to measure their ability to displace the radioligands from their binding sites. The total reaction volume was 500 μl. The assay was terminated by the addition of 5 ml ice-cold 10 mM Tris-HCl, pH 8.0, followed by two washes through glass fiber filters presoaked in 1% polethyleneimine for at least 45 min to minimize non-specific binding. Both these assays were run in duplicate. Counts were extracted from the filters using Ecoscint (National Diagnostics, Manville, NJ) for at least 8 h prior to counting. Ki values were calculated using the Cheng-Prusoff equation [35].
Research highlights.
Preparation and evaluation of a series of indoles as sigma receptor ligands
Identification of several sigma-2 selective derivatives
Extensive pharmacological profile characterization for the best compound
Acknowledgments
This project was supported by grants from The National Institute on Drug Abuse (DA023205, DA013978). The National Institute on Drug Abuse arranged for the in vitro receptor and transporter assays at Oregon Health and Science University (OHSU) and for the Novascreen assays through contractual agreement with the vendors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Quirion R, Bowen WD, Itzhak Y, Jumien JL, Musacchio JM, Rothman RB, Su TP, Tam SW, Taylor DP. Trends Pharmacol Exp Ther. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-a. [DOI] [PubMed] [Google Scholar]
- 2.Hanner M, Moebius FF, Flandorfer A, Knaus HG, Striessnig J, Kempner E, Glossmann H. Proc Natl Acad Sci USA. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seth P, Fei YJ, Li HW, Huang W, Leibach FH, Ganapathy V. J Neurochem. 1998;70:922–931. doi: 10.1046/j.1471-4159.1998.70030922.x. [DOI] [PubMed] [Google Scholar]
- 4.Matsumoto RR. Sigma receptors: historical perspective and background. In: Matsumoto RR, Bowen WD, Su TP, editors. Sigma receptors: chemistry, cell biology and clinical implications. Springer; New York: 2007. pp. 1–23. [Google Scholar]
- 5.Bourrie B, Bribes E, Derocq JM, Vidal H, Casellas P. Curr Opin Invest Drugs. 2004;5:1158–1163. [PubMed] [Google Scholar]
- 6.Volz HP, Stoll KD. Pharmacopsychiatry. 2004;37:S214–S220. doi: 10.1055/s-2004-832680. [DOI] [PubMed] [Google Scholar]
- 7.Maurice T, Su TP. Pharmacology & Therapeutics. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez C, Arnt J, Costall B, Kelly ME, Meier E, Naylor RJ, Perregaard J. J Pharmacol Exp Ther. 1997;283:1323–1332. [PubMed] [Google Scholar]
- 9.Villard V, Espallergues J, Keller E, Alkam T, Nitta A, Yamada K, Nabeshima T, Vamvakides A, Maurice T. Neuropsychopharmacology. 2009;34:1552–1566. doi: 10.1038/npp.2008.212. [DOI] [PubMed] [Google Scholar]
- 10.Guitart X, Codony X, Monroy X. Psychopharmacology. 2004;174:301–319. doi: 10.1007/s00213-004-1920-9. [DOI] [PubMed] [Google Scholar]
- 11.Matsumoto RR, Liu Y, Lerner M, Howard EW, Brackett DJ. Eur J Pharmacol. 2003;469:1–12. doi: 10.1016/s0014-2999(03)01723-0. [DOI] [PubMed] [Google Scholar]
- 12.Maurice T, Martin-Fardon R, Romieu P, Matsumoto RR. Neurosci Biobehav Rev. 2002;26:499–527. doi: 10.1016/s0149-7634(02)00017-9. [DOI] [PubMed] [Google Scholar]
- 13.Choi SR, Yang B, Plossl K, Chumpradit S, Wey SP, Acton PD, Wheeler K, Mach RH, Kung HF. Nucl Med Biol. 2001;28:657–666. doi: 10.1016/s0969-8051(01)00234-7. [DOI] [PubMed] [Google Scholar]
- 14.Vilner BJ, John CS, Bowen WD. Cancer Res. 1995;55:408–413. [PubMed] [Google Scholar]
- 15.Rowland DJ, Tu Z, Xu J, Ponde D, Mach RH, Welch MJ. J Nucl Med. 2006;47:1041–1048. [PubMed] [Google Scholar]
- 16.Tu Z, Xu J, Jones LA, Li S, Dumstorff C, Vangveravong S, Chen DL, Wheeler KT, Welch MJ, Mach RH. J Med Chem. 2007;50:3194–3204. doi: 10.1021/jm0614883. [DOI] [PubMed] [Google Scholar]
- 17.Mach RH, Smith CR, al-Nabulsi I, Whirett BR, Childers SR, Wheeler KT. Cancer Res. 1997;57:156–161. [PubMed] [Google Scholar]
- 18.Ostenfeld MS, Fehrenbacher N, Hoyer-Hansen M, Thomsen C, Farkas T, Jaattela M. Cancer Res. 2005;65:8975–8983. doi: 10.1158/0008-5472.CAN-05-0269. [DOI] [PubMed] [Google Scholar]
- 19.Crawford KW, Bowen WD. Cancer Res. 2002;62:313–322. [PubMed] [Google Scholar]
- 20.Narayanan S, Bhat R, Mesangeau C, Poupaert JH, McCurdy CR. Future Med Chem. 2011;3:79–94. doi: 10.4155/fmc.10.279. [DOI] [PubMed] [Google Scholar]
- 21.Maeda DY, Williams W, Bowen WD, Coop A. Bioorg Med Chem Lett. 2000;10:17–18. doi: 10.1016/s0960-894x(99)00590-9. [DOI] [PubMed] [Google Scholar]
- 22.Maier CA, Wunsch B. J Med Chem. 2002;45:4923–4930. doi: 10.1021/jm020889p. [DOI] [PubMed] [Google Scholar]
- 23.Yous S, Wallez W, Belloir M, Caignard DH, McCurdy CR, Poupaert JH. Med Chem Res. 2005;14:158–168. [Google Scholar]
- 24.Gassiot AC, Charton J, Girault-Mizi S, Gilleron P, Debreu-Fontaine MA, Sergheraert C, Melnyk P. Bioorg Med Chem Lett. 2005;15:4828–4832. doi: 10.1016/j.bmcl.2005.07.039. [DOI] [PubMed] [Google Scholar]
- 25.Hajipour AR, Fontanilla D, Chu UB, Arbabian M, Ruoho AE. Bioorg Med Chem. 2010;18:4397–4404. doi: 10.1016/j.bmc.2010.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perregaard J, Moltzen EK, Meier E, Sanchez C. J Med Chem. 1995;38:1998–2008. doi: 10.1021/jm00011a019. [DOI] [PubMed] [Google Scholar]
- 27.Mach RH, Huang Y, Freeman RA, Wu L, Vangveravong S, Luedtke RR. Bioorg Med Chem Lett. 2004;14:195–202. doi: 10.1016/j.bmcl.2003.09.083. [DOI] [PubMed] [Google Scholar]
- 28.Mesangeau C, Narayanan S, Green AM, Shaikh J, Kaushal N, Viard E, Xu YT, Fishback JA, Poupaert JH, Matsumoto RR, McCurdy CR. J Med Chem. 2008;51:1482–1486. doi: 10.1021/jm701357m. [DOI] [PubMed] [Google Scholar]
- 29.Standard conditions for Suzuki were followed ; see for example: Jacquemard U, Routier S, Dias N, Lansiaux A, Goossens JF, Bailly C, Merour JY. Eur J Med Chem. 2005;40:1087–1095. doi: 10.1016/j.ejmech.2005.04.011.
- 30.Nguyen EC, McCracken KA, Liu Y, Pouw B, Matsumoto RR. Neuropharmacology. 2005;49:638–645. doi: 10.1016/j.neuropharm.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 31.Matsumoto RR, Bowen WD, Tom MA, Vo VN, Truong DD, De Costa BR. Eur J Pharmacol. 1995;280:301–10. doi: 10.1016/0014-2999(95)00208-3. [DOI] [PubMed] [Google Scholar]
- 32.Matsumoto RR, Shaikh J, Wilson LL, Vedam S, Coop A. Eur Neuropsychopharmacol. 2008;18:871–81. doi: 10.1016/j.euroneuro.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsumoto RR, Pouw B. Eur J Pharmacol. 2000;401:155–160. doi: 10.1016/s0014-2999(00)00430-1. [DOI] [PubMed] [Google Scholar]
- 34.Fishback JA, Mesangeau C, Poupaert JH, McCurdy CR, Matsumoto RR. Eur J Pharmacol. 2011;653:1–7. doi: 10.1016/j.ejphar.2010.10.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099–108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]