

Abstract
During the cell cycle, gap junction communication, morphology and distribution of connexin43 (Cx43)-containing structures change dramatically. As cells round up in mitosis, Cx43 labeling is mostly intracellular and intercellular coupling is reduced. We investigated Cx43 distributions during mitosis both in endogenous and exogenous expressing cells using optical pulse-chase labeling, correlated light and electron microscopy, immunocytochemistry and biochemical analysis. Time-lapse imaging of GFP/tetracysteine tagged Cx43 (Cx43-GFP-4C) expressing cells revealed an early disappearance of gap junctions, progressive accumulation of Cx43 in cytoplasmic structures, and an unexpected subset pool of protein concentrated in the plasma membrane surrounding the midbody region in telophase followed by rapid re-appearance of punctate plaques upon mitotic exit. These distributions were also observed in immuno-labeled endogenous Cx43-expressing cells. Photo-oxidation of ReAsH-labeled Cx43-GFP-4C cells in telophase confirmed that Cx43 is distributed in the plasma membrane surrounding the midbody as apparent connexons and in cytoplasmic vesicles. We performed optical pulse-chase labeling and single label time-lapse imaging of synchronized cells stably expressing Cx43 with internal tetracysteine domains through mitosis. In late telophase, older Cx43 is segregated mainly to the plasma membrane while newer Cx43 is intracellular. This older population nucleates new gap junctions permitting rapid resumption of communication upon mitotic exit.
Keywords: connexin43, mitosis, tetracysteine tag, live cell imaging, correlated light and electron microscopy
Introduction
Gap junctions are dynamic structures, consisting of hundreds to thousands of channels, made up of connexins, organized in quasi-crystalline arrays (1). These intercellular structures permit adjacent cells to engage in direct communication by allowing the passage of ions and small metabolites (2–5). The level of gap junction communication (GJC) between cells is well regulated via multiple mechanisms. These include alterations in the permeability of connexin channels as well as the rapid assembly and disassembly of the structures themselves. In vivo rapid disassembly occurs in heart in response to hypoxia (6, 7), and rapidly growing tumor cells are often deficient in GJC (8). In vitro, cells decrease GJC and disassemble junctions in response to phorbol esters (9), growth factors and oncogenes (10). Even in the absence of stimuli, connexins undergo a rapid and constant turnover as illustrated by a short half-life for Connexin43 (Cx43) of 1–5h (11) during which Cx43 becomes phosphorylated at a dozen or more residues. In fact, this constant turnover of Cx43 has been proposed to be one way in which cells regulate their levels of intercellular communication (12). Previous studies have also emphasized the role of phosphorylation in channel gating and assembly (13–15). Some of these events result in phosphorylation dependent conformational changes detectable as electrophoretically distinct species when analyzed by SDS-PAGE; these comprise a fast migrating form (referred to as P0 or NP) that includes the non-phosphorylated isoform, and multiple slower migrating forms (P1 and P2) (16) and a form present during mitosis (called Pm or P3) (17).
One of the most striking instances of gap junction remodeling occurs during mitosis, when GJC ceases and the majority of Cx43 appears as large intracellular accumulations (17–20). This type of dramatic rearrangement of organelles during mitosis is well documented. For example, in studies of organelle inheritance/fate for the Golgi apparatus, Shorter and Warren show data that support a model of fragmentation of the Golgi structure during prometaphase to anaphase, followed by both de-novo synthesis and recycling of components upon Golgi apparatus reassembly during telophase (21). It has also been shown that these events can be regulated via phosphorylation of specific Golgi resident proteins, such as GRASP65 (22). In the case of Cx43, internalization during mitosis is coincident with an increase in total Cx43 phosphorylation (18, 23), including increased apparent phosphorylation at S255, possibly S262 (17, 18) and S368 (24), and the formation of the unique slow migrating Pm phospho-isoform of Cx43. As cells exit mitosis, the Pm form disappears coincident with an increase in P0 and P1, though total Cx43 is decreased (17), and GJC is rapidly resumed (19). Thus, several steps in the Cx43 lifecycle are regulated through mitosis, including channel closure, gap junction disassembly, internalization and degradation as well as re-initiation of GJC between cells after mitotic exit. These events can occur in the absence of protein synthesis (19), suggesting that the re-initiation of GJC may involve a pool of Cx43 that is retained/recycled through mitosis to be reassembled into functional junctions.
In this study, we investigated the trafficking and recycling of Cx43 during mitosis both in endogenous and exogenous expressing cells using in vivo time-lapse imaging, optical pulse-chase labeling and correlated light (LM) and electron microscopy (EM) in combination with immunocytochemistry and biochemical analysis. Tetracysteine technology (25) and antibodies specific for different organelle markers allowed us to define the origin and localizations of different cellular pools of Cx43 and determine how different Cx43 subpopulations are trafficked. We used the current generation of the tetracysteine tag (26), called 4C, for in vivo optical pulse-chase experiments (27–30) that allowed us to spatially discriminate new versus old proteins during the various stages of mitosis. This was correlated with the labeling of various organelle markers in cells in telophase to dissect out the identity of new and old Cx43 containing structures in order to examine partitioning of Cx43 during mitosis and to determine whether post-mitotic resumption of GJC occurs via de novo synthesis of gap junctions or from connexons found in the mother cell.
Results
Cx43 is redistributed to intracellular and plasma membranes in mitotic cells
We analyzed the distribution of Cx43 in endogenous Cx43-expressing NRK and RAT1 cells (Figure 1). As shown previously (17–19), cells in interphase show typical punctate plaques while in metaphase and anaphase we observed an increase in cytoplasmic Cx43 staining, especially apparent in RAT1 cells. Notably, we found that as cells progress to telophase there is a subset of protein that appears to accumulate at the cleavage furrow area between the two daughter cells, clear in both RAT1 and NRK cells.
Figure 1. Distribution of endogenous Cx43 during different phases of mitosis.

Illustration of the stages of mitosis shown with chromosome and tubulin-based mitotic apparatus as characteristic hallmarks of each stage (left column). Middle and right column show 3D volumetric representations of confocal image stacks of endogenously expressing Cx43 RAT-1 fibroblasts and NRK cells at each of these stages. For cells in telophase note the labeling of Cx43 at the cleavage furrow. Immunolabels include anti-alpha tubulin-Cy5 (white), anti-Cx43-FITC (green), DAPI (blue). Scale bar, 15 μm.
In co-localization studies with different organelles markers in NRK cells (Figure 2), we found that Cx43 showed essentially no co-localization with a Golgi marker (GM130) in prophase to anaphase with some degree of overlay in telophase; minimal co-localization with the lysosome marker (LAMP1); however it became increasingly associated with early endosomes (EEA1) throughout mitosis starting at anaphase with the strongest co-localization residing in the cytoplasm during late telophase.
Figure 2. Co-labeling of endogenous Cx43 and various organelle markers in NRK cells during various phases of the cell cycle.

Images represent 3D reconstructions from confocal image stacks: anti-Cx43-CY5 (red), DAPI (blue), and GM130, Lamp1, EEA1- FITC (green). Scale bar, 10 μm.
To further examine the dynamic changes in Cx43 localization through mitosis we performed time-lapse microscopy on MDCK cells stably expressing Cx43 with a Green Fluorescent Protein and tetracysteine tag at the C-terminus (Cx43-GFP-4C). In order to image several cells progressing through mitosis, we synchronized cells using a combination of serum starvation and application of aphidicolin, an inhibitor of DNA synthesis (31), to create an enriched population of cells undergoing mitosis suitable for imaging (diagrammed in Figure 3A). Specifically, confluent cells were trypsinized and grown for about 20 hours in the presence of minimal serum and aphidicolin, resulting in a G1/S block. The drug was then washed out, normal serum was restored and cells were allowed to progress through S phase to mitosis. After 7 hrs, when cells begin entering mitosis, time-lapse imaging of the intrinsic GFP fluorescence was performed (Figure 3B, Supplemental movie1). Similar to the immunofluorescence images from endogenous Cx43 (Figures 1–2), in interphase these cells showed the typical punctate gap junction plaques (indicated by white arrowheads in Figure 3B). As they entered mitosis we observed an early disappearance of gap junction plaques, progressive accumulation of Cx43 in cytoplasmic structures in metaphase and anaphase (indicated by yellow arrowheads in Figure 3B), followed by re-appearance of the protein at the cell surface in telophase with a more diffuse (non-punctate) pool of Cx43 concentrated in the plasma membrane surrounding the cleavage furrow at the midbody region (indicated by white arrows in Figure 3B). This localization at the cleavage furrow is transient, observed in only a few time points of the time-lapse video where the interval between each point is 6 minutes. As cells progress to cytokinesis and exit the mitotic phase, formation of punctate gap junctions occurs rapidly (indicated by white arrowheads in Figure 3B).
Figure 3. Distribution of recombinant Cx43-GFP-4C during mitosis.
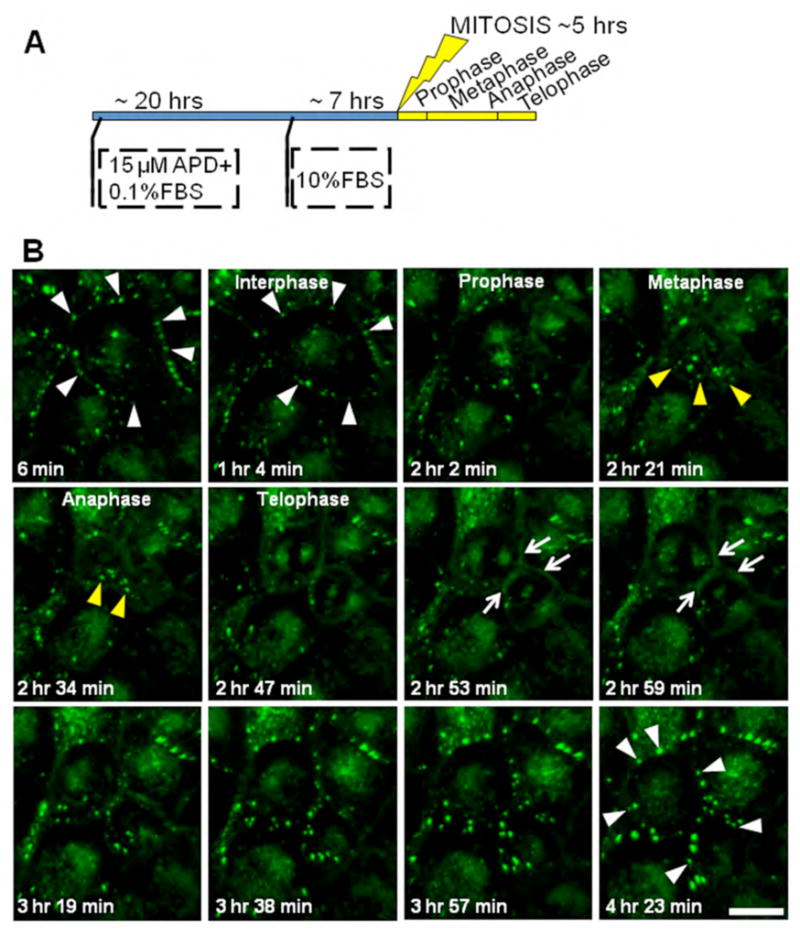
(A) Schematic of the synchronization protocol adopted for the imaging experiments. A combination of serum starvation (0.1% fetal bovine serum, FBS) and application of the drug aphidicolin (APD, an inhibitor of DNA synthesis that prevents cells in G1 phase from entering the DNA synthesis phase) was used. After 20 hours of incubation with the drug, regular media with 10% FBS is restored and after 7 hrs cells start entering in mitosis. (B) Time-lapse imaging of GFP fluorescence in Cx43-GFP-4C expressing MDCK cells. Images represent 3D volume reconstructions from confocal image stacks over time. In interphase cells show the typical punctate gap junction plaques (white arrowheads). As they progress into mitosis they reveal an early disappearance of gap junction plaques, progressive accumulation of Cx43-GFP-4C in cytoplasmic structures in metaphase and anaphase (yellow arrowheads), followed by the re-appearance of the protein at the cell surface in telophase with a more diffuse (non-punctate) pool of Cx43-GFP-4C concentrated in the plasma membrane surrounding the cleavage furrow at the midbody region (white arrows). As cells progress to cytokinesis and exit the mitotic phase, formation of punctate gap junction plaques is quickly restored (white arrowheads). Scale bar, 20 μm.
Cx43 is found in the cleavage furrow membrane and intracellular vesicles during telophase
To explore the origin and cellular distribution of Cx43 during mitosis, we used the tetracysteine technology (26, 32) that previously established that gap junction channels are added to plaque edges and internalized from the center of the plaque, providing an insight into the spatio-temporal dynamics of new versus older Cx43 populations (30). The advantages of this technology are the ability to specifically stain proteins in living cells without compromising cellular ultrastructure allowing fluorescent imaging via LM of spatio-temporal expression changes followed by fixation and specific visualization of Cx43 by EM to obtain high resolution information on its distribution in subcellular domains and interactions with other cytoplasmic compartments. Briefly, a biarsenical non-fluorescent membrane permeant compound ReAsH-EDT2 (ReAsH complexed with two EDT molecules) is applied to the outside of the cells, crosses the plasma membrane, finds the 4C motif inserted in the target protein within the cell, binds covalently to it, and becomes brightly fluorescent making it suitable for live cell imaging. This same bound fluorescent molecule can also be used for EM imaging due to its ability to be excited by light to produce singlet oxygen that can locally polymerize diaminobenzidine (DAB) producing an osmophilic reaction product that is electron dense. Since the labeling is done with the profluorescent ReAsH-EDT2 when the cells are still alive, and only subsequently fixed, the preservation of the ultrastructure is optimal. Figure 4 shows the FRET-mediated photo-oxidation of ReAsH-labeled MDCK Cx43-GFP-4C cells in telophase (2 pairs, labeled 1 and 2) by correlated LM/EM. A confocal image (Figure 4A) and corresponding EM (Figure 4B) show the diffuse, non-punctate concentration of Cx43 in the plasma membrane surrounding the cleavage furrow. Figure 4C is a higher magnification of the cleavage furrow from pair 2 shown in Figures 4A and B, showing a high concentration of ReAsH/Cx43 staining in the plasma membrane surrounding and proximal to the midbody. Figure 4D shows labeling of vesicles scattered throughout the cytoplasm of the same dividing cell, pair 2.
Figure 4. EM localization of Cx43 in telophase.
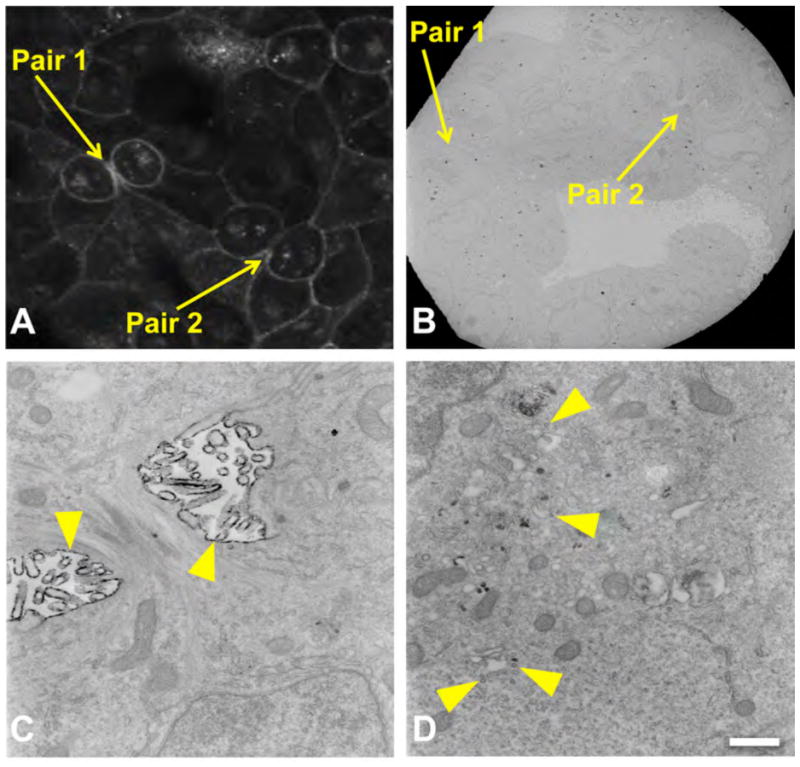
FRET-mediated photo-oxidation of ReAsH-labeled MDCK Cx43-GFP-4C cells in telophase (A, pair 1 and 2) and correlated image at low power EM (B). High resolution EM of pair 2 (C, D) shows the concentration of Cx43 in the plasma membrane surrounding the cleavage furrow (yellow arrowhead, C) as well labeling of vesicles in the cytoplasm (yellow arrowhead, D). Scale bar, 500 nm.
Overall these results have shown that there appears to be several distinct subcellular populations of Cx43 during mitosis, including cytoplasmic vesicles; during telophase some cytoplasmic vesicles co-localize with an early endosomal marker, and a distinct plasma membrane fraction is concentrated at the cleavage furrow. In order to further characterize the origin and trafficking of these subpopulations, we performed optical pulse-chase experiments using the tetracysteine technology.
The tetracysteine label can be used as an internal tag
Since it has been established that the fusion of GFP to the Cx43 C-terminus can interfere with ZO-1 binding, and can result in unusually large plaques (33, 34), we generated a Cx43 construct containing internal tetracysteine tags. We first inserted the 4C at position 337. Unfortunately, following labeling with the biarsenical ligands the signal was weak and therefore, was not suitable for our imaging experiments (data not shown). A second construct was generated with 2 internal 4C sequences, one at 309 and the other at 337 (Figure 5A), which produced sufficient fluorescence and exhibited normal plaque size and number per cell when stably expressed in MDCK cells (Figure 5B). We found that the labeling obtained with FlAsH (green) or ReAsH (red) was specific for Cx43 as indicated by almost complete overlap of staining using a Cx43 polyclonal antibody (Figure 5B).
Figure 5. Characterization of the new internally tagged Cx43.
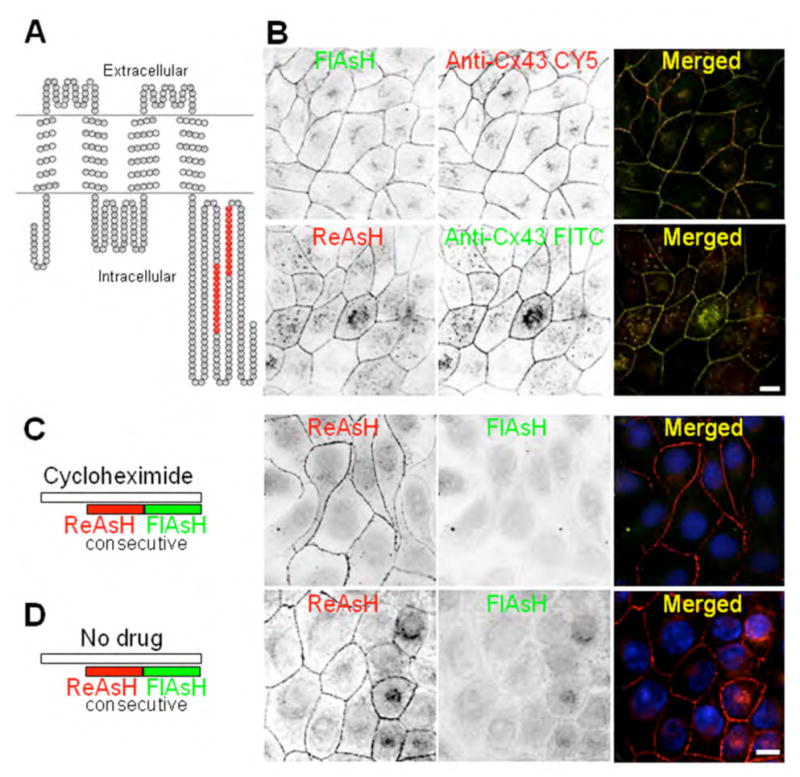
(A) Construct generated with 2 internal 4C sequences, one at position 309 and the other at 337 of Cx43 (Cx43-4C309/337). (B) MDCK cells stably expressing Cx43-4C309/337. We expressed these Cx43 constructs in MDCK cells that lack Cx43 expression and therefore, gap junction plaques. The labeling obtained with FlAsH-EDT2 or ReAsH-EDT2 (images are displayed with an inverted color table, black is the highest signal while white represents no signal, i.e. background) is specific as indicated by the co-localization with the Cx43 antibody staining (merged images). Scale bar, 10 μm. (C, D) Control experiments for pulse-chase protocol: cells expressing Cx43-4C309/337 were labeled with ReAsH (red) over a 1 hr period, washed with BAL and then immediately labeled with FlAsH (green) to check for complete saturation of labeling with ReAsH (red) of pre-existing Cx43-4C309/337. (C) Consecutive labelings with ReAsH (red) and then FlAsH (green) in the presence of a protein synthesis inhibitor (cycloheximide, 1hr before and during labeling). All pre-existing Cx43-4C309/337 were labeled with ReAsH (red) while FlAsH staining was negative. (D) Similar experiment as in (C), but in the absence of cycloheximide. All pre-existing Cx43-4C309/337 were also labeled with ReAsH (red) while FlAsH staining is showing the few new proteins synthesized during the 20 minutes period for washes in between the two consecutive labelings and the 1 hour period for the labeling with the second biarsenical FlAsH.
A major advantage of the tetracysteine/biarsenical labeling methodology is the ability to temporally and spatially distinguish new versus old proteins by using spectrally distinct fluorescent biarsenical ligands that only fluoresce when bound to the genetically appended tetracysteine protein tag (30). We refer to this as in vivo optical pulse-chase labeling. The internally tagged construct was appropriate for this type of pulse-chase experiment by sequential labeling with ReAsH (red) and FlAsH (green) in the presence or absence of the protein synthesis inhibitor, cycloheximide (Figure 5C, D). Note in the absence of cycloheximide (Figure 5D) “new” protein synthesized during the incubation time (in total 1 hour for the labeling with the biarsenical, and 20 minutes for the wash) was labeled by FlAsH while in the presence of the drug (Figure 5C) only “old” protein is present, as indicated by exclusive ReAsH labeling.
We performed functional studies to assess gap junction coupling and ZO-1 interaction in cells expressing the internally tagged Cx43. We carried out scrape-load dye transfer assay in communication-deficient MDCK cells that served as negative control, in MDCK cells stably expressing the internally tagged Cx43 (Cx43-4C309/337), and in NRK cells endogenously expressing Cx43 (Figure 6A). Transfer of Lucifer Yellow was observed in cells expressing the internally tagged Cx43 at comparable levels to the NRK cells endogenously expressing Cx43 but not in the communication-deficient cells (Figure 6B). The slightly higher level of dye transfer in Cx43-4C309/337 expressing MDCK cells is likely more due to cell volume differences between MDCK and NRK cells rather than functional differences between the WT and tagged Cx43 species. Moreover, these MDCK cells stably expressing Cx43-4C309/337 maintained ZO-1 interactions as opposed to Cx43-GFP-4C MDCK cells (Figure 6C).
Figure 6. Internally tagged Cx43 gap junctions transfer Lucifer Yellow as efficiently as the wild-type gap junctions.

(A) Representative Lucifer Yellow transfer images from endogenous expressing Cx43 NRK cells, MDCK cells stably expressing Cx43-4C309/337, and MDCK communication-deficient cells (negative control). Top row is LY fluorescence while the bottom row shows the LY fluorescence superimposed on the DIC image to highlight cell monolayers. Scale bar is 40 μm. (B) Summary of intercellular Lucifer Yellow transfer reported as number of Lucifer Yellow positive cells for the various groups (n=11). The internally tagged Cx43 functions at comparable levels to the endogenous Cx43. (C) Co-immunoprecipitation of ZO-1 with Cx43 antibody. Cx43 was immunoprecipitated from NRK cells or MDCK cells expressing Cx43-4C309/337 or Cx43-GFP-4C and the <100 kDa range was blotted with an N-terminal antibody to Cx43 and the >100kDa range was blotted with an antibody to ZO-1. Note, in NRK cells the NT1 antibody recognizes a band migrating at a similar molecular weight as Cx43-GFP-4C. This band appeared in a subset of experiments using these cells and could be a dimer of Cx43.
Cx43 is phosphorylated on S255 and S262 during mitosis
It has been previously shown that mitotic cells are more highly phosphorylated than interphase cells and exhibit a slow migrating isoform (18). Two specific serines in Cx43, S255 and/or S262, have been described as being substrates for kinases active during mitosis (17, 18). To confirm whether one, the other or both were active in our studies, immunoblot analysis was performed (Figure 7) on lysates from asynchronous (A) and mitosis synchronized cells (M). Note that this protocol only results in a few fold enrichment in cells, not a pure mitotic population. Several previous studies have shown that all Cx43 from a pure mitotic population shows a large phosphorylation-dependent shift in migration (17, 18). To determine whether Cx43 phosphorylation was affected in the cell lines and constructs used in these studies, we examined their SDS-PAGE mobility in immunoblots (Cx43-4C309/337 corresponds to the internally tagged construct, and GFP-4C to the Cx43 C-terminal fusion protein). Figure 7 shows signal for total Cx43, detected by an antibody to the N-terminus (NT1). Rat1, NRK and MDCK cells expressing wild type Cx43 (43WT) show multiple migratory phospho-isoforms (indicated by brackets) typical for Cx43 in both asynchronous (indicated by A) and a population enriched for mitotic cells (indicated by M). As previously reported, the mitotic isoform show a rather dramatic shift in migration that can be eliminated by phosphatase treatment (17, 18). Note a low level of the apparent mitotic form can be seen even in the asynchronous WT43 cells. This may indicate a greater number of mitotic cells in this cell line or may just reflect differences in the cell lines used. The internally tagged Cx43 (Cx43-4C309/337) exhibits an anomalous migration pattern, presumably due to the addition of the two tetracysteine sequences (each resulting in 11 additional amino acids), but it still exhibits specific phosphorylation-dependent conformation changes detectable via SDS-PAGE. Formation of a mitosis specific isoform (Pm) is also seen in the Cx43-GFP-4C construct. While migratory isoforms have been shown to be an indicator of changes in Cx43 phosphorylation, our interest is to examine specific phosphorylation events regulating gap junction biology. Previous studies using phosphotryptic peptide analysis indicated that Cx43 from mitotic cells could be phosphorylated on a peptide containing S255 and S262 (17, 18). To further these studies and determine whether either or both residues are phosphorylated, we utilized phospho-specific antibodies for these sites and found that, indeed, both of these antibodies bound to slow migrating bands with increased levels in the mitotic enriched populations in all cell lines and constructs used in these studies (pS255-middle panel and pS262-bottom panel). The phospho-antibodies, pS255 in particular, do show some bands, especially at high molecular weights, that do not appear to be related to Cx43 since no co-migrating bands were detected with the NT1 and phospho-antibodies using the Odyssey imaging system. However, the dominant NT1 and pS255 bands show complete overlay in the molecular weight range of Cx43 and these bands are absent in parental cells that do not contain Cx43 (data not shown). We conclude that Cx43 can, in fact, be phosphorylated at both sites in mitotic cells and, importantly, that addition of the 4C or GFP-4C sequences did not appreciably affect the site specific phosphorylation of Cx43 during mitosis.
Figure 7. Cx43 is phosphorylated on S255 and S262 during mitosis.
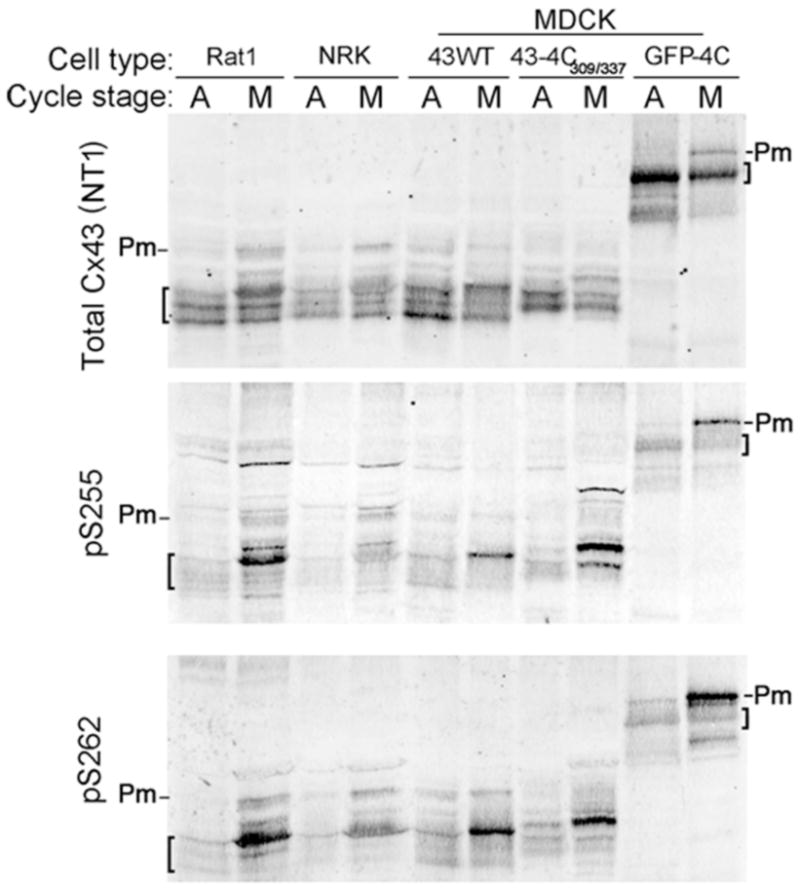
(A) Cell lysates from asynchronous (A) or synchronized mitotic (M) cells from various cell lines were analyzed via immunoblotting. Top panel shows total Cx43 from each cell line used in these studies and migratory isoforms are noted by brackets. Middle and bottom panel show Cx43 phosphorylated on S255 (pS255) and S262 (pS262), respectively. Pm indicates the band representing phosphorylated species that occurs during mitosis.
Spatial discrimination of proteins made before and during mitosis
Both static immunofluorescence images and dynamic live cell microscopy movies indicate the presence of Cx43 in distinct subcellular localizations that change throughout mitosis. Previous biochemical analyses indicated that there may be multiple fates for different subpopulations of Cx43, where some protein may be degraded while some may undergo de-phosphorylation and reutilization (17). The potential for reutilization has also been shown functionally, as daughter cells are able to quickly resume gap junction communication after mitosis, even in the absence of protein synthesis and trafficking through the reassembled Golgi (18, 19). We performed in vivo optical pulse-chase experiments to distinguish the origin and fate of different pools of Cx43 during mitosis, in particular, Cx43 synthesized before mitotic entry compared to the more newly synthesized material during cell division. The protocol for these experiments is shown in Figure 8A, where the first labeling with ReAsH (red) was performed before mitosis, followed by a chase time of 3 hrs, followed by a second labeling with FlAsH (green) at the end of mitosis. The cells were then fixed and imaged. This method allows spatial discrimination of older proteins synthesized and trafficked to the plasma membrane before mitosis (ReAsH labeled in red) from the nascent pool synthesized as cells entered and progressed through mitosis (FlAsH labeled in green).
Figure 8. In vivo optical pulse-chase labeling with ReAsH (red) and FlAsH (green) as cells progressed towards and through mitosis.

(A) Schematic of the synchronization protocol adopted for the pulse-chase experiments. MDCK cells stably expressing Cx43-4C309/337 were synchronized in G1/S then released in order to perform the labeling. The first labeling with ReAsH (red) was performed before mitosis, followed by a chase time of 3hrs, and the second labeling with FlAsH (green) at the end of mitosis. (B) 3D volume reconstructions from confocal image stacks: at the end of mitosis, the older pool (red) is mainly localized at the plasma membrane, while the newer pool (green) is intracellular. Scale bar, 10 μm. (C) XY, YZ and XZ optical sections through the volume along the indicated axes (white dashed lines) reveals the localization of older proteins (red) at the plasma membrane in the cleavage furrow region and the newer proteins (green) in the cytoplasm. Scale bar, 20 μm. (D, E) 3D volume rendering showing the segregation of older and newer proteins in telophase (D) and interphase cells (E). Inset in E (higher magnification) shows the typical gap junction formation/removal where new gap junction channels (green) are added at the edges of the plaque.
These data suggest that “old” Cx43 in the plasma membrane could be reutilized to form new gap junctions upon mitotic exit. As shown in Figure 8B–D, at the end of mitosis, the new and old fractions of Cx43 are clearly discriminated as indicated by a lack of co-localization in Figure 8B, merged (cell in late telophase). Instead we observe a pool of old Cx43 (red) concentrated largely at the perimeter of the cell while the newer pool (green) is intracellular. This is especially apparent in the 3D volume reconstructions and rendering (Figure 8D). Interestingly, there appeared to be 2 separate pools of Cx43 located in or around the midbody. An old fraction is localized to the plasma membrane at the cleavage furrow (Figure 8B, ReAsH) while new Cx43 appears to be concentrated in the cytoplasm (Figure 8B, FlAsH). The plasma membrane fraction is consistent with the immunostaining observed for endogenous Cx43 (Figures 1–2) and with the recombinant Cx43-GFP-4C in the live cell imaging (Figure 3, Supplemental movie1) as well as at the EM level (Figure 4). The more newly synthesized material is found within the midbody region as shown in the merged xz panel (Figure 8C), where the FlAsH signal is clearly surrounded by the ReAsH. As a control, MDCK cells in interphase underwent the same pulse chase protocol and show canonical, discrete gap junctions, where the newer pool (green) of Cx43 is seen at the edges of the plaque (Figure 8E). It should be noted that the general mechanism of gap junction accretion demonstrated by Gaietta et al. (30), and Lauf et al. (35) for Cx43, and recently for Cx26 (36), whereby channels are added to the plaque edges, is also observed in the cell lines used here, and this mechanism is independent of plaque size. Thus, plaque edge accretion and plaque center degradation is a conserved mechanism for gap junction plaque growth and dynamics among different cell types and isoforms.
To test whether the “old” Cx43 proteins labeled before mitosis could be reutilized to form new gap junctions upon the mitotic phase exit, we performed time-lapse imaging of green FlAsH-labeled Cx43-4C309/337 and followed their fate during and after completion of mitosis. As shown in the Figure 9, Supplemental movie2 the initial staining of the cells in interphase shows the typical punctate appearance of gap junction plaques (indicated by white arrowheads). As the cell enter mitosis and progress from prophase to anaphase some of the pre-existing Cx43 forming gap junction plaques (white arrowheads in prophase and metaphase) are clearly internalized (yellow arrowheads of the same regions in metaphase, anaphase and telophase) to reappear at the cell surface only in late telophase. Following the mitotic phase the “old” FlAsH-labeled Cx43 is reutilized to form new punctate gap junction plaques by the daughter cells (white arrowheads in the bottom row of images). A similar time-lapse movie of ReAsH-labeled Cx43-4C309/337 is shown in Supplemental movie3.
Figure 9. Time-lapse imaging of green FlAsH-labeled Cx43-4C309/337 in MDCK cells before, during and after completion of mitosis.

Following FlAsH labeling cells were imaged every 6 minutes. FlAsH fluorescence is superimposed to the differential interference contrast images. Images represent 3D volume reconstructions from confocal image stacks over time. The cell in interphase shows the typical punctate appearance of gap junction plaques (indicated by white arrowheads). Following the mitotic phase the “old” FlAsH-labeled Cx43 is reutilized to form new punctate gap junction plaques by the daughter cells (white arrowheads). The yellow arrowheads show Cx43 accumulated intracellularly. Scale bar is 20 μm.
To discriminate the subcellular compartments where different pools of Cx43 are distributed, we combined the optical pulse-chase labeling using red ReAsH and green FlAsH with immunofluorescence of various organelles markers (Figure 10). The degree of overlap between the marker and FlAsH or ReAsH is graphed as the Pearson coefficient (PC, a measure of the correlation between two variables) multiplied by 100 in Figure 10B with value of zero corresponding to non-overlapping images and value of 100 reflecting 100% co-localization. We observed some overlap of Cx43 with the Golgi marker, giantin, with FlAsH showing more overlay (18%± 8, mean ± SEM, n=4) than ReAsH (13%±4, mean ± SEM, n=4), indicating that FlAsH-labeled Cx43 (new protein) was en route to the plasma membrane through the Golgi apparatus. Conversely, significant overlap of the endosomal marker EEA1 occurred with older Cx43 than new Cx43 populations (26%± 4, mean ± SEM with n=8 for ReAsH, and 6% ± 2 with n=8 for FlAsH), suggesting that older protein had already been trafficked to the plasma membrane and had undergone endocytosis. These results are consistent with the distribution of endogenous Cx43 observed in NRK cells (Figure 2) that showed minimal co-localization with the lysosomal marker LAMP1, and a strong overlay with early endosomes in telophase.
Figure 10. Co-labeling of ReAsH (red, old protein) and FlAsH (green, new protein) with various organelle markers in cells in telophase.

(A) Images displayed with an inverted color table (black is highest signal while white represents no signal, i.e. background) are maximum intensity projections of confocal image stacks. The organelle markers Giantin, Lamp1, and EEA1 were labeled with CY5-conjugated secondary antibodies (blue in merged images). Cyan represents the overlap of new Cx43 (green) and the marker (blue), magenta represents the overlap of old Cx43 (red) and the marker (blue) and white indicates overlap of all three. Scale bar, 10 μm. (B) Co-localization analysis of the various markers with ReAsH or FlAsH was quantified using the Pearson coefficient (PC) with value of zero corresponding to non-overlapping images, and value of 1 reflecting complete co-localization. In these graphs, the PC was multiplied by 100 as an indicator of percent co-localization. Presented here are the average values ± standard error (S.E.) of n=4 cells for Giantin, n=7 for Lamp1, and n=8 cells for EEA1.
Discussion
The shape and total surface of a cell and its daughters change during cell division. Cells initially round up during prophase and metaphase, then reacquire their flattened shape during cytokinesis. It has been shown that the amount of plasma membrane and the surface expression of membrane-bound proteins diminishes at the beginning of mitosis but recovers rapidly at the end (37). Gap junctions are rapidly assembled and disassembled during their life cycle. In particular, there are large-scale changes during mitosis, including the disappearance of the network of gap junctions that typically encircle and join cells. However, rapid resumption of gap junction communication and presumably gap junction plaque assembly occurs at mitotic exit. These observations led us to pose the question: “is de novo synthesis of connexin during cell division the sole source of new gap junctions?” In this study, we answered this question using in vivo time-lapse imaging, optical pulse chase methods and photoconversion for correlative LM and EM, and we demonstrate that in addition to de novo synthesis, Cx43 originally located in the plasma membrane redistributes and recycles during cell division. A key finding from this investigation is that while the majority of Cx43 protein is internalized during the early stages of mitosis (progressively from prophase to anaphase), and new protein synthesis makes up the intracellular pools during the later stages, there is also a subset of Cx43 that reappears at the cell surface during the later stages of mitosis (late telophase), apparently being recycled, to nucleate the formation of gap junction plaques as they redistribute in the plasma membrane of the daughter cells. These “nucleating connexons” would be critical for cells to re-establish gap junction mediated intercellular communication in a time efficient manner following cell division. The use of a coalesced nucleating pool may be especially critical in the formation of clusters of gap junction channels that exclude other integral membrane proteins (especially glycoproteins) thereby minimizing the amount of surface area necessary to bring the two cells into close apposition (38).
Recycling of Cx43 occurs during mitosis
During interphase, Cx43 is observed at cell-cell contact regions with noticeable localizations occurring in distinct synthetic cellular compartments such as the Golgi or in degradative cellular compartments such as lysosomes. In contrast, Cx43 in mitotic cells appears to be concentrated in intracellular structures. The majority of protein is internalized during the early stages of mitosis as observed by time-lapse imaging of Cx43-GFP-4C or Cx43-4C309/337 expressing cells. As cells progress through telophase the Cx43 proteins start reappearing at the cell surface, and our results, show for the first time that ‘old’ pools synthesized before mitosis can be recycled and reutilized to form new gap junction plaques in the daughter cells. Furthermore, we show the localization at EM resolution of a subset of Cx43 at the plasma membrane in the cleavage furrow in telophase, confirming previous observations from immunofluorescence analysis that some Cx43 is located near the plasma membrane (19). This distribution appears to be in the form of connexons since typical gap junction plaques were not observed with EM imaging. The dynamic rearrangement of cell surface proteins is a common feature of cytokinesis (39, 40). In MDCK cells, proteins associated with the plasma membrane accumulate in the cleavage furrow during cell division, then rapidly disperse in interphase, and this rearrangement appears to be essential for cytokinesis. Based on our results Cx43 present at the cell surface seems to follow the same reorganization. A model has been proposed that the global change in protein density affects the mechanical balance between the equatorial and polar regions and play an essential role in the initial phase of cytokinesis (41, 42).
We have shown that new Cx43 protein synthesis makes up the intracellular pools during the later stages of mitosis. During telophase older Cx43 is mainly retained at the plasma membrane with a small subset of these older proteins localized in early endosomal compartments. Based on our time-lapse imaging we propose that the older Cx43 reappearing at the cell surface acts as nucleation sites for formation of new gap junction plaques of the daughter cells. These “seeding channels” would serve to speed up the process of re-establishing cell-cell communication after exit from telophase than if gap junction plaques were formed by only newly synthesized channels. The localization of Cx43 in the early endosome is consistent with the mechanism proposed by Boucrot and Kirchhausen (37) by which modulation of endosomal recycling controls cell area and surface expression of membrane-bound proteins during mitosis. They reported that when cells round up at the beginning of mitosis the decrease in total surface area is due mainly to a shutdown in membrane recycling from endosomal compartments back to the cell surface. Then, following anaphase, endosomes participate in plasma membrane recovery, which is essential for cell division. Based on their model, during cell rounding there is formation of an internal membrane reservoir that stores membrane components to be subsequently released later in cell division. Our results from the live cell imaging of old proteins labeled before mitosis in conjunction with their predominant co-localization with an endosomal marker in telophase suggest that the reappearance of Cx43 at the cell surface in the final stages of mitosis might be mediated by a recycling of Cx43 from an endosomal compartment back to the cell surface (see Figure 9, Supplemental movie2 and 3). More in vivo imaging experiments targeting specific subsets of Cx43 during cell division will be important in assessing the role of endosomal compartments in recycling back the protein to the cell surface in reestablishing gap junction communication after mitotic exit.
How are phosphorylation events and Cx43 recycling related?
The changes in gap junction distribution during mitosis are correlated with a reduction in gap junctional communication (20) and increase in phosphorylation of Cx43 (17–19). However, the relationship and process by which gap junction plaques and cytoplasmic structures are disassembled and reassembled are not clear, nor are the signaling pathways involved. Phosphorylation induced by p34cdc2/cyclin B kinase has been proposed as a possible factor that may lead to internalization of Cx43 and loss of gap junction structures during cell division (17). In agreement with these findings, our results suggest that the trafficking of Cx43 during mitosis is not just a large-scale internalization of cell surface proteins and recycling, but is a highly coordinated process with different populations of Cx43 phospho-isoforms localized to specific cellular compartments. Cx43 contains 12–13 serines that get phosphorylated at key steps in its life cycle (43). We have confirmed here with phospho-specific antibodies that two of these serines, S255 and S262, are phosphorylated during mitosis, independent of the addition or insertion of any tag; however, whether these specifically phosphorylated serines act as tags, gate keepers or signals to other proteins is unknown. Future experiments will use phosphorylation specific antibodies to expand on the role of phosphorylation in targeting Cx43 to specific compartments and the timing and sequence of these phosphorylation events during the cell cycle.
Interactions/conformation of the C-terminus appears to be unaffected by internal insertions of small tags at two specific locations
A unique facet of our study was the design and implementation of a Cx43 construct with two 4C internal tags in the C-terminus for visualizing protein age and movements within cells while maintaining known C-terminal interaction domains. This is the first study to insert two non-contiguous 4C tags into a protein sequence. Examples of single insertions include the host-encoded cellular prion protein (PrPC) (44); the GAG protein in HIV-1 (45); TEM-1 beta-lactamase (46) and the α(2A)-adrenergic G protein-coupled receptor (47).
We previously showed, using monoclonal antibodies, that the largely unordered Cx43 C-terminus likely adapts locally organized conformations that are coordinated by specific phosphorylation events (48). Here, we inserted two 4C peptide domains into regions of the C-terminus where we suspected additions would have little or no impact on trafficking or phosphorylation. These insertions were after N308 and D336 in areas of the C-terminus several amino acids up- and downstream from known phosphorylated serines or tyrosines or known binding partner domains, and on either side of a putative alpha-helix (49). However, for unknown reasons, the C-terminus neighborhoods surrounding each tetracysteine domain must have slightly different local conformations or accessibilities resulting in different biarsenical binding affinities. The increase in fluorescence with two 4C segments is unlikely to be due to a cooperativity effect since these two 4C domains are far enough apart that they should act independently. If they did interact, we most likely would have observed a decrease in fluorescence from quenching (50). While we do see differences in the signal strength and variations of mobility via immunoblotting, both protein trafficking and phosphorylation during mitosis appears the same for Cx43-WT, Cx43-4C309/337 or Cx43-GFP-4C when each is stably expressed in MDCK cells. The fact that we could insert small tags into the C-terminus and maintain normal trafficking, function and ZO-1 binding verifies the highly flexible and non-globular nature of the carboxy terminus, confirming that any ordered structure within the C-terminus is made from short stretches of primary sequence and may be transient allowing for the binding of cytoplasmic partners (51, 52). What still needs to be investigated is the molecular control switches that regulate these processes.
Materials and Methods
Plasmids and Mutagenesis
After determining that this construct did not yield sufficient fluorescence with ReAsH labeling protocol for live-cell imaging and photo-oxidation, a second 4C sequence was inserted. To do this, a DNA fragment corresponding to Q304ASEQNFLNCCPGCCMEPWANY313 was synthesized. After PCR of a construct representing the amino acids AQ3 1–308, this product and the synthetic fragment were digested with Nhe1, gel purified and ligated together. Following purification of this product, it was ligated to a PCR fragment representing the rest of the C-terminal end of Cx43 at the Hha1 site. The resulting sequence full-length Cx43 containing the two 4C inserts was verified and inserted into a pIREShygro vector using BamH1 and Not1 restriction sites.
Cell Culture and Transfections
Normal rat kidney (NRK), RAT-1 fibroblasts and Madin-Darby canine kidney (MDCK) cells were maintained at 37°C, and 10% CO2 in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (GIBCO-BRL, Invitrogen, Carlsbad, CA). Transductions were carried out using a retroviral system according to the protocols from the Nolan laboratory (www.stanford.edu/group/nolan) as we have previously published (27). Experiments were conducted on endogenous expressing or stably Cx43-expressing cell lines generated by transduction followed by selection with the antibiotic hygromycin (Gibco-BRL, Invitrogen).
Antibodies
For immunofluorescence we used the following primary antibodies: rabbit polyclonal anti-Cx43 (1:400, Sigma, St. Louis, MO, Cat.# C6219); mouse monoclonal anti-Cx43 (1:250, Chemicon, Temecula, CA, Cat.# MAB3067); rabbit polyclonal anti-giantin (1:100, Abcam, Cambridge, MA, Cat.# ab24586); mouse monoclonal anti-EEA1 (1:250, BD Biosciences, San Jose, CA, Cat.# 610456); rabbit polyclonal anti-Lamp1 (1:250, Abcam, Cambridge, MA, Cat.# ab24170); mouse monoclonal anti-ZO-1 (1:1000, Invitrogen, Carlsbad, Cat. # 339100); mouse monoclonal anti-alpha tubulin (1:250, Sigma, St. Louis, MO, Cat.# T6074); mouse monoclonal anti- GM130 (1:200, BD Biosciences, San Jose, CA, Cat.# 610823). For co-immunoprecipitation mouse monoclonal antibodies, IF1 and CT1 (both isotype IgG2a) (Fred Hutchinson Cancer Research Center Hybridoma Facility, Seattle, WA, described in (43)) were used. For immunoblotting: mouse monoclonal anti-Cx43 (NT1) (1:1000, Fred Hutchinson Cancer Research Center Hybridoma Facility, Seattle, WA, described in (43)) and rabbit polyclonal anti-pS255 Cx43 and anti-pS262 (Santa Cruz Biotechnology, Inc, Santa Cruz, CA, Cat.# sc-12899-R and sc-22267-R, respectively).
Immunocytochemistry and Confocal Microscopy
Cells were grown on poly-D-lysine–coated glass coverslips, fixed in 4% paraformaldehyde/phosphate buffered saline for 20 min, washed, and labeled for immunofluorescence. Primary antibodies were mixed and diluted with blocking buffer diluted five fold in phosphate buffered saline (PBS). The secondary antibodies were diluted 1:100 in the same buffer. Data acquisition was done with an Olympus FluoView1000 laser-scanning confocal microscope (Olympus, Center Valley, PA), with a 60X1.42 NA lens. Image analysis of z-scan was done using the Imaris Software (Bitplane AG, St. Paul, MN) and the Image J software (rsb.info.nih.gov/ij). The usual color table for some images was inverted with a continuous gray scale but black represents strong fluorescence and white represents no fluorescence (e.g. background), allowing better discrimination of punctate expression patterns. The Pearson Coefficient, a measure of the correlation between two variables, was calculated using the Image J software for quantification of degree of overlap for various stainings.
Gel Electrophoresis and Immunoblotting
Whole cell lysates were prepared in sample buffer containing 50 mM NaF, 500 μM Na3VO4, 2 mM PMSF, and 1x complete protease inhibitor (Roche), sonicated and cellular proteins were separated by SDS-PAGE (10% polyacrylamide). For co-immunoprecipitation, cells were lysed and Cx43 was immunoprecipitated as previously described (53). Protein was transferred to nitrocellulose, the membrane was blocked, and antibodies were incubated as previously indicated (54). For co-immunoprecipitation, the blot was cut into a higher (>100 kDa) and lower piece for incubation with ZO-1 and Cx43 antibodies, respectively. pS255 and pS262 were detected with IRDye800 donkey anti–rabbit (Rockland Immunochemicals, Gilbertsville, PA) and total Cx43 (NT1) and ZO-1 were detected with IR800 rabbit anti–mouse IgG1 (Rockland Immunochemicals, Gilbertsville, PA) antibodies (all extensively cross-reacted against other species) and directly quantified using the Li-Cor Biosciences Odyssey infrared imaging system and associated software. Images were converted from 16-bit to 8-bit images after maximizing the dynamic range of pixel intensity using the “Levels” function in Adobe Photoshop. Images were inverted to present them in the conventional black-on-white manner.
Optical In Vivo pulse-chase labeling with ReAsH-EDT2 and FlAsH-EDT2
MDCK cells stably expressing two internal 4C domains in the C-terminus (FLNCCPGCCME)–tagged Cx43 (Cx43-4C309/337) were labeled for 1 h at 37°C with 180 nM ReAsH-EDT2/12.5 μM EDT in Hanks’ Balanced Salt Saline (HBSS). Free and nonspecifically bound ReAsH was removed by washing with 2,3-Dimercapto-1-Propanol (BAL, 500 μM, 20 min at 37°C in HBSS). Cells were then incubated for 3 h (chase time) in the presence of complete medium at 37°C. The newly synthesized recombinant proteins were stained at the end of the chase time with the second label (1.2 μM FlAsH-EDT2). After washing with BAL (250 μM, 20 min at 37°C in HBSS), cells were fixed with 4% paraformaldehyde or methanol for light microscopic imaging as previously described (27, 28, 30).
Photoconversion of Diaminobenzidine and Electron Microscopy
After 20 min fixation, cells were rinsed in cacodylate buffer (0.1 M, pH 7.4) and blocked for 30 min on ice with 10 mM KCN, 20 mM aminotrizole, 0.01 % hydrogen peroxide, 50mM glycine in cacodylate buffer (0.1 M, pH 7.4) to reduce nonspecific background. For photo-oxidation, DAB (1mg/ml) in oxygenated cacodylate buffer was added to the plate and cells were irradiated with 585nm light from a Xenon lamp (without neutral density filters) for 8–12 minutes until a brownish reaction product appeared in place of the red fluorescence due to ReAsH. For FRET-based fluorescent photo-oxidation, GFP was excited at 450–490 nm. EM preparation and imaging were performed as previously described (27).
Live cell imaging
Time-lapse imaging was conducted using an Olympus FluoView1000 confocal microscope equipped with a temperature-controlled chamber (at 37°C) and a 60X1.42 NA objective. Medium: Opti-MEM supplemented with 5% FBS covered with a glass coverslip. For Supplemental movie1 the GFP fluorescence was collected, 488 nm excitation/494–535 nm emission. Images were recorded every 6 minutes for a total duration of 4 hrs and 23 minutes, 0.21 μm/pixel, image size 1024X1024 pixels in 21 z stacks (step size 0.5 μm), 12bits/pixel, sampling speed 2μs/pixel. For Supplemental movie2 both the FlasH fluorescence and the differential interference contrast were collected, 515 nm excitation/530–630 nm emission. Images were recorded every 6 minutes for a total duration of about 5 hours, 0.276 μm/pixel, image size 512X512 pixels in 11 z stacks (step size 1 μm), 12 bits/pixel, sampling speed 2 μs/pixel. For Supplemental movie3 both the ReAsH fluorescence and the differential interference contrast were collected, 568 nm excitation/580–600 nm emission. Images were recorded every 5 minutes for a total duration of about 4 hours, 0.207 μm/pixel, image size 512X512 pixels in 13 z stacks (step size 1 μm), 12 bits/pixel, sampling speed 2 μs/pixel. Image processing such as 3D volume rendering and 4D movie generation was done with the software Imaris 6 (Bitplane AG, Zurich, Switzerland). Full resolution movies and images are available for downloading from the Cell Centered Database (55) (http://ccdb.ucsd.edu/CCDBWebSite/index.html) under project ID number P2076.
Supplementary Material
Synchronized populations of MDCK cells expressing Cx43-GFP-4C reveal the dynamic re-distribution of Cx43 during mitosis. Images (1024 X 1024 pixels) of GFP fluorescence (488 nm excitation/494–535 nm emission) were recorded every 6 minutes for a total duration of 4 hours and 23 minutes. 3D volume reconstructions from confocal image stacks over time are displayed (two frames per minute).
Time-lapse imaging of FlAsH-labeled MDCK cells expressing Cx43-4C309/337 showing their re-arrangement and fate during and after mitosis. Images (512 X 512 pixels) of FlAsH fluorescence (515 nm excitation/530–630 nm emission) as well as the differential interference contrast images were recorded every 6 minutes for a total duration of about 5 hours. 3D volume reconstructions from confocal image stacks over time are displayed (two frames per minute).
Time-lapse imaging of ReAsH-labeled MDCK cells expressing Cx43-4C309/337 showing their re-arrangement and fate during and after mitosis. Images (512 × 512 pixels) of ReAsH fluorescence (568 nm excitation/580–600 nm emission, pseudo-colored in green) as well as the differential interference contrast images were recorded every 5 minutes for a total duration of about 4 hours. 3D volume reconstructions from confocal image stacks over time are displayed (two frames per minute).
Acknowledgments
We thank Ohkyung Kwon and Masako Terada for their assistance with the data processing of the time-lapse movies. Funding for this research was contributed by National Institutes of Health grants GM55632 (PDL), GM072881 (GES) and GM065937 (GES) and National Institutes of Health Grant RR04050 supports the National Center for Microscopy and Imaging Research at San Diego (Mark Ellisman). The CCDB is supported by National Institutes of Health grant DA016602 (Maryann Martone).
Footnotes
Supplemental information. Full resolution 4D movies (5 MB Supplemental Movie1 and 5 MB Supplemental Movie2), original image data and metadata are available from the Cell Centered Database (CCDB) under Project ID P2076 (http://ccdb.ucsd.edu/CCDBWebSite/index.html).
References
- 1.Sosinsky G, Gaietta G, Giepmans B. Gap junction morphology and dynamics in situ. In: Harris ALD, editor. Connexins: A Guide. Totowa, NJ: Humana Press; 2008. pp. 241–261. [Google Scholar]
- 2.White T, Paul D. Genetic diseases and gene knockouts reveal diverse connexin functions. Ann Rev Physiology. 1999;61:283–310. doi: 10.1146/annurev.physiol.61.1.283. [DOI] [PubMed] [Google Scholar]
- 3.Sohl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res. 2004;62(2):228–232. doi: 10.1016/j.cardiores.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 4.Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83(4):1359–1400. doi: 10.1152/physrev.00007.2003. [DOI] [PubMed] [Google Scholar]
- 5.Laird DW. Life cycle of connexins in health and disease. Biochem J. 2006;394(Pt 3):527–543. doi: 10.1042/BJ20051922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, Kleber AG, Schuessler RB, Saffitz JE. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87(8):656–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 7.Schulz R, Heusch G. Connexin 43 and ischemic preconditioning. Cardiovasc Res. 2004;62(2):335–344. doi: 10.1016/j.cardiores.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 8.Naus CC, Laird DW. Implications and challenges of connexin connections to cancer. Nat Rev Cancer. 2010;10(6):435–441. doi: 10.1038/nrc2841. [DOI] [PubMed] [Google Scholar]
- 9.Lampe PD. Analyzing phorbol ester effects on gap junctional communication: a dramatic inhibition of assembly. J Cell Biol. 1994;127(6 Pt 2):1895–1905. doi: 10.1083/jcb.127.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lau AF, Kanemitsu MY, Kurata WE, Danesh S, Boynton AL. Epidermal growth factor disrupts gap-junctional communication and induces phosphorylation of connexin43 on serine. Mol Biol Cell. 1992;3(8):865–874. doi: 10.1091/mbc.3.8.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laird DW, Puranam KL, Revel JP. Turnover and phosphorylation dynamics of connexin43 gap junction protein in cultured cardiac myocytes. Biochem J. 1991;273(Pt 1):67–72. doi: 10.1042/bj2730067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Musil LS, Le AC, VanSlyke JK, Roberts LM. Regulation of connexin degradation as a mechanism to increase gap junction assembly and function. J Biol Chem. 2000;275(33):25207–25215. doi: 10.1074/jbc.275.33.25207. [DOI] [PubMed] [Google Scholar]
- 13.Lampe PD, Lau AF. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol. 2004;36(7):1171–1186. doi: 10.1016/S1357-2725(03)00264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lau AF, Kurata WE, Kanemitsu MY, Loo LW, Warn-Cramer BJ, Eckhart W, Lampe PD. Regulation of connexin43 function by activated tyrosine protein kinases. J Bioenerg Biomembr. 1996;28(4):359–368. doi: 10.1007/BF02110112. [DOI] [PubMed] [Google Scholar]
- 15.Solan JL, Lampe PD. Connexin phosphorylation as a regulatory event linked to gap junction channel assembly. Biochim Biophys Acta. 2005;1711(2):154–163. doi: 10.1016/j.bbamem.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 16.Musil LS, Goodenough DA. Biochemical analysis of connexin43 intracellular transport, phosphorylation and assembly into gap junctional plaques. J Cell Biol. 1991;115:1357–1374. doi: 10.1083/jcb.115.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lampe PD, Kurata WE, Warn-Cramer BJ, Lau AF. Formation of a distinct connexin43 phosphoisoform in mitotic cells is dependent upon p34cdc2 kinase. J Cell Sci. 1998;111 ( Pt 6):833–841. doi: 10.1242/jcs.111.6.833. [DOI] [PubMed] [Google Scholar]
- 18.Kanemitsu MY, Jiang W, Eckhart W. Cdc2-mediated phosphorylation of the gap junction protein, connexin43, during mitosis. Cell Growth Differ. 1998;9(1):13–21. [PubMed] [Google Scholar]
- 19.Xie H, Laird DW, Chang TH, Hu VW. A mitosis-specific phosphorylation of the gap junction protein connexin43 in human vascular cells: biochemical characterization and localization. J Cell Biol. 1997;137(1):203–210. doi: 10.1083/jcb.137.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stein LS, Boonstra J, Burghardt RC. Reduced cell-cell communication between mitotic and nonmitotic coupled cells. Exp Cell Res. 1992;198(1):1–7. doi: 10.1016/0014-4827(92)90141-t. [DOI] [PubMed] [Google Scholar]
- 21.Shorter J, Warren G. Golgi architecture and inheritance. Annu Rev Cell Dev Biol. 2002;18:379–420. doi: 10.1146/annurev.cellbio.18.030602.133733. [DOI] [PubMed] [Google Scholar]
- 22.Preisinger C, Korner R, Wind M, Lehmann WD, Kopajtich R, Barr FA. Plk1 docking to GRASP65 phosphorylated by Cdk1 suggests a mechanism for Golgi checkpoint signalling. EMBO J. 2005;24(4):753–765. doi: 10.1038/sj.emboj.7600569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419(2):261–272. doi: 10.1042/BJ20082319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solan JL, Fry MD, TenBroek EM, Lampe PD. Connexin43 phosphorylation at S368 is acute during S and G2/M and in response to protein kinase C activation. J Cell Sci. 2003;116(Pt 11):2203–2211. doi: 10.1242/jcs.00428. [DOI] [PubMed] [Google Scholar]
- 25.Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312(5771):217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- 26.Martin BR, Giepmans BN, Adams SR, Tsien RY. Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat Biotechnol. 2005;23(10):1308–1314. doi: 10.1038/nbt1136. [DOI] [PubMed] [Google Scholar]
- 27.Boassa D, Ambrosi C, Qiu F, Dahl G, Gaietta G, Sosinsky G. Pannexin1 channels contain a glycosylation site that targets the hexamer to the plasma membrane. J Biol Chem. 2007;282(43):31733–31743. doi: 10.1074/jbc.M702422200. [DOI] [PubMed] [Google Scholar]
- 28.Boassa D, Qiu F, Dahl G, Sosinsky G. Trafficking dynamics of glycosylated pannexin 1 proteins. Cell Commun Adhes. 2008;15(1):119–132. doi: 10.1080/15419060802013885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ju W, Morishita W, Tsui J, Gaietta G, Deerinck TJ, Adams SR, Garner CC, Tsien RY, Ellisman MH, Malenka RC. Activity-dependent regulation of dendritic synthesis and trafficking of AMPA receptors. Nat Neurosci. 2004;7(3):244–253. doi: 10.1038/nn1189. [DOI] [PubMed] [Google Scholar]
- 30.Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296(5567):503–507. doi: 10.1126/science.1068793. [DOI] [PubMed] [Google Scholar]
- 31.Shen MR, Chou CY, Ellory JC. Swelling-activated taurine and K+ transport in human cervical cancer cells: association with cell cycle progression. Pflugers Arch. 2001;441(6):787–795. doi: 10.1007/s004240000476. [DOI] [PubMed] [Google Scholar]
- 32.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J Am Chem Soc. 2002;124(21):6063–6076. doi: 10.1021/ja017687n. [DOI] [PubMed] [Google Scholar]
- 33.Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16(12):5686–5698. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hunter AW, Jourdan J, Gourdie RG. Fusion of GFP to the carboxyl terminus of connexin43 increases gap junction size in HeLa cells. Cell Commun Adhes. 2003;10(4–6):211–214. doi: 10.1080/cac.10.4-6.211.214. [DOI] [PubMed] [Google Scholar]
- 35.Lauf U, Giepmans BN, Lopez P, Braconnot S, Chen SC, Falk MM. Dynamic trafficking and delivery of connexons to the plasma membrane and accretion to gap junctions in living cells. Proc Natl Acad Sci U S A. 2002;99(16):10446–10451. doi: 10.1073/pnas.162055899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ambrosi C, Boassa D, Pranskevich J, Smock A, Oshima A, Xu J, Nicholson BJ, Sosinsky GE. Analysis of four connexin26 mutant gap junctions and hemichannels reveals variations in hexamer stability. Biophys J. 2010;98(9):1809–1819. doi: 10.1016/j.bpj.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boucrot E, Kirchhausen T. Endosomal recycling controls plasma membrane area during mitosis. Proc Natl Acad Sci U S A. 2007;104(19):7939–7944. doi: 10.1073/pnas.0702511104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braun J, Abney JR, Owicki JC. How a gap junction maintains its structure. Nature. 1984;310(5975):316–318. doi: 10.1038/310316a0. [DOI] [PubMed] [Google Scholar]
- 39.Bauer T, Motosugi N, Miura K, Sabe H, Hiiragi T. Dynamic rearrangement of surface proteins is essential for cytokinesis. Genesis. 2008;46(3):152–162. doi: 10.1002/dvg.20377. [DOI] [PubMed] [Google Scholar]
- 40.Yonemura S, Nagafuchi A, Sato N, Tsukita S. Concentration of an integral membrane protein, CD43 (leukosialin, sialophorin), in the cleavage furrow through the interaction of its cytoplasmic domain with actin-based cytoskeletons. J Cell Biol. 1993;120(2):437–449. doi: 10.1083/jcb.120.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang YL. The mechanism of cortical ingression during early cytokinesis: thinking beyond the contractile ring hypothesis. Trends Cell Biol. 2005;15(11):581–588. doi: 10.1016/j.tcb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 42.Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol. 2006;7(1):9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 43.Solan JL, Lampe PD. Key connexin 43 phosphorylation events regulate the gap junction life cycle. J Membr Biol. 2007;217(1–3):35–41. doi: 10.1007/s00232-007-9035-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coleman BM, Nisbet RM, Han S, Cappai R, Hatters DM, Hill AF. Conformational detection of prion protein with biarsenical labeling and FlAsH fluorescence. Biochem Biophys Res Commun. 2009;380(3):564–568. doi: 10.1016/j.bbrc.2009.01.120. [DOI] [PubMed] [Google Scholar]
- 45.Gousset K, Ablan SD, Coren LV, Ono A, Soheilian F, Nagashima K, Ott DE, Freed EO. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 2008;4(3):e1000015. doi: 10.1371/journal.ppat.1000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Erster O, Eisenstein M, Liscovitch M. Ligand interaction scan: a general method for engineering ligand-sensitive protein alleles. Nat Methods. 2007;4(5):393–395. doi: 10.1038/nmeth1046. [DOI] [PubMed] [Google Scholar]
- 47.Hoffmann C, Gaietta G, Bunemann M, Adams SR, Oberdorff-Maass S, Behr B, Vilardaga JP, Tsien RY, Ellisman MH, Lohse MJ. A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat Methods. 2005;2(3):171–176. doi: 10.1038/nmeth742. [DOI] [PubMed] [Google Scholar]
- 48.Sosinsky GE, Solan JL, Gaietta GM, Ngan L, Lee GJ, Mackey MR, Lampe PD. The C-terminus of connexin43 adopts different conformations in the Golgi and gap junction as detected with structure-specific antibodies. Biochem J. 2007;408(3):375–385. doi: 10.1042/BJ20070550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sorgen PL, Duffy HS, Spray DC, Delmar M. pH-dependent dimerization of the carboxyl terminal domain of Cx43. Biophys J. 2004;87(1):574–581. doi: 10.1529/biophysj.103.039230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Engelenburg SB, Nahreini T, Palmer AE. FACS-Based Selection of Tandem Tetracysteine Peptides with Improved ReAsH Brightness in Live Cells. Chembiochem. 2010;11:489–93. doi: 10.1002/cbic.200900689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sorgen PL, Duffy HS, Sahoo P, Coombs W, Delmar M, Spray DC. Structural changes in the carboxyl terminus of the gap junction protein connexin43 indicates signaling between binding domains for c-Src and zonula occludens-1. J Biol Chem. 2004;279(52):54695–54701. doi: 10.1074/jbc.M409552200. [DOI] [PubMed] [Google Scholar]
- 52.Sosinsky GE, Nicholson BJ. Structural organization of gap junction channels. Biochim Biophys Acta. 2005;1711(2):99–125. doi: 10.1016/j.bbamem.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 53.Singh D, Solan JL, Taffet SM, Javier R, Lampe PD. Connexin 43 interacts with zona occludens-1 and -2 proteins in a cell cycle stage-specific manner. J Biol Chem. 2005;280(34):30416–30421. doi: 10.1074/jbc.M506799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of Connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119(Pt 16):3435–3442. doi: 10.1242/jcs.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martone ME, Gupta A, Wong M, Qian X, Sosinsky G, Ludascher B, Ellisman MH. A cell-centered database for electron tomographic data. J Struct Biol. 2002;138(1–2):145–155. doi: 10.1016/s1047-8477(02)00006-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synchronized populations of MDCK cells expressing Cx43-GFP-4C reveal the dynamic re-distribution of Cx43 during mitosis. Images (1024 X 1024 pixels) of GFP fluorescence (488 nm excitation/494–535 nm emission) were recorded every 6 minutes for a total duration of 4 hours and 23 minutes. 3D volume reconstructions from confocal image stacks over time are displayed (two frames per minute).
Time-lapse imaging of FlAsH-labeled MDCK cells expressing Cx43-4C309/337 showing their re-arrangement and fate during and after mitosis. Images (512 X 512 pixels) of FlAsH fluorescence (515 nm excitation/530–630 nm emission) as well as the differential interference contrast images were recorded every 6 minutes for a total duration of about 5 hours. 3D volume reconstructions from confocal image stacks over time are displayed (two frames per minute).
Time-lapse imaging of ReAsH-labeled MDCK cells expressing Cx43-4C309/337 showing their re-arrangement and fate during and after mitosis. Images (512 × 512 pixels) of ReAsH fluorescence (568 nm excitation/580–600 nm emission, pseudo-colored in green) as well as the differential interference contrast images were recorded every 5 minutes for a total duration of about 4 hours. 3D volume reconstructions from confocal image stacks over time are displayed (two frames per minute).