

Abstract
Cortical spreading depression (CSD) is associated with severe hypoperfusion in mice. Using minimally invasive multimodal optical imaging, we show that severe flow reductions during and after spreading depression are associated with a steep decline in cerebral metabolic rate of oxygen. Concurrent severe hemoglobin desaturation suggests that the oxygen metabolism becomes at least in part supply limited, and the decrease in cortical blood volume implicates vasoconstriction as the mechanism. In support of oxygen supply-demand mismatch, cortical nicotinamide adenine dinucleotide (NADH) fluorescence increases during spreading depression for at least 5 minutes, particularly away from parenchymal arterioles. However, modeling of tissue oxygen delivery shows that cerebral metabolic rate of oxygen drops more than predicted by a purely supply-limited model, raising the possibility of a concurrent reduction in oxygen demand during spreading depression. Importantly, a subsequent spreading depression triggered within 15 minutes evokes a monophasic flow increase superimposed on the oligemic baseline, which markedly differs from the response to the preceding spreading depression triggered in naive cortex. Altogether, these data suggest that CSD is associated with long-lasting oxygen supply-demand mismatch linked to severe vasoconstriction in mice.
Keywords: cerebral metabolic rate of oxygen, laser speckle flowmetry, mouse, multispectral reflectance imaging, spreading depression
Introduction
Cortical spreading depression (CSD) is a wave of neuronal and glial depolarization characterized by transient loss of transmembrane ionic gradients (Leao, 1944). The massive rise in extracellular K+ and glutamate concentrations, usually lasting less than a minute, is believed to be critical for the contiguous spread in gray matter at a rate of ∼3 mm/min. A large body of evidence suggests that CSD is the electrophysiological substrate of migraine aura, and may trigger headache as well (Ayata, 2010). Underscoring the pathophysiological importance of CSD in diverse neurologic disorders, CSD-like depolarization waves have been detected with high frequency in ischemic stroke, trauma and intracranial hemorrhage, and aggravate tissue injury (Dohmen et al, 2008; Dreier et al, 2006; Hashemi et al, 2009; Strong et al, 2007).
In most species, CSD evokes a large hyperemic response coupled to a marked stimulation of oxidative metabolism. In mice, however, CSD causes severe vasoconstriction, during which cerebral blood flow (CBF) and oxygenation decrease to levels that can be considered ischemic (Ayata et al, 2004b; Brennan et al, 2007; Chang et al, 2010). Recently, CSD was shown to be associated with a large and spatially heterogeneous increase in cortical nicotinamide adenine dinucleotide (NADH) levels in mice. However, these changes appeared to occur in the absence of detectable vasoconstriction in mice (Takano et al, 2007), implicating increased O2 demand as the primary mechanism for the mismatch.
Notably, most studies have utilized invasive techniques and instrumentation, such as cranial windows and implanted electrodes for pO2 and electrophysiological recordings, which are known to alter the cortical physiology and the vascular response to CSD (Chang et al, 2010). We, therefore, employed minimally invasive full-field laser speckle flowmetry and multispectral reflectance imaging through intact skull to investigate the impact of CSD on CBF and volume, oxyhemoglobin (oxyHb) and deoxyhemoglobin (deoxyHb) concentrations, hemoglobin saturation, and cerebral metabolic rate of O2 (CMRO2) simultaneously in mouse cortex, and used them to gain insight into tissue oxygen supply-demand mismatch during CSD (Gjedde et al, 2005). In addition, to further assess the cellular redox state as a measure of relative rates of glycolytic and oxidative metabolism and O2 supply-demand mismatch, we imaged changes in NADH fluorescence using two-photon microscopy through a closed cranial window. Data show that profound hemoglobin desaturation and NADH increase accompanies vasoconstriction and severe hypoperfusion both during and after CSD, that O2 metabolism becomes at least in part supply limited, and that the response to CSD in naive cortex dramatically differs from cortex previously exposed to a CSD.
Materials and methods
The care and handling of the animals and experimental protocols were in accordance with the National Institutes of Health guidelines and approved by the institutional animal care and use committee.
Multispectral Reflectance and Laser Speckle Imaging
General surgical preparation
Wild-type (C57BL/6J, n=5 males and 6 females) mice were anesthetized (1% isoflurane in 70% N2O plus 30% O2), intubated, and mechanically ventilated (SAR830, CWE). Arterial blood pressure (78±9 mm Hg) was monitored continuously, and arterial blood gas and pH every 15 minutes (pH 7.33±0.04, pCO2 35±4 mm Hg, pO2 159±5 mm Hg) via a femoral artery catheter. Rectal temperature was monitored and maintained at 37°C via a servo-controlled heating pad. Mice were placed on a stereotaxic frame, scalp reflected after a midline incision, and intact skull overlying the right hemisphere was covered with a thin layer of mineral oil to prevent drying and enhance transparency. A 1-mm burr hole was drilled under saline cooling over the frontal cortex (2 mm anterior, 1 mm lateral from bregma). Absence of a mechanical CSD induction during drilling was confirmed by laser speckle flowmetry monitoring.
Imaging system
The multimodal optical imaging system provides simultaneous laser speckle flowmetry and multispectral reflectance imaging as previously described (Ayata et al, 2004a; Dunn et al, 2003; Jones et al, 2008). Laser speckle imaging of CBF was performed using a near infrared laser diode (785 nm, 75 mW) and a CCD camera (Cohu 4600, San Diego, CA, USA, 640 × 480 pixels). Light for multispectral reflectance imaging was provided by a quartz tungsten halogen fiber-optic illuminator (Techniquip R150; Capra Optical, Natick, MA, USA). Before illuminating the sample, light was guided through a filter wheel with six 10-nm-wide bandpass filters (560 to 610 nm) and the diffuse reflectance images were captured by a second CCD camera (Coolsnap fx; Roper Scientific, Tucson, AZ, USA; 1,300 × 1,030 pixels with 3 × 3 binning, resulting in 434 × 343 image size for multispectral reflectance). The illumination light for both imaging modalities was delivered to the sample with an oblique angle, collected by a variable magnification objective ( × 0.75 to × 3; Edmund Optics, Barrington, NJ, USA), split by a dichroic mirror, and acquired simultaneously by CCD cameras. The final imaging field was positioned over the right hemisphere (7 × 6 mm2; Figure 1). Raw speckle frames were continuously acquired at 2.5 Hz, and multispectral reflectance image frames at the filter wheel rotation frequency of 1.6 Hz. The data were subsequently interpolated to the common time base using the recorded filter wheel angular positions and exposure times of both cameras.
Figure 1.

Experimental design. (A) Imaging field (7 × 6 mm2) was positioned over the right hemisphere. Cortical spreading depression (CSD) was induced by epidural KCl (300 mmol/L) application through a small burr hole overlying the prefrontal cortex (circle). Hemodynamic and metabolic changes were quantified by placing regions of interest (ROI; 0.25 mm2 squares) within the middle or anterior cerebral artery (MCA and ACA) territories. (B) A typical oxyhemoglobin (oxyHb) tracing is shown to demonstrate the method of measurement of hemodynamic and metabolic changes during CSD. The abrupt ‘onset' of hypoperfusion response was taken as time 0. Blue circles indicate the four deflection points defining the multiphasic portion of the response. Both the magnitude of these deflection points, and their latency from response onset were measured in each mouse (e.g., horizontal and vertical dotted lines shown for ‘b'), averaged within each experimental group, and plotted along with additional fixed time points (baseline, and 3, 5, 7, 9, and 11 minutes after response onset, red circles) to reconstruct the waveform.
Image analysis
Absolute values of parenchymal oxyHb and deoxyHb were obtained using an algorithm based on nonlinear least square fitting of multimodal optical parameters with assumed baseline oxyHb and deoxyHb concentrations (60 and 40 μmol/L, respectively) and scattering coefficient of 150/cm (Jones et al, 2008). Relative CBF and absolute tissue oxyHb and deoxyHb concentrations were then used to calculate hemoglobin O2 saturation (satHb=oxyHb/(oxyHb+deoxyHb)), and changes in cerebral blood volume (ΔCBV ≈ΔoxyHb+ΔdeoxyHb) and cerebral metabolic rate of O2 (ΔCMRO2=ΔCBF × ΔOEF). Relative oxygen extraction fraction (ΔOEF) was estimated assuming that venous satHb is equal to satHb in parenchyma and that arterial satHb is equal to one, as previously described (Dunn et al, 2005; Jones et al, 2008). The time courses of CBF, CBV, oxyHb, deoxyHb, SatHb, and CMRO2 were quantified using two regions of interest (ROI; 0.25 mm2) placed within the middle and anterior cerebral artery (MCA and ACA) territories avoiding visible pial vessels (Figure 1A, squares). To faithfully represent the multiphasic changes during and after a CSD, we measured and averaged across animals the magnitudes and latencies of the following deflection points (Figure 1B, circles, oxyHb shown as an example): baseline, the onset of hypoperfusion, trough (a), transient normalization (b), second trough, second rise (c), and 3, 5, 7, 9, and 11 minutes after CSD onset. We then interpolated the averaged deflection points to reconstruct the response to a CSD.
Experimental protocol
After surgical preparation, multimodal optical imaging was started and continued uninterrupted for a total of 30 minutes. After baseline imaging for 2 minutes, a CSD was induced by briefly touching a 1-mm diameter cotton ball soaked in 300 mmol/L KCl solution on the prefrontal cortex through the burr hole, immediately followed by careful saline wash. Fifteen minutes later, this procedure was repeated to trigger a second CSD.
Two-Photon Fluorescence Imaging
For cranial window preparation, mice (C57BL/6J, n=7 females) were anesthetized with isoflurane (1% in 70% N2O plus 30% O2) through a nose cone and placed on a stereotaxic frame. After shaving and laterally reflecting the scalp, a midline craniectomy 5 mm in diameter was carefully drilled under saline cooling to remove most of the frontal and parietal bones bilaterally. A closed cranial window was constructed using a glass cover slip, filled with phosphate-buffered saline, and sealed with dental cement. Two small occipital burr holes were drilled posterolaterally one on each hemisphere for CSD induction. After window preparation, mice were allowed to rest for >1 hour to allow for the recovery of cerebrovascular effects of any accidentally induced CSD during drilling. Arterial blood gases were monitored during two-photon imaging (pH 7.33±0.02, pCO2 43±3 mm Hg, pO2 160±17 mm Hg).
Two-photon imaging was performed after intravenous injection of fluorescein isothiocyanate (FITC)-conjugated dextran using a commercial four-channel two-photon microscope (Prairie Technologies, Middleton, WI, USA). A × 20 objective (Olympus XLUMPLFL20XW/IR-SP, 0.95 NA, 2 mm working distance, Center Valley, PA, USA) was used. Images consisting of 512 × 512 pixels were acquired over a field of view of either 466 × 466 μm2 or 233 × 233 μm2 during passage of the CSD. The estimated thickness of the image plane was a few microns. The frame rate was 0.74 frames/s. The laser (Spectra Physics Mai Tai HP, Irvine, CA, USA) wavelength was set to 740 nm to excite both NADH and FITC-conjugated dextran. NADH was detected using a filter with a 460-nm center wavelength and a bandwidth of 50 nm. FITC-dextran was detected using a filter with a 525-nm center wavelength and a bandwidth of 50 nm.
The imaging plane was within 50 μm of the cortical surface, and placed within the MCA–ACA watershed zone. The large diameter of cranial window allowed the imaging field to be placed on either hemisphere. Because CSD never propagates into the contralateral cortex (Eikermann-Haerter et al, 2011), we studied one CSD in each hemisphere in succession. In a subset of mice with normal systemic physiology after the completion of imaging in the second hemisphere, we returned to the first hemisphere and repeated the protocol one more time, always allowing >1 hour between the two CSDs in the same hemisphere.
NADH signal changes were quantified in the capillary bed (ROI at least 50 μm away from large vessels) and in the vicinity of parenchymal penetrating arterioles (ROI within 50 μm of the vessel wall). Relative NADH signal time courses were computed by normalizing the integrated signal at each time point within the ROI to the integrated signal within the ROI averaged over all images before CSD arrival. Both capillary and periarteriolar ROIs avoided any shadows casted by large vessels above the imaging plane, as well as the occasional cortical regions with sharply demarcated high baseline NADH signal; CSD-induced NADH changes in the latter were also measured using separate ROIs. When present in the imaging field, changes in arteriolar diameter were also measured and temporally coregistered with NADH changes. Before display, images were averaged spatially by convolution with a two-dimensional Gaussian kernel with a standard deviation of ∼2.5 μm. Temporal averaging of three consecutive images was used for all movies. For figures (Figures 5 and 6) in the main text, temporal averaging was performed as indicated. For overlaying vasculature on NADH images, a transparency mask was constructed at each time point based on the FITC-dextran fluorescence images. Relative NADH signal change images were obtained by normalizing each NADH image to the average of all images before CSD arrival. Absence of significant brain movement in or out of the imaging plane was confirmed by observing the capillaries.
Statistical Analysis
One- or two-way repeated measures analysis of variance was used to compare the time course of each measured parameter within and between groups. Data are presented as mean±standard error.
Results
Epidural application of KCl onto the frontal cortex triggered a CSD in all mice that propagated throughout the ipsilateral cortex, but never spread to the contralateral hemisphere. In the MCA territory, all measured parameters showed a multiphasic response to CSD (Figures 2 and 3; Supplementary Movies 1 and 2), which started with an abrupt and profound decrease in CBF to 31%±3% of baseline (Figure 3Ba). Coupled to the hypoperfusion, oxyHb decreased to 39%±5% of baseline, whereas deoxyHb showed little change, presumably because of the reduction in total Hb concentration (i.e., CBV) to 64%±3% of baseline offsetting the expected increase in deoxyHb. As a result, satHb was reduced to 59%±6% of baseline, corresponding to a 62% increase in OEF, suggesting a mismatch between O2 supply and demand. Concurrent with the hemoglobin desaturation, CMRO2 decreased to 51%±3% of baseline. At the temporal resolution of our imaging system (1.6 Hz), we did not detect an increase in any of the measured parameters preceding the onset of hypoperfusion (Figure 3B, inset). The vasoconstrictive phase reached a nadir 20 to 25 seconds after its onset, followed by a transient recovery peaking within 1 minute (Figure 3Bb). A second smaller rise appeared ∼2 minutes after the CSD onset (Figure 3Bc). This was followed by a longer lasting vasoconstrictive phase (i.e., post-CSD oligemia; Figure 3Bd), during which all measured parameters except deoxyHb once again decreased, and OEF increased by ∼50% compared with resting state. In the ACA territory, the changes during the initial hypoperfusion phase were smaller in both magnitude and duration (Figure 4A), but otherwise did not significantly differ from the MCA territory. None of the hemodynamic and metabolic parameters significantly differed between male and female mice (Figure 4B). Moreover, nearly identical CBF changes were recorded in the FVB mouse strain, suggesting that the hemodynamic response to CSD is not strain dependent (male mice, n=8, data not shown).
Figure 2.

Time-lapse images of hemodynamic and metabolic changes during cortical spreading depression (CSD). Representative images from a female mouse show changes in six measured parameters during two consecutive CSDs 15 minutes apart propagating in anteroposterior direction across the imaging field. The first CSD was associated with multiphasic changes (A), whereas the subsequent one superimposed on the post-CSD oligemia after the preceding CSD appeared monophasic (B). Red dashed lines indicate CSD wavefront based on the onset of hypoperfusion. The time after the first KCl application is shown on top. All parameters, including hemoglobin O2 saturation (satHb), are shown as percent change relative to their pre-CSD baseline as indicated in the color bar. Supplementary Movies 1 and 2 show the hemodynamic and metabolic responses to the first and second CSDs in relative and absolute values, respectively, with high spatiotemporal resolution.
Figure 3.

Time course of hemodynamic and metabolic changes during cortical spreading depression (CSD). (A) Representative tracings showing changes in six hemodynamic and metabolic parameters during two consecutive CSDs measured within the middle cerebral artery (MCA) territory in a female mouse. Vertical dashed lines represent the CSD onset. All six parameters are shown relative to their pre-CSD baseline. †, calculated using CBF, oxyHb and deoxyHb. (B) The averaged time course of hemodynamic and metabolic response to the first CSD in female mice (n=6). Inset shows the onset of CSD response (gray-shaded box) on expanded time scale. The profound hypoperfusion and deoxygenation (a), the transient recovery (b), the second small rise (c), and post-CSD oligemia (d), as well as the transient dip between b and c (*) are indicated. (C) The averaged time course of hemodynamic and metabolic response to the second CSD in female mice. The response to second CSD was mainly monophasic. Please note that changes in hemoglobin O2 saturation (SatHb) are shown as percent of baseline, rather than actual percent saturation. Solid curves indicate measured, dashed curves indicate calculated parameters. Vertical dashed line represents the CSD onset. Error bars are omitted for clarity and can be seen in Figure 4C.
Figure 4.

The hemodynamic and metabolic changes during cortical spreading depression (CSD) in different arterial territories, in male and female mice, and in response to a second consecutive CSD. (A) The hemodynamic and metabolic responses to the first CSD measured within the middle and anterior cerebral artery (MCA and ACA) territories are shown superimposed for direct comparison (n=6 females). Both the magnitude and the duration of hypoperfusion were smaller within the ACA territory. In addition, the second smaller peak observed in MCA territory was virtually absent within the ACA territory. P<0.05 versus MCA in all measured parameters. (B) The hemodynamic and metabolic responses to the first CSD did not differ between male and female mice shown superimposed for direct comparison (n=5 and 6, respectively). Responses were recorded within the MCA territory, and did not differ statistically. (C) The hemodynamic and metabolic responses to two consecutive CSDs are shown superimposed for direct comparison (n=6 females). Responses were recorded within the MCA territory. Note that the ostensibly monophasic response to the second CSD was superimposed on post-CSD oligemia and closely resembled the first CSD with the exception of the initial hypoperfusion. The standard errors of both the magnitudes and the latencies of measured time points are also shown in each panel. Statistical comparisons were not done because of different baselines for the first and second CSDs for all measured parameters.
The hemodynamic and metabolic response to a second CSD evoked 15 minutes after the first one differed radically from the response to the first CSD, presumably because the response to the second CSD was superimposed on the oligemic phase after the first CSD (Figures 3C and 4C; Supplementary Movies 1 and 2). During the second CSD, all measured parameters except deoxyHb showed a monophasic increase that lasted about 3 minutes; however, the deflection points corresponding to those of the first CSD were still recognizable. After the CSD, all parameters returned to their oligemic levels after the first CSD.
To obtain further insight into tissue metabolism and oxygenation, we performed cortical NADH fluorescence imaging using two-photon microscopy (Figure 5). The NADH response to CSD differed between the capillary bed and periarteriolar tissue. In the capillary bed, NADH fluorescence abruptly increased to a peak (13%±1% increase), often preceded by an initial small dip (n=15 CSDs in seven mice; Figure 5A and C, blue tracing). NADH levels then decreased to a plateau and remained elevated for over 5 minutes. The peak NADH increase was spatially heterogeneous without a consistent relationship to individual capillaries, but became more homogeneous during the plateau phase (Figure 5A; Supplementary Movie 3). In the proximity of a penetrating parenchymal arteriole, the initial NADH dip was larger (8%±4% decrease), the peak increase was diminished (5%±3% increase), and the long-lasting elevated plateau was absent (n=5 CSDs in four mice; Figure 5B, green tracing; Supplementary Movie 4). Simultaneous with the onset of NADH rise, the diameters of pial and penetrating arterioles abruptly decreased by 23%±6% and remained relatively constricted for over 5 minutes (n=7 CSDs in four mice; Figure 5C), consistent with the previously reported pronounced hypoperfusion in this species (Ayata et al, 2004b; Chang et al, 2010).
Figure 5.

Time course of nicotinamide adenine dinucleotide (NADH) fluorescence and arteriolar diameter changes during cortical spreading depression (CSD) measured by two-photon imaging. (A) Representative raw (upper row) and relative (lower row) images show the NADH changes in capillary bed (upper group) and periarteriolar tissue (lower group) to CSD. Vessels, labeled by fluorescein isothiocyanate (FITC)-dextran, are superimposed on the raw images. The NADH increase in capillary bed was spatially heterogeneous without a consistent relationship to capillaries, whereas the periarteriolar response in the vicinity of penetrating arterioles (asterisks) was more muted. The timings of images (a–f) are shown in panel B. Red rectangle shows a typical periarteriolar region of interest (ROI). The apparent NADH increase overlying the larger arteriole (arrows) represents movement of the vessel out of the imaging plane, and shrinking of the shadow of the vessel above the imaging plane (yellow arrowhead), due to vasoconstriction; such artifacts were routinely avoided during ROI placement. (B) The averaged time course of NADH signal changes in capillary bed (blue, n=15 CSDs in seven mice) and periarteriolar tissue (green, n=5 CSDs in four mice) during a CSD are shown together with changes in arteriolar diameter (red, n=7 CSDs in four mice). In the capillary bed, CSD was associated with an abrupt increase in NADH fluorescence preceded by a small and brief dip. After the peak, NADH levels remained elevated for at least 5 minutes. In contrast, periarteriolar NADH showed a bigger initial dip and a smaller peak increase, followed by a transient undershoot below baseline; there was no persistent NADH increase in periarteriolar tissue after a CSD (P<0.05 versus capillary bed). The abrupt onset of vasoconstriction temporally corresponded to the initial NADH dip in all mice (inset shows a representative experiment). Arterioles remained mildly constricted for at least 5 minutes after a CSD, closely resembling the cerebral blood flow (CBF) and cerebral blood volume (CBV) changes obtained using laser speckle and multispectral imaging (Figures 3 and 4). Gray-shaded regions (a–i) indicate the approximate timing of images shown in panels A and C. Periarteriolar NADH was measured only when a penetrating parenchymal arteriole was present in the imaging field, whereas diameter changes were measured when a pial or penetrating arteriole was present. Data are mean±standard error (thick and thin lines, respectively). (C) A representative pial arteriole (red arrowhead) shows the vasoconstrictive response to CSD. In contrast, capillary or venous diameters (blue arrowhead) did not change during CSD, although red blood cell flow arrest was visible in some vessels during the trough. Calibration bars=50 μm. Representative experiments are shown in Supplementary Movies 3 and 4.
Of note, we encountered sharply demarcated ‘islets' of elevated NADH fluorescence at baseline that varied between 50 and 250 μm in longest diameter and showed 33%±9% higher NADH signal than surrounding cortex (Figure 6). These islets resembled the recently described cortical NADH patterns (Kasischke et al, 2010); however, they were present in only 30% of all fields of view studied, and did not show a consistent spatial relationship to nearby arterioles, venules, or capillaries. The predominant response to CSD within these islets was an overall decrease in NADH signal, although the initial dip and subsequent peak were still recognizable (n=6 CSDs in five mice; Figure 6).
Figure 6.

Cortical regions with elevated baseline nicotinamide adenine dinucleotide (NADH) fluorescence. Representative raw NADH images (upper row) show a sharply demarcated ‘islet' with high baseline fluorescence signal in the vicinity of a cortical arteriole (upper left corner), observed in ∼30% of the cortical fields studied. Arrowhead indicates the shadow of the large vessel above the imaging plane. Relative images (lower row) show that NADH did not significantly increase in these islets during cortical spreading depression (CSD), and the response in periarteriolar tissue (asterisks) was once again muted. The averaged NADH response in these islets with high baseline NADH was triphasic, followed by a persistent decrease in NADH signal after CSD (n=6 CSDs in five mice). Red rectangle shows the typical region of interest (ROI) placement to quantify NADH changes within these regions. Gray-shaded regions (a–c) on the graph indicate the approximate timing of NADH images. The apparent NADH increase overlying the large arteriole (arrows) is once again an artifact of vasoconstriction visible on the angiogram (b). Data are mean±standard error (thick and thin lines, respectively). Calibration bars=50 μm.
Discussion
The profound hypoperfusion response to CSD is characteristic in the mouse and predicts severe tissue hypoxia (Ayata et al, 2004b; Chang et al, 2010). Using simultaneous laser speckle and multispectral reflectance imaging minimally invasively through intact skull, we found an abrupt reduction in satHb to 60% of baseline during and in the wake of CSD. Because deoxygenation was tightly coupled to reductions in CBF and CBV, vasoconstriction (confirmed by two-photon microscopy) appeared to be the dominant mechanism, and severely limited the O2 availability before any increase in CMRO2 was detected. Indeed, at the onset of CSD, CMRO2 declined near-simultaneously with CBF, CBV, and satHb, and never increased above baseline at any stage during and after CSD. Because calculated CMRO2 reflects O2 consumption rather than demand, we performed additional studies measuring cortical NADH changes. Coincident with vasoconstriction and hemoglobin desaturation, cortical tissue NADH increased abruptly, suggesting mitochondrial O2 demand exceeding supply and/or rapid glycolytic activation. Importantly, the increase in NADH was more pronounced and prolonged in regions far from parenchymal arterioles. Recent work showed that penetrating arterioles deliver oxygen to surrounding tissue (Kasischke et al, 2010; Sakadzic et al, 2010), suggesting luxury oxygen supply and higher tissue pO2 (pO2,t) near arterioles. Therefore, more pronounced and prolonged NADH increase away from cortical arterioles suggests more severe reduction in mitochondrial oxidative phosphorylation in these regions during and after CSD. Recent data from hippocampal slices also suggest that when O2 availability is reduced, mitochondrial oxidative phosphorylation can become O2 supply limited (Galeffi et al, 2011). Altogether, these data support the conclusion that reduced CMRO2 during CSD in mice is due, at least in part, to O2 supply limitation.
The multispectral data further suggest, however, that the CMRO2 decrease during CSD in mice may be more than would be expected from O2 supply limitation alone, and that additional factors interfering with mitochondrial oxidative phosphorylation may exist. Previous calculations showed that CMRO2 becomes supply limited when pO2,t in the vicinity of mitochondria falls below a critical level of ∼4 mm Hg (Kasischke et al, 2010). Although we did not quantify pO2,t, it can be estimated from CBF and CMRO2 changes measured in our studies, using the formalism of Gjedde et al (2005). The model states that assuming an arterial satHb of 100%, pO2,t is related to baseline OEF (EO), relative CBF (rCBF) and O2 consumption (rCMRO2), and baseline capillary and tissue pO2 (pO2,co and pO2,to, respectively) in the following manner:
 |
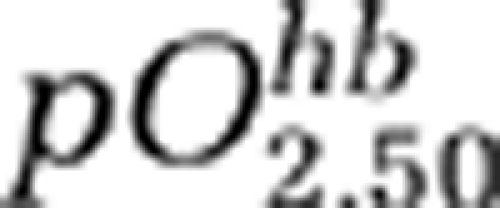 is the partial pressure of O2 when satHb=50%, and h is the Hill's coefficient (we use
is the partial pressure of O2 when satHb=50%, and h is the Hill's coefficient (we use 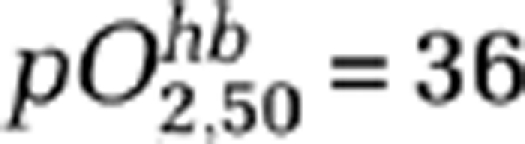 mm Hg and h=2.7). We have also included a term rKO to account for relative changes in O2 diffusibility from the blood to the mitochondria, which is a function of the surface area of flowing blood tissue interface, O2 diffusion coefficient, and number of capillaries perfused with moving red blood cells. According to this model, if baseline pO2,t=17 mm Hg, EO=40% (Kasischke et al, 2010; Yaseen et al, 2011) and rKO=1, then a 70% reduction in rCBF during CSD predicts a 30% to 40% reduction in rCMRO2, if reduced O2 supply (i.e., pO2,t=0 to 4 mm Hg) is the only mechanism limiting CMRO2. Therefore, it appears unlikely that >50% CMRO2 reduction we obtained in mice during CSD can be explained solely on the basis of supply limitation (Figure 3Ba). Additional mechanisms that could further limit O2 consumption during CSD is mitochondrial depolarization and swelling (Zhou et al, 2010), which may interfere with the electron transport chain by altering the proton-motive potential, and a decrease in rKO (e.g., reduced O2 diffusion coefficient). Noninvasive pO2,t recordings might help to resolve whether mechanisms other than supply limitation (e.g., mitochondrial block) contribute to the CMRO2 drop during CSD in mice (Sakadzic et al, 2009, 2010).
mm Hg and h=2.7). We have also included a term rKO to account for relative changes in O2 diffusibility from the blood to the mitochondria, which is a function of the surface area of flowing blood tissue interface, O2 diffusion coefficient, and number of capillaries perfused with moving red blood cells. According to this model, if baseline pO2,t=17 mm Hg, EO=40% (Kasischke et al, 2010; Yaseen et al, 2011) and rKO=1, then a 70% reduction in rCBF during CSD predicts a 30% to 40% reduction in rCMRO2, if reduced O2 supply (i.e., pO2,t=0 to 4 mm Hg) is the only mechanism limiting CMRO2. Therefore, it appears unlikely that >50% CMRO2 reduction we obtained in mice during CSD can be explained solely on the basis of supply limitation (Figure 3Ba). Additional mechanisms that could further limit O2 consumption during CSD is mitochondrial depolarization and swelling (Zhou et al, 2010), which may interfere with the electron transport chain by altering the proton-motive potential, and a decrease in rKO (e.g., reduced O2 diffusion coefficient). Noninvasive pO2,t recordings might help to resolve whether mechanisms other than supply limitation (e.g., mitochondrial block) contribute to the CMRO2 drop during CSD in mice (Sakadzic et al, 2009, 2010).
In most studies, CSD was associated with a transient reduction in pO2,t; however, its timing, magnitude, and shape (i.e., monophasic, biphasic with an initial reduction, or triphasic with an initial increase) have been quite variable possibly reflecting the baseline physiological state (e.g., blood pressure), single versus multiple CSDs, species-dependent hemodynamic response, and the experimental techniques employed in each study (Lukyanova and Bures, 1967; Marshall, 1959; Mayevsky and Sclarksy, 1983; Piilgaard and Lauritzen, 2009; Sakadzic et al, 2009; Takano et al, 2007; Van Harreveld and Stamm, 1952). The mitochondrial redox response to CSD has also been variable. A monophasic decrease in tissue NADH levels (i.e., oxidation), as well as a biphasic response with an initial decrease and subsequent increase have both been reported during CSD (Dora et al, 1984; Haselgrove et al, 1990; Lothman et al, 1975; Mayevsky et al, 1974; Rex et al, 1999; Rosenthal and Somjen, 1973). When tissue perfusion or oxygenation was compromised, however, NADH levels showed a monophasic increase often with an initial brief dip (Mayevsky et al, 1982; Schechter et al, 2009), resembling the NADH responses we and others observed in mice (Takano et al, 2007). Similarly, peri-infarct depolarization waves increase tissue NADH in hypoperfused penumbra, and decrease it when they propagate into the nonischemic cortex (Strong et al, 1996, 2000). These data support the conclusion that O2 supply is limited during CSD in mice, presumably by the vasoconstrictive response, leading to an abrupt CMRO2 reduction sharply contrasting the persistent increase in CMRO2 calculated in other species (Dunn et al, 2003; Piilgaard and Lauritzen, 2009; Sakadzic et al, 2009). Taken together, the abrupt and long-lasting hemoglobin deoxygenation and NADH increase coupled to severe vasoconstriction suggest that the CMRO2 response and the degree of O2 supply-demand mismatch during CSD are dominated by the species-dependent hemodynamic response in mice. Of note, hemodynamic responses to CSD did not differ within a physiological range of arterial pressures (70 to 100 mm Hg), pH (7.30 to 7.40), pCO2 (30 to 40 mm Hg), and pO2 (100 to 160 mm Hg) values in additional experiments (data not shown).
Two-photon excited NADH fluorescence imaging has been used previously as an indicator of cellular redox state (Kasischke et al, 2004). However, the profound cellular and vascular changes occurring during spreading depression, if not properly accounted for, may confound the interpretation of the detected fluorescence signal. First, intracellular organelle swelling and dendritic beading increase tissue scattering during spreading depression (Somjen, 2001). Scattering reduces two-photon excitation at the focal plane (Theer and Denk, 2006), thereby decreasing the amount of detected fluorescence. For this reason, we confined our measurements to within 50 μm of the cortical surface. Nevertheless, we cannot rule out the possibility that the observed initial dip in the NADH response is caused by increased scattering of excitation light accompanying the wave of cell depolarization. Since the duration of cell depolarization is typically <1 minute, we do not expect the relative NADH signal levels minutes after CSD to be confounded by scattering changes. Second, vasoconstriction during CSD reduces both intravascular scattering of excitation light and intravascular absorption of emission light, thereby increasing the amount of detected fluorescence. For this reason, we carefully positioned our imaging planes and ROIs to avoid large vessel shadows. Third, photobleaching and detector noise are possible factors, which may contribute to underestimation of relative changes. For these reasons, we set both the laser power and the detector gain to minimize these effects, and we checked the imaging field of view for possible evidence of bleaching after each CSD. Lastly, while NADH fluorescence changes are generally assumed to reflect mitochondrial NADH dynamics in neurons (Shuttleworth, 2009), we note that NADH fluorescence can represent contributions from multiple species in different cellular compartments with different lifetimes (Chia et al, 2008).
Using in vivo two-photon microscopy, Takano et al (2007) observed that the NADH increase during CSD showed a unique spatial relationship to capillaries. In the vicinity of capillaries, NADH increase was preceded by a transient ‘dip' lasting ∼30 seconds, whereas tissue farther away from capillaries showed a rapid and persistent NADH increase. In our experiments, we also found a spatially heterogeneous NADH increase, but there was no consistent relationship to capillaries. Instead, we observed that the NADH increase was consistently diminished in the vicinity of arterioles. The reasons for this discrepancy between the studies are unclear, but may involve differences in methodology, or systemic or cortical physiology (see below). In a small subset of imaging fields, we also observed sharply demarcated islets of elevated NADH fluorescence at baseline, similar to those recently described in mice (Kasischke et al, 2010). But once again we did not observe a consistent spatial relationship between the islets and the cortical vasculature. The biological mechanisms responsible for these NADH islets are unclear; however, it is possible that they represent direct cortical injury or secondary vascular thrombosis because of the closed cranial window preparation. Nevertheless, because CSD was able to propagate into these islets, and caused a long-lasting reduction in NADH fluorescence (Figure 6), data suggest that the tissue was still viable.
The advantage of the multimodal optical imaging system is the complete temporal and spatial overlap of all measured hemodynamic and metabolic parameters. Previous studies, including the multiparametric electrode/optode assemblies, relied upon multiple single-site measurements presumed to reflect spatiotemporally matched phases of a complex phenomenon. Moreover, the impact of craniectomy and electrode insertion on cortical physiology is of particular concern in mice. This is because mouse cortex is highly susceptible to CSD, making it likely to mechanically evoke accidental CSDs during the craniectomy procedure and electrode insertions. The hemodynamic and metabolic response to the first CSD in naive cortex dramatically differs from the response to subsequent CSDs (Figure 2), in part because subsequent CSDs occur on a background of severe post-CSD oligemia (CBF 30% to 40% of baseline), which usually takes over an hour to recover (Ayata et al, 2004b). Therefore, a CSD evoked during experimental preparation may confound the response to a subsequent intentional CSD and its interpretation, if it is unnoticed and/or sufficient time is not allowed for tissue recovery. This may explain the absence of a vasoconstrictive response during CSD in mice in one study (Takano et al, 2007). When studied minimally invasively as done here, the vasoconstrictive response to CSD is a constant finding, highly reproducible and conserved across different mouse strains (Ayata et al, 2004b; Chang et al, 2010). For the same reason, it is possible that the previously reported spatial relationship of CSD-induced NADH changes to cortical capillaries (Takano et al, 2007) might also have been due to a preceding undetected CSD causing severe hypoperfusion and hypoxia upon which the subsequently imaged CSDs were superimposed. This possibility can be circumvented by allowing the cortex to rest for more than an hour after window preparation before inducing a CSD, as was routinely done in our study.
In summary, a unique vasoconstrictive response creates a marked O2 supply-demand mismatch during and in the wake of CSD in mice, severely limiting O2 availability for consumption. More work is needed to ascertain whether there is a concurrent reduction in mitochondrial O2 demand during intense neuronal depolarization. The striking differences between mice and other species in the hemodynamic and metabolic response to CSD have pathophysiological implications for the interpretation and extrapolation of data obtained from mice in disease states where CSD-like injury depolarizations occur with high frequency and impact the outcome.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This study was supported by the National Institutes of Health (NS061505, Ayata; NS055104, Lo; NS057476, Boas; K99NS067050, Srinivasan) and the American Heart Association (10SDG2610275, Eikermann-Haerter; 11IRG5440002, Srinivasan).
Supplementary Material
References
- Ayata C. Cortical spreading depression triggers migraine attack: pro. Headache. 2010;50:725–730. doi: 10.1111/j.1526-4610.2010.01647.x. [DOI] [PubMed] [Google Scholar]
- Ayata C, Dunn AK, Gursoy OY, Huang Z, Boas DA, Moskowitz MA. Laser speckle flowmetry for the study of cerebrovascular physiology in normal and ischemic mouse cortex. J Cereb Blood Flow Metab. 2004a;24:744–755. doi: 10.1097/01.WCB.0000122745.72175.D5. [DOI] [PubMed] [Google Scholar]
- Ayata C, Shin HK, Salomone S, Ozdemir-Gursoy Y, Boas DA, Dunn AK, Moskowitz MA. Pronounced hypoperfusion during spreading depression in mouse cortex. J Cereb Blood Flow Metab. 2004b;24:1172–1182. doi: 10.1097/01.WCB.0000137057.92786.F3. [DOI] [PubMed] [Google Scholar]
- Brennan KC, Beltran-Parrazal L, Lopez-Valdes HE, Theriot J, Toga AW, Charles AC. Distinct vascular conduction with cortical spreading depression. J Neurophysiol. 2007;97:4143–4151. doi: 10.1152/jn.00028.2007. [DOI] [PubMed] [Google Scholar]
- Chang JC, Shook LL, Biag J, Nguyen EN, Toga AW, Charles AC, Brennan KC. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain. 2010;133:996–1012. doi: 10.1093/brain/awp338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia TH, Williamson A, Spencer DD, Levene MJ. Multiphoton fluorescence lifetime imaging of intrinsic fluorescence in human and rat brain tissue reveals spatially distinct NADH binding. Opt Express. 2008;16:4237–4249. doi: 10.1364/oe.16.004237. [DOI] [PubMed] [Google Scholar]
- Dohmen C, Sakowitz OW, Fabricius M, Bosche B, Reithmeier T, Ernestus RI, Brinker G, Dreier JP, Woitzik J, Strong AJ, Graf R. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann Neurol. 2008;63:720–728. doi: 10.1002/ana.21390. [DOI] [PubMed] [Google Scholar]
- Dora E, Gyulai L, Kovach AG. Determinants of brain activation-induced cortical NAD/NADH responses in vivo. Brain Res. 1984;299:61–72. doi: 10.1016/0006-8993(84)90788-1. [DOI] [PubMed] [Google Scholar]
- Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, Lehmann TN, Sarrafzadeh A, Willumsen L, Hartings JA, Sakowitz OW, Seemann JH, Thieme A, Lauritzen M, Strong AJ. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129:3224–3237. doi: 10.1093/brain/awl297. [DOI] [PubMed] [Google Scholar]
- Dunn AK, Devor A, Bolay H, Andermann ML, Moskowitz MA, Dale AM, Boas DA. Simultaneous imaging of total cerebral hemoglobin concentration, oxygenation, and blood flow during functional activation. Opt Lett. 2003;28:28–30. doi: 10.1364/ol.28.000028. [DOI] [PubMed] [Google Scholar]
- Dunn AK, Devor A, Dale AM, Boas DA. Spatial extent of oxygen metabolism and hemodynamic changes during functional activation of the rat somatosensory cortex. Neuroimage. 2005;27:279–290. doi: 10.1016/j.neuroimage.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Yuzawa I, Qin T, Wang Y, Baek K, Kim YR, Hoffmann U, Dilekoz E, Waeber C, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C. Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J Neurosci. 2011;31:5755–5763. doi: 10.1523/JNEUROSCI.5346-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeffi F, Somjen GG, Foster KA, Turner DA. Simultaneous monitoring of tissue PO(2) and NADH fluorescence during synaptic stimulation and spreading depression reveals a transient dissociation between oxygen utilization and mitochondrial redox state in rat hippocampal slices. J Cereb Blood Flow Metab. 2011;31:626–639. doi: 10.1038/jcbfm.2010.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjedde A, Johannsen P, Cold GE, Ostergaard L. Cerebral metabolic response to low blood flow: possible role of cytochrome oxidase inhibition. J Cereb Blood Flow Metab. 2005;25:1183–1196. doi: 10.1038/sj.jcbfm.9600113. [DOI] [PubMed] [Google Scholar]
- Haselgrove JC, Bashford CL, Barlow CH, Quistorff B, Chance B, Mayevsky A. Time resolved 3-dimensional recording of redox ratio during spreading depression in gerbil brain. Brain Res. 1990;506:109–114. doi: 10.1016/0006-8993(90)91205-u. [DOI] [PubMed] [Google Scholar]
- Hashemi P, Bhatia R, Nakamura H, Dreier JP, Graf R, Strong AJ, Boutelle MG. Persisting depletion of brain glucose following cortical spreading depression, despite apparent hyperaemia: evidence for risk of an adverse effect of Leao's spreading depression. J Cereb Blood Flow Metab. 2009;29:166–175. doi: 10.1038/jcbfm.2008.108. [DOI] [PubMed] [Google Scholar]
- Jones PB, Shin HK, Boas DA, Hyman BT, Moskowitz MA, Ayata C, Dunn AK. Simultaneous multispectral reflectance imaging and laser speckle flowmetry of cerebral blood flow and oxygen metabolism in focal cerebral ischemia. J Biomed Opt. 2008;13:044007. doi: 10.1117/1.2950312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasischke KA, Lambert EM, Panepento B, Sun A, Gelbard HA, Burgess RW, Foster TH, Nedergaard M. Two-photon NADH imaging exposes boundaries of oxygen diffusion in cortical vascular supply regions. J Cereb Blood Flow Metab. 2010;31:68–81. doi: 10.1038/jcbfm.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Leao AAP. Spreading depression of activity in cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Lothman E, Lamanna J, Cordingley G, Rosenthal M, Somjen G. Responses of electrical potential, potassium levels, and oxidative metabolic activity of the cerebral neocortex of cats. Brain Res. 1975;88:15–36. doi: 10.1016/0006-8993(75)90943-9. [DOI] [PubMed] [Google Scholar]
- Lukyanova LD, Bures J. Changes in pO2 due to spreading depression in the cortex and nucleus caudatus of the rat. Physiol Bohemoslov. 1967;16:449–455. [PubMed] [Google Scholar]
- Marshall WH. Spreading cortical depression of Leao. Physiol Rev. 1959;39:239–279. doi: 10.1152/physrev.1959.39.2.239. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Sclarksy DL. Correlation of brain NADH redox state, K+, PO2 and electrical activity during hypoxia, ischemia and spreading depression. Adv Exp Med Biol. 1983;159:129–141. doi: 10.1007/978-1-4684-7790-0_12. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Zarchin N, Friedli CM. Factors affecting the oxygen balance in the awake cerebral cortex exposed to spreading depression. Brain Res. 1982;236:93–105. doi: 10.1016/0006-8993(82)90037-3. [DOI] [PubMed] [Google Scholar]
- Mayevsky A, Zeuthen T, Chance B. Measurements of extracellular potassium, ECoG and pyridine nucleotide levels during cortical spreading depression in rats. Brain Res. 1974;76:347–349. doi: 10.1016/0006-8993(74)90467-3. [DOI] [PubMed] [Google Scholar]
- Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab. 2009;29:1517–15127. doi: 10.1038/jcbfm.2009.73. [DOI] [PubMed] [Google Scholar]
- Rex A, Pfeifer L, Fink F, Fink H. Cortical NADH during pharmacological manipulations of the respiratory chain and spreading depression in vivo. J Neurosci Res. 1999;57:359–370. [PubMed] [Google Scholar]
- Rosenthal M, Somjen G. Spreading depression, sustained potential shifts, and metabolic activity of cerebral cortex of cats. J Neurophysiol. 1973;36:739–749. doi: 10.1152/jn.1973.36.4.739. [DOI] [PubMed] [Google Scholar]
- Sakadzic S, Roussakis E, Yaseen MA, Mandeville ET, Srinivasan VJ, Arai K, Ruvinskaya S, Devor A, Lo EH, Vinogradov SA, Boas DA. Two-photon high-resolution measurement of partial pressure of oxygen in cerebral vasculature and tissue. Nat Methods. 2010;7:755–759. doi: 10.1038/nmeth.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakadzic S, Yuan S, Dilekoz E, Ruvinskaya S, Vinogradov SA, Ayata C, Boas DA. Simultaneous imaging of cerebral partial pressure of oxygen and blood flow during functional activation and cortical spreading depression. Appl Opt. 2009;48:D169–D177. doi: 10.1364/ao.48.00d169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter M, Sonn J, Mayevsky A. Brain oxygen balance under various experimental pathophysiologycal conditions. Adv Exp Med Biol. 2009;645:293–299. doi: 10.1007/978-0-387-85998-9_44. [DOI] [PubMed] [Google Scholar]
- Shuttleworth CW. Use of NAD(P)H and flavoprotein autofluorescence transients to probe neuron and astrocyte responses to synaptic activation. Neurochem Int. 2009;56:379–386. doi: 10.1016/j.neuint.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Harland SP, Meldrum BS, Whittington DJ. The use of in vivo fluorescence image sequences to indicate the occurrence and propagation of transient focal depolarizations in cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:367–377. doi: 10.1097/00004647-199605000-00003. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Hartings JA, Dreier JP. Cortical spreading depression: an adverse but treatable factor in intensive care. Curr Opin Crit Care. 2007;13:126–133. doi: 10.1097/MCC.0b013e32807faffb. [DOI] [PubMed] [Google Scholar]
- Strong AJ, Smith SE, Whittington DJ, Meldrum BS, Parsons AA, Krupinski J, Hunter AJ, Patel S, Robertson C. Factors influencing the frequency of fluorescence transients as markers of peri-infarct depolarizations in focal cerebral ischemia. Stroke. 2000;31:214–222. doi: 10.1161/01.str.31.1.214. [DOI] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10:754–762. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- Theer P, Denk W. On the fundamental imaging-depth limit in two-photon microscopy. J Opt Soc Am A Opt Image Sci Vis. 2006;23:3139–3149. doi: 10.1364/josaa.23.003139. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Stamm JS. Vascular concomitants of spreading cortical depression. J Neurophysiol. 1952;15:487–496. doi: 10.1152/jn.1952.15.6.487. [DOI] [PubMed] [Google Scholar]
- Yaseen MA, Srinivasan VJ, Sakadzic S, Radhakrishnan H, Gorczynska I, Wu W, Fujimoto JG, Boas DA. Microvascular oxygen tension and flow measurements in rodent cerebral cortex during baseline conditions and functional activation. J Cereb Blood Flow Metab. 2011;31:1051–1063. doi: 10.1038/jcbfm.2010.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N, Gordon GR, Feighan D, MacVicar BA. Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb Cortex. 2010;20:2614–2624. doi: 10.1093/cercor/bhq018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.