

Abstract
Nucleophilic imine additions with vinyl organometallics have developed into efficient, high yielding, and robust methodologies to generate structurally diverse allylic amines. We have used the hydrozirconation-transmetalation-imine addition protocol in the synthesis of allylic amine intermediates for peptide bond isosteres, phosphatase inhibitors, and mitochondria-targeted peptide mimetics. The gramicidin S-derived XJB-5-131 and JP4-039 and their analogs have been prepared on up to 160 g scale for preclinical studies. These (E)-alkene peptide isosteres adopt type II′ β-turn secondary structures and display impressive biological properties, including selective reactions with reactive oxygen species (ROS) and prevention of apoptosis.
Keywords: Imine additions, allylic amines, alkene peptide isosteres, mitochondrial targeting, gramicidin S, XJB-5-131, JP4-039
INTRODUCTION
Allylic Amines
Analogous to allylic alcohols, allylic amines represent useful functionalized 3-carbon building blocks for the synthesis of heterocycles and bioactive amines. In addition to oxygenations and aminations, metathesis reactions of allylic amines have been used to generate more complex derivatives (Figure 1). Several protocols are available for the synthesis of α-chiral allylic amines,1 including the rearrangement of allylic trichloroacetimidates,1b Ni(0)-mediated allylic amination,1c C,H-bond activation, and C-C bond-forming hydrogenation.1k Currently, nucleophilic additions to imines represent the most versatile strategy to prepare chiral allylic amines.1d-k In addition to the direct addition of vinyl organometallic species,1g-i the reductive coupling of alkynes1d-f and the acylvinyl anion addition1j provide diastereo- and/or enantiomerically enriched products.
Figure 1.
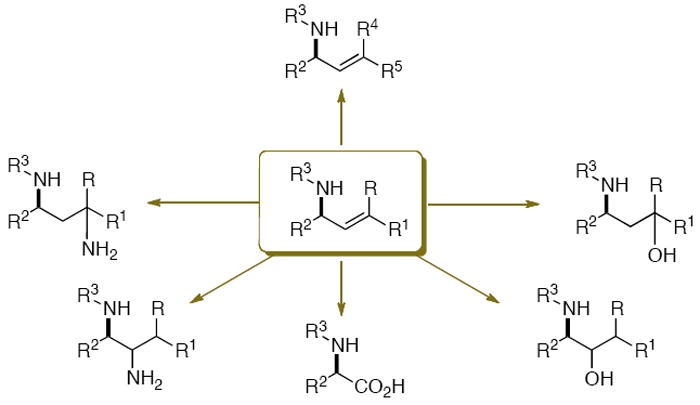
Representative synthetic transformations of allylic amine building blocks.
Jamison and coworkers developed an enantioselective synthesis of tetrasubstituted allylic amines by intermolecular coupling of a disubstituted alkyne, an imine, and triethylborane (Scheme 1).1e The conversion was catalyzed by a chiral [Ni(cod)2]/ferrocenyl phosphane complex (3). A (tert-butyldimethylsilyloxy)ethyl group was used to enhance reactivity and selectivity in the reaction of the achiral imine (1), and this auxiliary group could be removed from products 4-6 via a two-step protocol without decrease in enantiomeric excess.
Scheme 1.
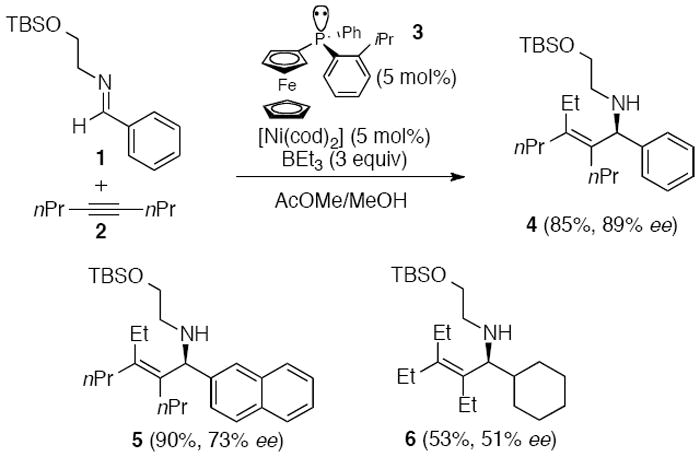
Ni-catalyzed intermolecular coupling to prepare enantiomerically enriched allylic amines.1e
The Krische group was able to form allylic amines via an asymmetric iridium-catalyzed C-C bond forming hydrogenation process related to hydroformylation reactions (Scheme 2). Hydrogenation of internal alkyne 8 in the presence of either aryl or alkyl aldimines such as 7 and chiral catalyst 9 resulted in the enantiomerically enriched trisubstituted allylic sulfonylamine 10. The regioselectivity of the reaction with unsymmetrically substituted internal alkynes was also high.
Scheme 2.
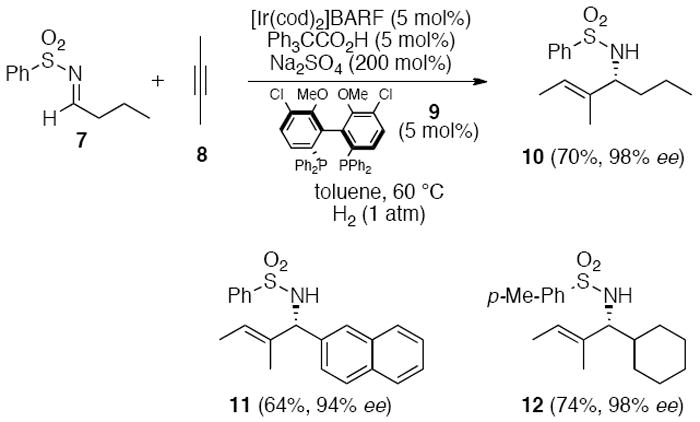
Reductive coupling of alkynes with N-arylsulfonyl imines.1f
The reaction of α-hydroxypropargylsilanes with chiral sulfinylimines to form trisubstituted allylic amines was applied by the Scheidt group (Scheme 3).1j Brook rearrangement of the α-hydroxypropargylsilane 13 to form the lithium allenolate was achieved with n-BuLi. The α-acylvinyl anion equivalent reacted with sulfinylimine (R)-14 to give the β-substituted aza-Morita-Baylis-Hillman product 15 in good yield and diastereoselectivity. The (Z)-alkene was the preferred product in all cases.
Scheme 3.
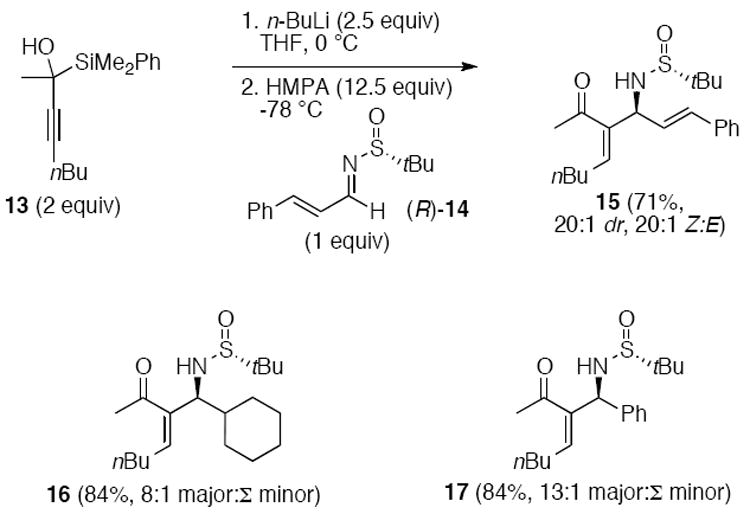
α-Acylvinyl anion addition to imines (major:Σminor refers to the ratio of the major product as drawn to the sum of all minor products).1j
Chiral sulfinyl imines were also utilized by Ellman and coworkers to form di-, tri- or tetrasubstituted allylic amines (Scheme 4).1i The sulfinyl amine (R)-18 was coupled with potassium trifluoroborates 19 via Rh(I)-catalysis. Air-stable [Rh(OH)(cod)]2 along with 1,2-bis-(diphenylphosphinoyl)benzene (dppbenz) gave the best yield and diastereoselectivity of the allylic sulfinylimines 20-22.
Scheme 4.
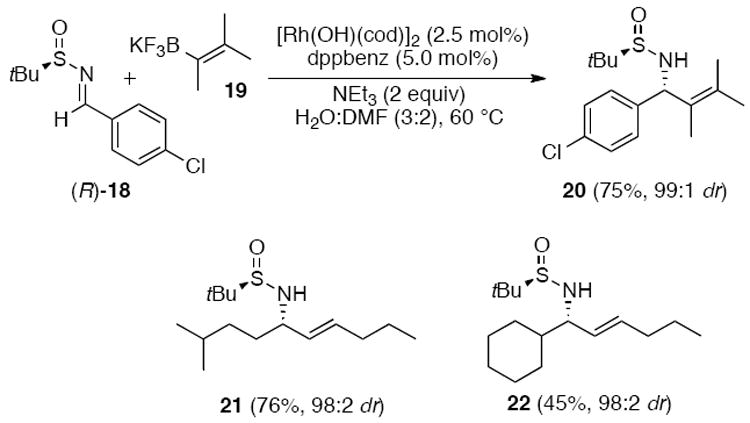
Rh(I)-catalyzed addition of alkenyl trifluoroborates to N-tert-butylsulfinylimines.1i
In the development of the hydrozirconation-transmetalation-imine addition protocol for the asymmetric synthesis of (E)-allylic amines, our group was able to take advantage of the ease of access to functionalized alkyne starting materials. Furthermore, the carboalumination/water-accelerated imine addition chemistry allowed for the preparation of terminally disubstituted allylic amines.2 Figure 2 provides an overview of the products available with these methodologies.
Figure 2.
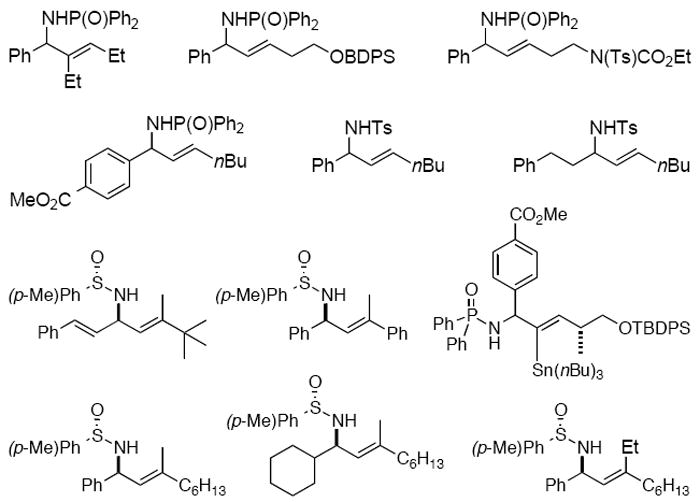
Representative allylic amine building blocks obtained with alkyne hydro(carbo)metalation-imine addition methods.
Peptide Isosteres
The use of peptides as pharmaceuticals is of considerable interest, since oligo- and polypeptides are natural ligands for receptors and enzymes. However, peptide-based orally administered drug formulations are still rare and tend to exhibit poor absorption and cell permeability as well as low bioavailability. The undesirable PK properties of peptides as well as their propensity for rapid in vivo degradation by peptidases have encouraged the pursuit of peptide mimetics as alternative therapeutic agents. Peptide isosteres contain a nonhydrolyzable group in place of the amide bond. In order for a rational design of these compounds to be effective, they must be able to adopt secondary structures typical for native peptides.3 Among others, methylene amine,4 ketomethylene,5 hydroxyethylamine,6 hydroxymethylene and hydroxyethylene,7 dihydroxyethylene,8 cyclopropylalkylamine,9 methyl- and (trifluoromethyl)alkenes,10 and β-amino acids11 have been used for this purpose. Our group has studied (E)-alkenes9b,10b,12 as peptide bond replacements (Figure 3),3b,13 and the synthesis and applications of these isosteres as mitochondrial targeting compounds14 is a major focus of our program.
Figure 3.
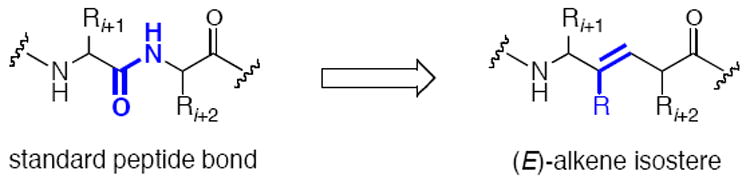
Structure of (E)-alkene peptide isosteres as peptide bond replacements.
Mitochondrial Targeting Agents
The cyclodecapeptide antibiotic gramicidin S displays significant resistance to peptide-cleaving proteases (Figure 4).15 It has been shown to interact with microbial membrane lipids, and this interaction as well as its general biological profile are correlated to its secondary structure composed of an amphipathic anti-parallel β-sheet and two type II′ β-turns. Our group has studied the conformational and biological effects of isosteric substitutions with both di- and trisubstituted alkenes in the gramicidin S backbone.13,16
Figure 4.
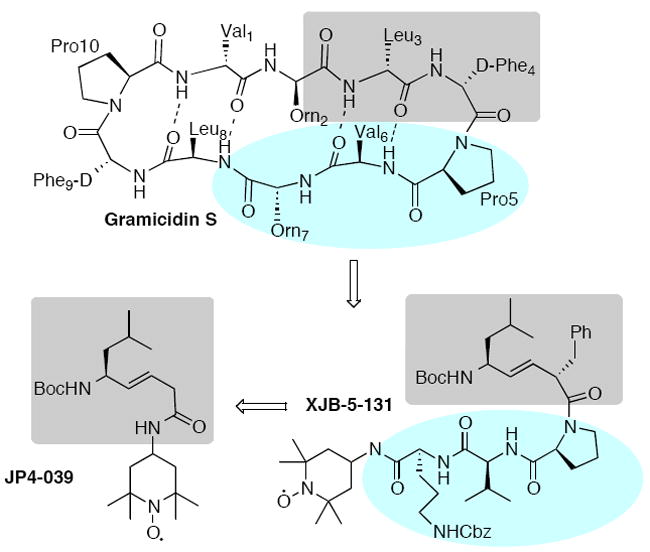
Mitochondrial targeting agents based on the antibiotic gramicidin S.
The design of the mitochondrial targeting agents XJB-5-131 and JP4-039 was inspired by the microbial membrane affinity of gramicidin S and guided by its characteristic secondary structural features. Replacement of an internal amide bond with an (E)-alkene moiety was envisioned to increase both the rigidity of the peptide mimetic as well as enhance its membrane permeability by removing a polar hydrogen bond donor/acceptor function. XJB-5-131, a first generation analog of gramicidin S, retained the Leu-DPhe-Pro-Val-Orn segment of the parent structure to fully encompass the II′ β-turn motif.9a A 4-amino-TEMPO (4-AT) “pay load” was added at the C-terminus to serve as a scavenger of reactive oxygen species (ROS) formed in mitochondria. The structurally simplified JP4-039 possesses an alkene dipeptide isostere segment comprised of leucine and glycine residues. XJB-5-131 and JP4-039 were found to be enriched in mitochondria by factors of 600 and 30, respectively, over their cytosolic concentrations.14c,17
We attribute the ability of these peptide mimetics to cross cellular membranes and concentrate in mitochondria to their β-turn preference as well as their affinity for the mitochondrial lipid, cardiolipin.14c,17 For example, the X-ray analysis of JP4-039 displays a type II′ β-turn (Figure 5). The distance in the i→i+3 intramolecular H-bond between the carbonyl of the Boc group and the nitrogen of 4-AT measures 3.30 Å. Similarly, a CD-analysis of XJB-5-131 and X-ray analyses of related biologically active peptide mimetics confirm the presence of analogous β-turns.
Figure 5.
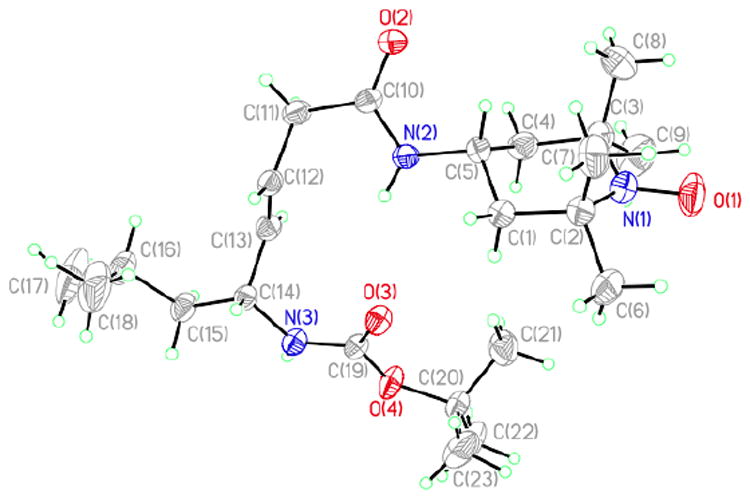
The crystal structure of JP4-039 reveals a type II′ β-turn.
METHODOLOGIES AND SYNTHETIC APPLICATIONS
Imine additions
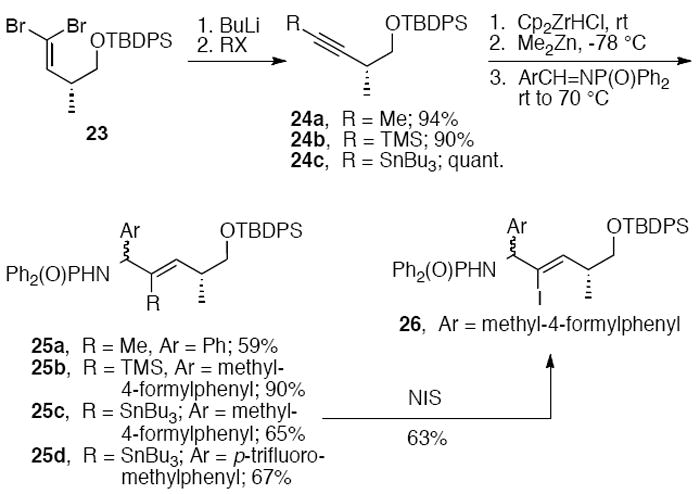
Early studies toward the synthesis of allylic amines in our group explored nucleophilic additions of vinyl zinc reagents to N,N-diphenylphosphinoylimines. Differentially substituted internal alkynes were synthesized by reaction of dibromoalkene 23 with butyl lithium followed by quenching with electrophiles. Methyl, trimethylsilyl, and tributylstannyl alkynes 24a-c were then subjected to hydrozirconation2d,18 with zirconocene hydrochloride,19 followed by transmetalation with dimethylzinc. Addition of phosphinoylimines to the reaction mixture gave alkenes 25a-d as a 1:1 mixture of diastereomers, which were easily separable by chromatography on SiO2.9b,12b The vinyl stannane 25c was iodinated with N-iodosuccinimide to provide iodoalkene 26. These intermediates could then be reacted under various cross-coupling conditions to generate a diverse set of allylic phosphinoylamines. For example, Stille cross-coupling of vinyl stannane 25c under microwave conditions led to the trisubstituted aryl- and heteroarylalkenes 27-29 in good yields (Scheme 6). 9b
Scheme 6.
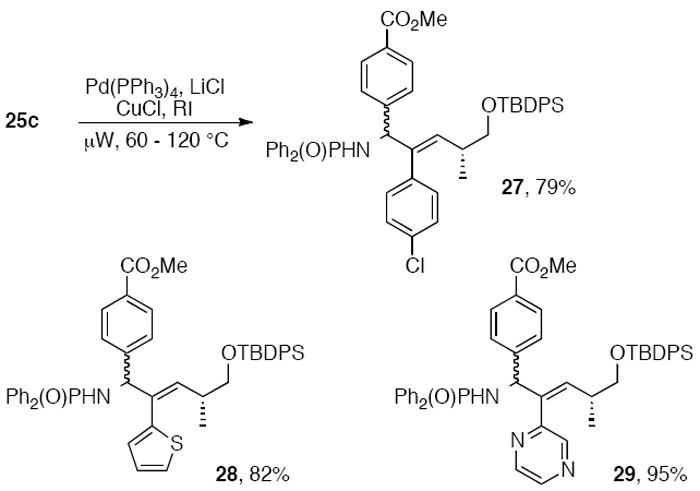
Microwave-accelerated Stille crosscouplings of 25c.9b
Vinyl iodide 26 was used to synthesize phenyl- and trifluoromethyl-substituted allylic amines 30 and 33, respectively. Negishi cross-coupling of 26 with phenylzinc bromide provided an 86% yield of 30 (Scheme 7). Alternatively, cross-coupling could be postponed until further elaboration of the C- and N-terminal functions. The phosphinoyl and silyl groups of 26 were removed with HCl(g), and the amine was acylated with CbzCl to give carbamate 31. Two-step oxidation of the primary alcohol to the acid followed by coupling to 2-naphthylamine provided 32, which was converted with methyl fluorosulfonyldifluoroacetate20 and copper thiophene carboxylate to the chromatographically separable trifluoromethyl alkenes 33a and 33b.
Scheme 7.
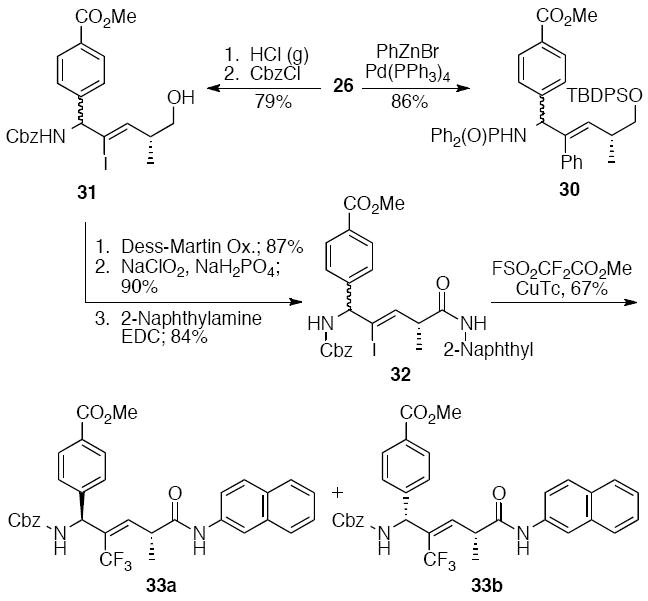
Pd-catalyzed arylation of iodoalkene 26 and trifluoromethylation of iodoalkene 32.9b
Our group also developed an accelerated carboalumination of alkynes in the presence of catalytic Cp2ZrCl2 and stoichiometric H2O.21 The intermediate vinyl alanes reacted with enantiomerically enriched sulfinyl imines to provide chiral allylic amines.2a For example, the vinyl alane formed from terminal alkyne 34 converted sulfinyl imine (R)-35 to the trisubstituted (E)-alkene 36 in good yield and >95% de (Scheme 8). The diastereoselectivity could be explained by a 4-membered Felkin-Anh-type transition state model.1h,22 This methodology was extended to the synthesis of cyclopropyl alkylamines.23 Furthermore, allylic amines could be obtained via a dimethylzinc-mediated addition of alkenyl zirconocenes to imino esters24 and by hydrozirconation followed by transmetalation to aluminum and addition to sulfinyl imines.2c
Scheme 8.
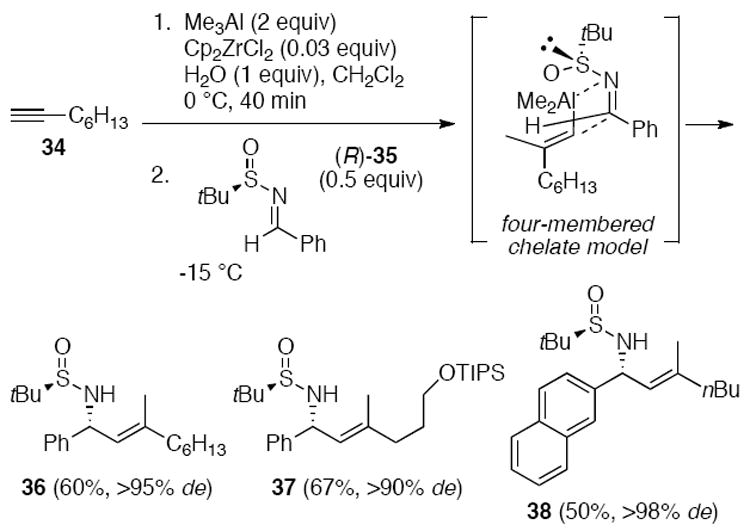
Alkyne carboalumination/sulfinyl imine addition as a stereoselective entry to (E)-allylic amines.2a
PEPTIDE MIMETICS BASED ON ALLYLIC AMINES
Synthesis of a Cdc25 Inhibitor
The imine addition methodology was optimized and applied to the synthesis of new (E)-alkene dipeptide isosteres. Our target design was based on the crystal structure of an active site peptide inhibitor of the dual-specificity phosphatase Cdc25.25 Monosilylation of the commercially available diol 39 followed by oxidation gave aldehyde 41 (Scheme 9). A Corey-Fuchs26 reaction provided dibromoalkene 42 which was treated with n-BuLi to form alkyne 43 after quenching with MeI. Hydrozirconation2d,18 and iodination of the internal alkyne led to the iodoalkene 44, which could be used as a common intermediate in the formation of other trisubstituted alkene peptide isosteres.
Scheme 9.
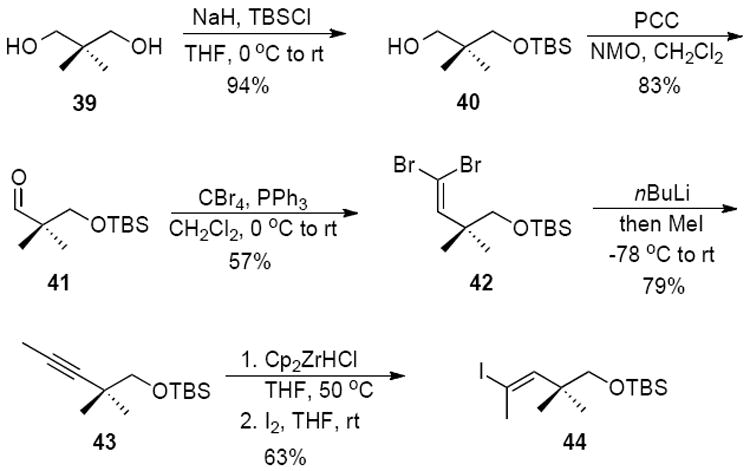
Synthesis of (E)-iodoalkene 44 by hydrozirconation of internal alkyne 43.
Building block 44 was used to synthesize intermediate 50 in the synthesis of isostere 57. Iodine-lithium exchange of 44, followed by transmetalation to magnesium1h and addition to sulfinyl imine (R)-45 gave 46 as a 2.5:1 mixture of diastereomers (Scheme 10). Protecting group removal, t-butyl carbamate formation, and oxidation of the primary alcohol led to acid 47. Acylation of amine 48 provided the trisubstituted alkene 49, and saponification, peptide coupling and removal of the Boc group led to the advanced intermediate 50.
Scheme 10.
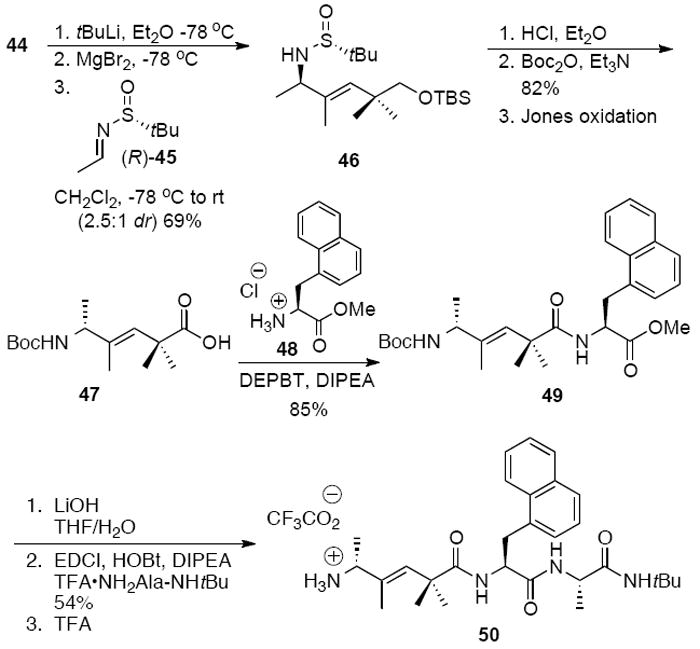
Synthesis of allylic amine 50.
The crystal structure of tetrapeptide mimetic 49 displayed a type II′ β-turn with prototypical dihedral angles (Figure 6). Interestingly, this compound crystallized in the chiral space group P6_s1 and formed an infinite one-dimensional hydrogen-bonded chain along the z-axis. The molecules assemble in a three-fold axis around long channels, which are filled with disordered solvent molecules (Figure 7). The diameter of the channel in the rhombi measures ca. 5.5 Å, and the sides have a length of ca. 9 Å.
Figure 6.
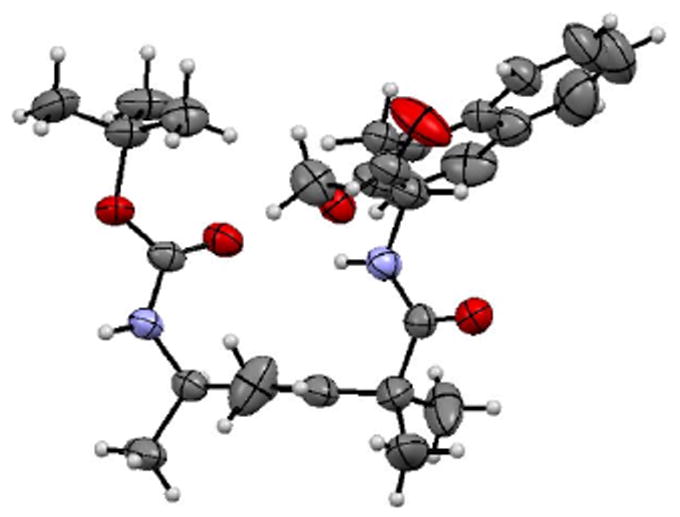
Crystal structure of 49.
Figure 7.
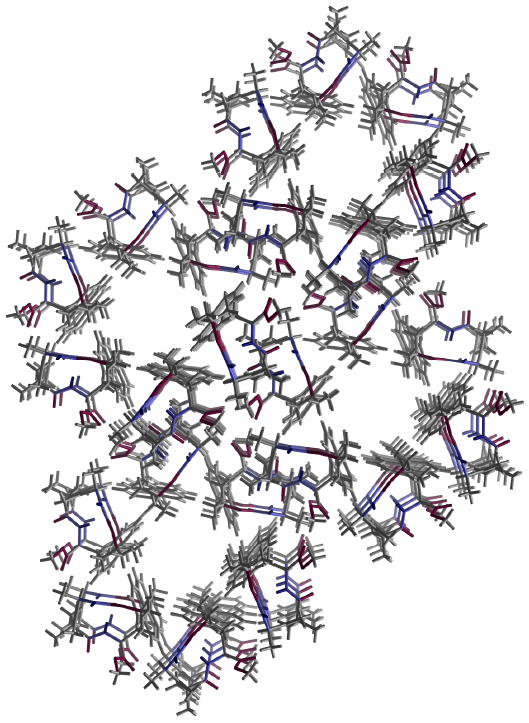
Crystal structure of 49 showing four solvent channels.
To complete the synthesis of 57, phenylalanine sulfonic acid derivative 54 was prepared by reaction of commercially available aldehyde 51 with Wittig reagent 52 and tetramethylguanidine (TMG) to give alkene 53. Stereoselective reduction of 53 and saponification provided acid 54. This acid was then coupled to amine 50 to provide 55. Removal of the Boc protecting group and subsequent coupling to carboxylic acid 56 gave 57, the protected trisubstituted alkene peptide isostere analog of the known25a peptidic Cdc25 phosphatase inhibitor.
Synthesis of Mitochondrial Targeting Agents
Both XJB-5-131 and JP4-039 adopt type II′ β-turn structures suitable for membrane passage. Because the trisubstituted alkene 49 was also shown to adopt this β-turn, an analog of JP4-039 with a trisubstituted alkene was synthesized from iodoalkene 44 (Scheme 12). Lithium-halogen exchange of 44 followed by transmetalation with cerium(III) and addition to sulfinyl amine (S)-58 provided allylic amine 59 in good yield as a 5:1 mixture of diastereomers that were separable by chromatography on SiO2. Deprotection of 59 followed by Boc-protection of the free amine gave 60. The primary alcohol was then oxidized to the acid and coupled to 4-AT to yield 61.
Scheme 12.
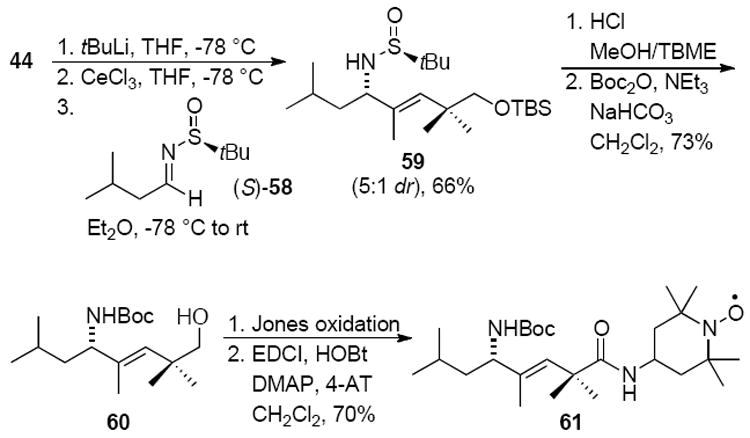
Synthesis of trisubstituted (E)-alkene peptide isostere 61 designed for mitochondrial targeting and scavenging of ROS.
Significantly, the hydrozirconation-transmetalation-imine addition proved to be a robust method for the scaleup of (E)-alkene gramicidin S analogs.27 The synthesis of (S)-65 began with the silylation of homopropargyl alcohol 62 (Scheme 13). Hydrozirconation of this alkyne on >150 g scale with Cp2ZrHCl,19 transmetalation with trimethylaluminum, and addition to sulfinyl imine (R)-5828 provided the highly diastereomerically enriched allylic sulfinyl amine (>20:1 de by 1H NMR analysis of the crude reaction mixture). The commercial availability of the hydrozirconating agent Cp2ZrHCl is limited, but the corresponding dichloride Cp2ZrCl2 can be obtained in bulk quantities and was converted by LAH reduction in ether to the hydrochloride. Removal of the sulfinyl group led to amine salt 64 in 96% yield. This amine was then Boc-protected and the silyl group was removed to reveal the homoallylic alcohol (S)-65 in 45% overall yield. (S)-65 was further used as a key intermediate to synthesize several simplified gramicidin S analogs, including XJB-5-131 and JP4-039.
Scheme 13.
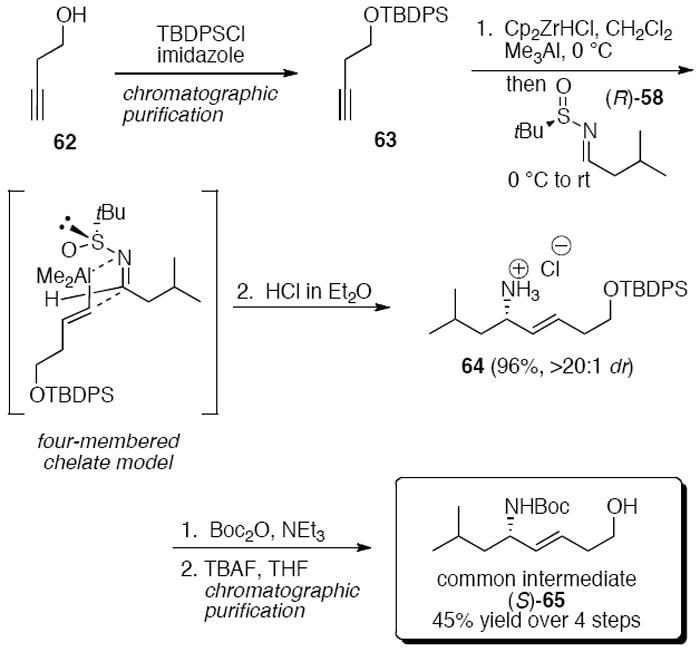
Large-scale synthesis of allylic amine intermediate (S)-65.27
Completion of the synthesis of JP4-039 from (S)-65 was accomplished in just two additional steps (Scheme 14). Jones oxidation of the primary alcohol followed by coupling to 4-AT gave the mitochondrial targeting agent in 33% overall yield from commercially available 62 in 99% purity and 99% ee. This synthetic sequence was completed on 160 g scale.
Scheme 14.
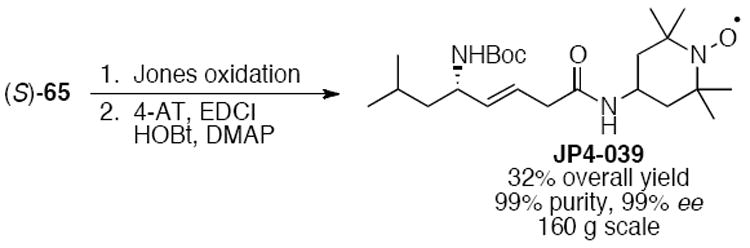
Completion of the enantioselective synthesis of JP4-039 from carbamate (S)-65.27
Gratifyingly, alkene peptide isosteres such as JP4-039 can demonstrate acceptable metabolic stabilities. In C57BL/6NHsd female mice, the half-life of JP4-039 was found to be 5.0 min in the blood, 10 min in the lungs, and 60 min in the liver, heart, and intestine. A difluorinated analog 68 was designed to further increase the pharmacokinetic properties of JP4-039 (Scheme 15). The synthesis of 68 originated from the common intermediate (S)-65. Jones oxidation followed by esterification with TMS-diazomethane gave methyl ester 66. Fluorination of 66 with N-fluoro-N-(phenylsulfonyl)benzenesulfonamide (NFSi) provided difluorinated ester 67. Saponification and subsequent coupling to 4-AT led to difluoro-JP4-039 (68). Metabolic stabilities in pooled male mouse liver microsomes (MMLM) revealed that 68 was indeed more stable than the parent compounds, XJB-5-131 and JP4-039. Specifically, 13% and 15% of XJB-5-131 and JP4-039 were detected by LCMS after 60 min in the presence of NADPH, with 98% and 93%, respectively, remaining in the absence of NADPH. Conversely, 27% of 68 was found after 60 min in the presence of NADPH, and 91% in its absence, demonstrating a roughly 2-fold improvement in stability of the fluorinated analog in the presence of activated cytochrome P450 isoforms.
Scheme 15.
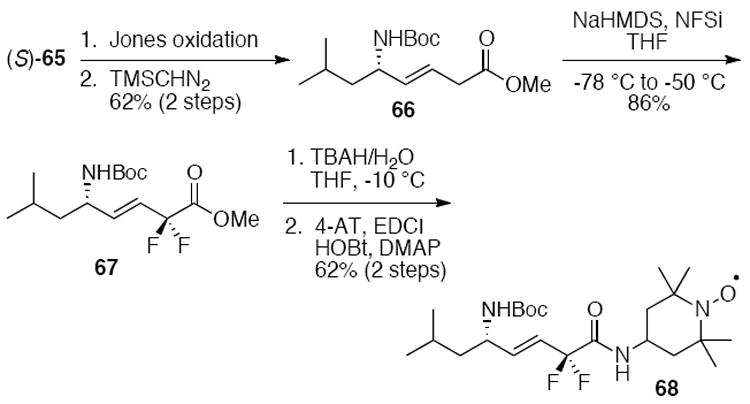
Stereoselective synthesis of the difluorinated analog 68 from (S)-65.27
The straightforward access to (S)-65 also allowed for a large-scale synthesis of XJB-5-131,29 representing a notable improvement over the 1st generation route.9a XJB-5-131 was obtained in 34% overall yield in a total of 12 steps for the longest linear sequence from commercially available starting materials (Scheme 16). Specifically, Jones oxidation of (S)-65, mixed anhydride formation with pivaloyl chloride, and condensation with benzyl oxazolidinone gave imide 70. The benzyl side chain in 71 was installed in >20:1 dr using an Evans asymmetric alkylation. Cleavage of the chiral auxiliary followed by coupling with tripeptide H-Pro-Val-Orn(Cbz)-OMe provided ester 72. Saponification and coupling with 4-AT completed the synthesis. This sequence allowed for the preparation of a ca. 2 g batch of XJB-5-131 as a single detectable stereoisomer. In addition to the improved scalability of this route, its efficiency vs. the earlier process was enhanced by the enantioselective approach to allylic amine 70, the use of a Jones oxidation for the one-step conversion of the alcohol to the acid, and the introduction of the benzyl side chain in the late-stage intermediate 70.
Scheme 16.
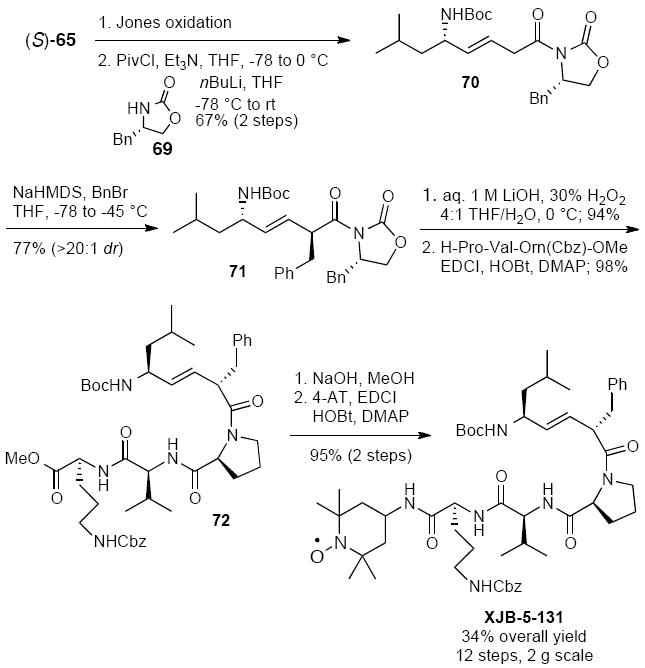
2nd Generation synthesis of XJB-5-131 from (S)-65.29
PERTINENT BIOLOGICAL ASSAYS
The ability of XJB-5-131 and JP4-039 to accumulate in mitochondria,14c,17 where they scavenge ROS and prevent hydroperoxidation of cardiolipin by cycling between nitroxide redox states, augurs well for their application in the treatment of a number of mitochondria-related diseases as well as for radiation countermeasures (Table 1).30,31
Table 1.
Overview of ongoing biological studies with XJB-5-131 and JP4-039.
| Biological Test | Results with XJB-5-131 | Results with JP4-039 |
|---|---|---|
| Lethal hemorrhagic shock | Significant prolonged survival in rats32 | n.d. |
| Radiation protection | n.d. | Accelerated bone wound healing in irradiated mice;34b increased survival in mice with esophagitis34a |
| Radiation mitigation | n.d. | Accelerated bone wound healing in irradiated mice;34b reduction in damage to irradiated 32Dcl cells at 24 h35 |
| Hyperoxic acute lung injury | Prevents CL oxidation; decreased apoptosis in concentration-dependent manner37 | n.d. |
| Acute brain injury | Reduced neuronal death in vitro and in vivo; reduced behavioral deficits in rats38 | n.d. |
MPEC: mouse pulmonary endothelial cell; CL: cardiolipin; n.d.: not determined.
Treatment with XJB-5-131 in rats subjected to lethal hemorrhagic shock prolonged survival even without resuscitation and injection with asanguinous fluids or blood.32 Both XJB-5-13133 and JP4-03934 demonstrated radioprotective properties in cells and in mice. Radioprotectants could prove useful in preventing radiation damage side effects in individuals undergoing cancer radiation therapy, for example. Furthermore, JP4-039 was shown to be a mitigator35 when it was administered to female C57Bl/6HNsd mice 24 h after the radiation event. These mice were found to have reduced peripheral blood lymphocytes, neutrophils, and bone marrow cellularity, demonstrating that JP4-039 was effective at mitigating hematopoietic syndrome.36 These and other potential therapeutic effects of these agents are currently under further investigation in a range of in vitro and in vivo systems.
CONCLUSIONS
The mitochondrial targeting agents JP4-039 and XJB-5-131 are examples of rationally designed, functional (E)-alkene peptide isosteres. The preparations and evaluations of these compounds were contingent on a robust synthetic access to allylic amine building blocks. The stereoselective synthesis of these compounds and other alkene peptide isosteres is concise, high yielding, and scalable to at least a half-kg level based on the hydro(carbo)metalation-transmetalation-imine addition methodologies developed in our laboratory. Specialized organometallic reagents, such as Cp2ZrCl2 and trimethylaluminum can be purchased on a multi-kilogram or even multi-ton scale. As it is the case for many new methodologies, however, applications at that scale will likely require extensive further optimizations by process chemists. For example, environmental impact, heavy metal usage, toxicity, hazard and cost of organometallic reagents, cryogenics, and chromatographic purifications will have to be resolved. Nonetheless, the authors hope that new synthetic methods such as the hydro(carbo)metalation-transmetalation-imine addition will inspire the design of structurally novel drug candidates and accelerate their move into Phase I studies.
Scheme 5.
Hydrozirconation/transmetalation/imine addition approach to trisubstituted allylic amines.9b,12b
Scheme 11.
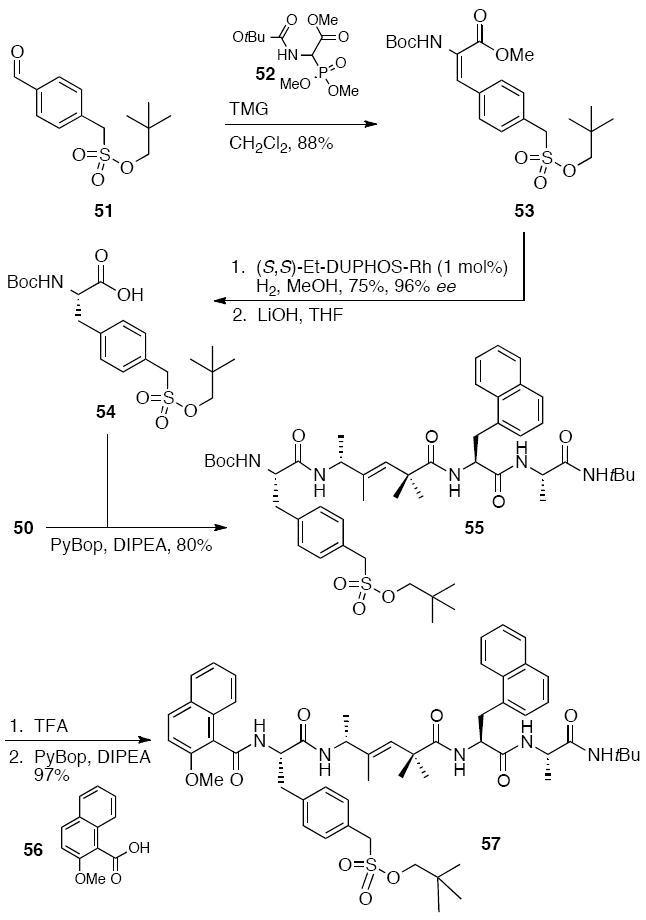
Completion of the synthesis of the protected alkene peptide isostere 57.
Acknowledgments
The authors thank the NSF and NIH for financial support. Drs. Corey Stephenson, Christopher Kendall, Jingbo Xiao, Jennifer Davoren, Joshua Pierce, and Marie-Céline Frantz in our laboratories at the University of Pittsburgh were valuable contributors to the synthetic work summarized in this Perspective, and Profs. Fink, Kagan, Greenberger, Epperly and Bayir, also at the University of Pittsburgh, were instrumental for the biological evaluations. We thank Dr. Steven Geib (University of Pittsburgh) for the X-ray analyses. Scale-up of the synthesis of JP4-039 was performed at Asymchem Inc.
ABBREVIATIONS
- 4-AT
4-amino TEMPO
- BARF
[B[3,5-(CF3)2C6H3]4]
- Cbz
carboxybenzyl
- cod
cyclooctadiene
- DEPBT
3-(diethylphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one
- DMAP
4-dimethylaminopyridine
- DIPEA
N,N-diisopropylethylamine
- dppbenz
1,2-bis(diphenylphosphino)benzene
- EDCI
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- HOBt
hydroxybenzotriazole
- NFSi
N-fluoro-N-(phenylsulfonyl)benzenesulfonamide
- NMO
N-methylmorpholine N-oxide
- PCC
pyridinium chlorochromate
- PyBop
benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate
- ROS
reactive oxygen species
- TBS
tert-butyldimethylsilyl
- TMG
tetramethylguanidine
References
- 1.(a) Johannsen M, Jørgensen KA. Chem Rev. 1998;98:1689. doi: 10.1021/cr970343o. [DOI] [PubMed] [Google Scholar]; (b) Anderson CE, Overman LE. J Am Chem Soc. 2003;125:12412. doi: 10.1021/ja037086r. [DOI] [PubMed] [Google Scholar]; (c) Berkowitz DB, Maiti G. Org Lett. 2004;6:2661. doi: 10.1021/ol049159x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Grossman RB, Davis WM, Buchwald SL. J Am Chem Soc. 1991;113:2321. [Google Scholar]; (e) Patel SJ, Jamison TF. Angew Chem Int Ed. 2004;43:3941. doi: 10.1002/anie.200460044. [DOI] [PubMed] [Google Scholar]; (f) Ngai M-Y, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:12644. doi: 10.1021/ja075438e. [DOI] [PubMed] [Google Scholar]; (g) Denmark SE, Weber T, Piotrowski DW. J Am Chem Soc. 1987;109:2224. [Google Scholar]; (h) Cogan DA, Liu G, Ellman J. Tetrahedron. 1999;55:8883. [Google Scholar]; (i) Brak K, Ellman JA. J Am Chem Soc. 2009;131:3850. doi: 10.1021/ja9002603. [DOI] [PubMed] [Google Scholar]; (j) Reynolds TE, Binkley MS, Scheidt KA. Org Lett. 2008;10:5227. doi: 10.1021/ol802227t. [DOI] [PubMed] [Google Scholar]; (k) Skucas E, Ngai M-Y, Komanduri V, Krische MJ. Acct Chem Res. 2007;40:1394. doi: 10.1021/ar7001123. [DOI] [PubMed] [Google Scholar]
- 2.(a) Wipf P, Nunes RL, Ribe S. Helv Chim Acta. 2002;85:3478. [Google Scholar]; (b) Wipf P, Kendall C, Stephenson CRJ. J Am Chem Soc. 2003;125:761. doi: 10.1021/ja028092a. [DOI] [PubMed] [Google Scholar]; (c) Wipf P, Pierce JG. Org Lett. 2006;8:3375. doi: 10.1021/ol0613057. [DOI] [PubMed] [Google Scholar]; (d) Wipf P, Kendall C. Top Organomet Chem. 2004;8:1. [Google Scholar]
- 3.(a) Vagner J, Qu H, Hruby VJ. Curr Opin Chem Biol. 2008;12:292. doi: 10.1016/j.cbpa.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wipf P, Xiao J, Stephenson CRJ. Chimia. 2009;63:764. doi: 10.2533/chimia.2009.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Li CS, Deschenes D, Desmarais S, Falgueyret J-P, Gauthier JY, Kimmel DB, Léger S, Massé F, McGrath ME, McKay DJ, Percival MD, Riendeau D, Rodan SB, Thérien M, Truong V-L, Wesolowski G, Zamboni R, Black WC. Bioorg Med Chem Lett. 2006;16:1985. doi: 10.1016/j.bmcl.2005.12.071. [DOI] [PubMed] [Google Scholar]; (b) Zanda M. New J Chem. 2004;28:1401. [Google Scholar]; (c) Leftheris K, Kline T, Vite GD, Cho YH, Bhide RS, Patel DV, Patel MM, Schmidt RJ, Weller HN, Andahazy ML, Carboni JM, Gullo-Brown JL, Lee FYF, Ricca C, Rose WC, Yan N, Barbacid M, Hunt JT, Meyers CA, Seizinger BR, Zahler R, Manne V. J Med Chem. 1996;39:224. doi: 10.1021/jm950642a. [DOI] [PubMed] [Google Scholar]
- 5.(a) Dondoni A, Perrone D. Tetrahedron Lett. 1992;33:7259. [Google Scholar]; (b) Vabeno J, Lejon T, Nielsen CU, Steffansen B, Chen W, Ouyang H, Borchardt RT, Luthman K. J Med Chem. 2004;47:1060. doi: 10.1021/jm031022+. [DOI] [PubMed] [Google Scholar]
- 6.Reid RC, Pattenden LK, Tyndall JDA, Martin JL, Walsh T, Fairlie DP. J Med Chem. 2004;47:1641. doi: 10.1021/jm030337m. [DOI] [PubMed] [Google Scholar]
- 7.(a) Myers AG, Barbay JK, Zhong B. J Am Chem Soc. 2001;123:7207. doi: 10.1021/ja010113y. [DOI] [PubMed] [Google Scholar]; (b) Aoyagi Y, Williams RM. Tetrahedron. 1998;54:10419. [Google Scholar]; (c) Lama T, Del Valle SE, Genest N, Lubell WD. Int J Pept Res Ther. 2007;13:355. [Google Scholar]
- 8.Righi G, Ronconi S, Bonini C. Eur J Org Chem. 2002;2002:1573. [Google Scholar]
- 9.(a) Wipf P, Xiao J, Jiang J, Belikova NA, Tyurin VA, Fink MP, Kagan VE. J Am Chem Soc. 2005;127:12460. doi: 10.1021/ja053679l. [DOI] [PubMed] [Google Scholar]; (b) Wipf P, Xiao J, Geib SJ. Adv Synth Catal. 2005;347:1605. [Google Scholar]
- 10.(a) Jakobsche CE, Peris G, Miller SJ. Angew Chem Int Ed. 2008;47:6707. doi: 10.1002/anie.200802223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wipf P, Henninger TC, Geib SJ. J Org Chem. 1998;63:6088. doi: 10.1021/jo981057v. [DOI] [PubMed] [Google Scholar]
- 11.(a) Price JL, Horne WS, Gellman SH. J Am Chem Soc. 2010;132:12378. doi: 10.1021/ja103543s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seebach D, Lukaszuk A, Patora-Komisarska K, Podwysocka D, Gardiner J, Ebert M-O, Reubi JC, Cescato R, Waser B, Gmeiner P, Hübner H, Rougeot C. Chem Biodivers. 2011;8:711. doi: 10.1002/cbdv.201100093. [DOI] [PubMed] [Google Scholar]
- 12.(a) Daly MJ, Ward RA, Thompson DF, Procter G. Tetrahedron Lett. 1995;36:7545. [Google Scholar]; (b) Wipf P, Xiao J. Org Lett. 2005;7:103. doi: 10.1021/ol0477529. [DOI] [PubMed] [Google Scholar]; (c) Jenkins CL, Vasbinder MM, Miller SJ, Raines RT. Org Lett. 2005;7:2619. doi: 10.1021/ol050780m. [DOI] [PubMed] [Google Scholar]; (d) Fu Y, Bieschke J, Kelly JW. J Am Chem Soc. 2005;127:15366. doi: 10.1021/ja0551382. [DOI] [PubMed] [Google Scholar]; (e) Tomita K, Narumi T, Niida A, Oishi S, Ohno H, Fujii N. Pept Sci. 2007;88:272. doi: 10.1002/bip.20676. [DOI] [PubMed] [Google Scholar]; (f) Wiktelius D, Luthman K. Org Biomol Chem. 2007;5:603. doi: 10.1039/b616906f. [DOI] [PubMed] [Google Scholar]; (g) Bandur NG, Harms K, Koert U. Synthesis. 2007;2007:2720. [Google Scholar]; (h) Wipf P, Fritch PC. J Org Chem. 1994;59:4875. [Google Scholar]; (i) Wipf P, Henninger TC. J Org Chem. 1997;62:1586. [Google Scholar]
- 13.Xiao J, Weisblum B, Wipf P. J Am Chem Soc. 2005;127:5742. doi: 10.1021/ja051002s. [DOI] [PubMed] [Google Scholar]
- 14.(a) Fink MP, Macias CA, Xiao J, Tyurina YY, Jiang J, Belikova N, Delude RL, Greenberger JS, Kagan VE, Wipf P. Biochem Pharmacol. 2007;74:801. doi: 10.1016/j.bcp.2007.05.019. [DOI] [PubMed] [Google Scholar]; (b) Kanai A, Zabbarova I, Amoscato A, Epperly M, Xiao J, Wipf P. Org Biomol Chem. 2007;5:307. doi: 10.1039/b613334g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jiang J, Kurnikov I, Belikova NA, Xiao J, Zhao Q, Amoscato AA, Braslau R, Studer A, Fink MP, Greenberger JS, Wipf P, Kagan VE. J Pharmacol Exp Ther. 2007;320:1050. doi: 10.1124/jpet.106.114769. [DOI] [PubMed] [Google Scholar]
- 15.Lee DL, Hodges RS. Pept Sci. 2003;71:28. [Google Scholar]
- 16.Xiao J, Weisblum B, Wipf P. Org Lett. 2006;8:4731. doi: 10.1021/ol0617704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kagan VE, Wipf P, Stoyanovsky D, Greenberger JS, Borisenko G, Belikova NA, Yanamala N, Samhan Arias AK, Tungekar MA, Jiang J, Tyurina YY, Ji J, Klein-Seetharaman J, Pitt BR, Shvedova AA, Bayir H. Adv Drug Delivery Rev. 2009;61:1375. doi: 10.1016/j.addr.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wipf P, Jahn H. Tetrahedron. 1996;52:12853. [Google Scholar]
- 19.(a) Hart DW, Schwartz J. J Am Chem Soc. 1974;96:8115. [Google Scholar]; (b) Schwartz J, Labinger JA. Angew Chem Int Ed. 1976;15:333. [Google Scholar]
- 20.(a) Chen Q-Y, Wu S-W. J Chem Soc Chem Commun. 1989:705. [Google Scholar]; (b) Fei X-S, Tian W-S, Chen Q-Y. J Chem Soc Perkin Trans. 1998;1:1139. [Google Scholar]
- 21.Wipf P, Lim S. Angew Chem Int Ed. 1993;32:1068. [Google Scholar]
- 22.(a) Shaw AW, deSolms SJ. Tetrahedron Lett. 2001;42:7173. [Google Scholar]; (b) Fujisawa T, Kooriyama Y, Shimizu M. Tetrahedron Lett. 1996;37:3881. [Google Scholar]; (c) Plobeck N, Powell D. Tetrahedron: Asymmetry. 2002;13:303. [Google Scholar]
- 23.(a) Wipf P, Kendall C, Stephenson CRJ. J Am Chem Soc. 2001;123:5122. doi: 10.1021/ja0157494. [DOI] [PubMed] [Google Scholar]; (b) Wipf P, Kendall C, Stephenson CRJ. J Am Chem Soc. 2002;125:761. doi: 10.1021/ja028092a. [DOI] [PubMed] [Google Scholar]; (c) Wipf P, Kendall C. Chem Eur J. 2002;8:1778. doi: 10.1002/1521-3765(20020415)8:8<1778::aid-chem1778>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 24.Wipf P, Stephenson CRJ. Org Lett. 2003;5:2449. doi: 10.1021/ol0347141. [DOI] [PubMed] [Google Scholar]
- 25.(a) Baeurle S, Blume T, Guenther J, Henschel D, Hillig RC, Husemann M, Mengel A, Parchmann C, Schmid E, Skuballa W. Bioorg Med Chem Lett. 2004;14:1673. doi: 10.1016/j.bmcl.2004.01.052. [DOI] [PubMed] [Google Scholar]; (b) Lazo JS, Aslan DC, Southwick EC, Cooley KA, Ducruet AP, Joo B, Vogt A, Wipf P. J Med Chem. 2001;44:4042. doi: 10.1021/jm0102046. [DOI] [PubMed] [Google Scholar]; (c) Brezak M-C, Quaranta M, Contour-Galcera M-O, Lavergne O, Mondesert O, Auvray PØ, Kasprzyk PG, Prevost GP, Ducommun B. Mol Cancer Ther. 2005;4:1378. doi: 10.1158/1535-7163.MCT-05-0168. [DOI] [PubMed] [Google Scholar]
- 26.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;13:3769. [Google Scholar]
- 27.Frantz M-C, Pierce JG, Pierce JM, Kangying L, Qingwei W, Johnson M, Wipf P. Org Lett. 2011;13:2318. doi: 10.1021/ol200567p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Staas DD, Savage KL, Homnick CF, Tsou NN, Ball RG. J Org Chem. 2002;67:8276. doi: 10.1021/jo0259313. [DOI] [PubMed] [Google Scholar]
- 29.Frantz M-C, Skoda EM, Davoren JE, Wang Z, Epperly MW, Stripay JL, Tyurin VA, Fink B, Greenberger JS, Bayir H, Rob-bins P, Niedernhofer L, Kagan VE, Wipf P. Manuscript in Preparation. 2011 [Google Scholar]
- 30.Hoye AT, Davoren JE, Wipf P, Fink MP, Kagan VE. Acc Chem Res. 2008;41:87. doi: 10.1021/ar700135m. [DOI] [PubMed] [Google Scholar]
- 31.Frantz M-C, Wipf P. Environ Mol Mutagen. 2010;51:462. doi: 10.1002/em.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macias CA, Chiao JW, Xiao J, Arora Devinder S, Tyurina YY, Delude RL, Wipf P, Kagan VE, Fink MP. Annals of Surg. 2007;245:305. doi: 10.1097/01.sla.0000236626.57752.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.(a) Jiang J, Belikova Natalia A, Hoye Adam T, Zhao Q, Epperly Michael W, Greenberger Joel S, Wipf P, Kagan Valerian E. Int J Radiat Oncol Biol Phys. 2008;70:816. doi: 10.1016/j.ijrobp.2007.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rwigema J-CM, Beck B, Wang W, Doemling A, Epperly MW, Shields D, Goff Julie P, Franicola D, Dixon T, Frantz M-C, Wipf P, Tyurina Y, Kagan VE, Wang H, Greenberger JS. Int J Radiat Oncol Biol Phys. 2011;80:860. doi: 10.1016/j.ijrobp.2011.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Epperly MW, Goff JP, Li S, Gao X, Wipf P, Dixon T, Wang H, Franicola D, Shen H, Rwigema J-CM, Kagan VE, Bernard M, Greenberger JS. In Vivo. 2010;24:811. [PMC free article] [PubMed] [Google Scholar]; (b) Gokhale A, Rwigema J-C, Epperly MW, Glowacki J, Wang H, Wipf P, Goff JP, Dixon T, Patrene K, Greenberger JS. In Vivo. 2010;24:377. [PMC free article] [PubMed] [Google Scholar]
- 35.Rajagopalan MS, Gupta K, Epperly MW, Franicola D, Zhang X, Wang H, Zhao H, Tyurin VA, Pierce JG, Kagan VE, Wipf P, Kanai AJ, Greenberger JS. In Vivo. 2009;23:717. [PMC free article] [PubMed] [Google Scholar]
- 36.Goff JP, Epperly MW, Dixon T, Wang H, Franicola D, Shields D, Wipf P, Li S, Gao X, Greenberger JS. In Vivo. 2011;25:315. [PMC free article] [PubMed] [Google Scholar]
- 37.Tyurina YY, Tyurin VA, Kaynar AM, Kapralova VI, Wasserloos K, Li J, Mosher M, Wright L, Wipf P, Watkins S, Pitt BR, Kagan VE. Am J Physiol. 2010;299:L73. doi: 10.1152/ajplung.00035.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ji J, Kline A, Cheng J, Wipf P, Tyurin VA, Alexander H, Kochanek PM, Kagan VE, Bayir HJ. J Neurotrauma. 2009;26:A3. [Google Scholar]