

Abstract
Regulatory sites that control gene expression are essential to the proper functioning of cells, and identifying them is critical for modeling regulatory networks. We have developed Magma (Multiple Aligner of Genomic Multiple Alignments), a software tool for multiple species, multiple gene motif discovery. Magma identifies putative regulatory sites that are conserved across multiple species and occur near multiple genes throughout a reference genome. Magma takes as input multiple alignments that can include gaps. It uses efficient clustering methods that make it about 70 times faster than PhyloNet, a previous program for this task, with slightly greater sensitivity. We ran Magma on all non-coding DNA conserved between Caenorhabditis elegans and five additional species, about 70 Mbp in total, in <4 h. We obtained 2,309 motifs with lengths of 6–20 bp, each occurring at least 10 times throughout the genome, which collectively covered about 566 kbp of the genomes, approximately 0.8% of the input. Predicted sites occurred in all types of non-coding sequence but were especially enriched in the promoter regions. Comparisons to several experimental datasets show that Magma motifs correspond to a variety of known regulatory motifs.
Key words: ChIP analysis, cis-regulatory elements, eukaryotic motif-finding, fast motif-finding, genome-wide motif-finding, motif-expression association, motif redundancy, transcription factor binding site discovery
1. Introduction
A key area of genomic research is understanding the cis-regulatory network that governs transcriptional regulation. Over the past two decades, many computational approaches have been developed to discover transcription factor (TF) binding sites in the genome by identifying recurring sequence motifs that bind a particular factor. Discovering such motifs is challenging, because they are usually short (5–12 bases) and degenerate.
Traditional algorithms to recognize motifs in genomic DNA take one of two basic approaches. The multiple gene, single species approach recognizes motifs because they recur with few changes in the promoters of multiple genes within a single genome (Stormo and Hartzell, 1989; Lawrence et al., 1993; Hertz and Stormo, 1999; Bailey et al., 2006; Elemento et al., 2007). In contrast, the single gene, multiple species—or phylogenetic footprinting—approach recognizes motifs in a single promoter region by their conservation across species, which is assumed to be greater than that of the surrounding background sequence (Gelfand, 1999; McGuire et al., 2000; McCue et al., 2001; Panina et al., 2001; Rajewsky et al., 2002; Frazer et al., 2003; Panina et al., 2003; Marchal et al., 2004). These methods work because binding sites are typically under selective pressure and therefore mutate more slowly than the surrounding sequence. Wang and Stormo (2003) combined these two approaches in their PhyloCon program, which uses alignments of orthologous promoter regions rather than individual DNA sequences. In this paradigm, a motif is required both to recur across different promoters and to be conserved across species in each of its occurrences. Other tools take a conceptually similar approach (Qin et al., 2003; Jensen et al., 2005; Monsieurs et al., 2006), all of which report results on bacterial promoters.
To scale PhyloCon's methods to discover motifs across an entire genome, the successor program PhyloNet (Wang and Stormo, 2005) implemented a BLAST-like seeded alignment algorithm to accelerate detection of putative motif instances across thousands of promoters. This allowed its application to all noncoding sequences of the yeast genome, but still at a high cost: >5 CPU-days on a 2.4-GHz workstation. The noncoding sequences of a higher eukaryotic genome represent tens to hundreds of times more sequences than yeast. Most phylogenetically based motif-finding algorithms scale quadratically with the input size, so the lengthy times expected for higher eukaryotic promoter analyses are a deterrent to genome-wide motif discovery.
This work describes Magma (Multiple Aligner of Genomic Multiple Alignments), a new algorithm for multi-gene, multi-species computational motif discovery. Magma significantly departs from the PhyloNet pipeline for accelerated operations, most substantially by introducing new algorithms to group putative TF binding sites into motifs and to reduce redundancy in its output. Magma also operates on gapped genomic sequence alignments. Using alignments of Saccharomyces promoters, Magma runs almost 70 times faster than PhyloNet with improved sensitivity. Magma scales to analyses of higher eukaryotes; it can analyze all proximal promoters in Drosophila in less time than that required by PhyloNet to analyze yeast. Although Magma's efficiency allows us to perform whole-genome motif-finding on higher eukaryotes, its motif-finding methods can sometimes produce many redundant, partially overlapping motifs. We alleviate this problem with a fast, greedy, set-covering approach (Chvatal, 1979).
We demonstrate Magma's motif discovery prowess using essentially all of Caenorhabditis elegans non-coding sequence: a 70-Mbp search space consisting of promoters, Un-Translated Regions (UTRs), introns, and downstream regions. To the best of our knowledge, this is the most comprehensive motif-finding effort to date in C. elegans. Furthermore, we show that these motifs and their conserved exemplar sites correspond to many known regulatory sites, are enriched in TF-bound regions, and are correlated with expression. Magma and all post-processing software and results are available at http://stormo.wustl.edu/∼nihuegbu/Magma/homepage.html.
2. Methods
Magma computation
Magma takes as input a collection of multiple sequence alignments or profiles such as the Multiple Alignment Format (MAF) blocks from University of California Santa Cruz (UCSC). These blocks are alignments of orthologous genomic sequences from different species. Its goal is to discover short motifs, which are approximate sequence patterns that occur in multiple instances, or exemplar sites, within each genome and appear distinct from the surrounding sequence. However, because Magma searches profiles rather than single sequences, each instance of a motif is itself a collection of aligned sequences exhibiting significant conservation across the species in its profile. Magma compares pairs of profiles using the average log-likelihood ratio (ALLR) score, a measure of similarity between columns of two multiple alignments (Wang and Stormo, 2003). The ALLR is well-defined for pairs of columns containing different total numbers of characters, so it may be applied to columns which have different number of bases due to gaps. For two motifs of equal length, their total ALLR score is simply the sum of the ALLR scores of their corresponding columns, ignoring gapped positions.
Magma discovers motifs by comparing one input profile, the query, to a database of all other profiles considering both possible orientations. Each profile in the input serves as the query in turn, until all profiles have been compared pairwise. Magma's search has two phases: generation of high-scoring segment pairs (HSPs), which locally align two profiles, and clustering of all HSPs involving a given query to form motifs. HSP generation is further subdivided into seed matching and extension.
An HSP is a local alignment of the query profile and a database profile, such that the total ALLR score of all aligned column pairs exceeds a user-defined threshold T. To reduce the computational cost of search, and to allow identification of multiple HSPs per profile pair, HSP generation uses a seeded alignment approach on a simplified representation of the input profiles. Each input profile is first quantized into a sequence over an alphabet of 15 symbols, each of which represents a particular vector of base counts, by mapping each profile column to the symbol whose vector has the most similar distribution (Wang and Stormo, 2005). The alignment score for a pair of symbols is the ALLR score for the corresponding pair of vectors. The quantized query and database profiles are scanned for seed matches, or pairs of fixed-length substrings with at least some minimum score, using a neighborhood hashing strategy analogous to that used by BLASTP for sequence alignment. Each seed match between two profiles is extended by dynamic programming into the best HSP passing through the match, and HSPs with scores exceeding T are retained. Whereas seed matching is done on the quantized profiles, extension is done in the original profiles using the full ALLR score.
Magma's clustering algorithm
The clustering phase collects and aligns putative motif instances from the HSPs generated by the previous phase. A cluster is a collection of HSPs, all of which overlap on a given query profile Pq. A cluster of n HSPs therefore defines intervals from at most n distinct profiles besides the query, all of which are aligned to Pq (and hence transitively to each other).
Clustering first groups all HSPs for a query, then reduces each cluster to a single motif, with each interval possibly contributing one motif instance. A motif may use only a subset of the cluster's intervals, and each interval must be adjusted so that all instances of the motif have the same length. Subsetting and length adjustment are performed so as to maximize the sum of ALLR scores between the instance drawn from Pq and each other instance in the motif.
Magma uses efficient clustering methods that offer strong performance and quality guarantees. Edges of an HSP overlap graph are determined by overlaps between intervals on the same profile, making this graph an interval graph. All maximal cliques in such a graph can easily be found in time linear in the number of HSPs and enumerated in time proportional to their total size (Gupta et al., 1982). Magma therefore uses interval clique finding to guarantee both maximality and exhaustive enumeration of clusters, with much better scalability than general clique finding. To avoid building clusters from HSPs that overlap by very little (e.g., a single base), it is desirable to enforce a minimum overlap of k positions to create an edge in the overlap graph. Magma enforces this criterion by reducing each interval's right endpoint by k-1 positions prior to clique finding.
To simplify conversion of clusters to motifs, Magma uses the following enumerative algorithm. For each HSP Hj in the cluster, let Pq (the query) and Pj be the profiles that it aligns, and let [lj, rj] and [l'j, r'j] be the intervals that it aligns from Pq and Pj, respectively. Let dj = l'j - lj be the diagonal of Hj, that is, the offset of its starting indices in the query and database profiles.
Suppose that the HSPs in a cluster have minj lj = L and maxj rj = R. For each left endpoint ℓ and right endpoint r, L ≤ ℓ ≤ r ≤ R, we find the best-scoring motif whose instance on Pq is the interval [ℓ, r]. The instance corresponding to HSP Hj is then [ℓ + dj, r + dj]. (If this instance runs off either end of Pj, then it is discarded for this choice of endpoints.) We then discard any instance whose ALLR score versus the query instance is negative and retain the total score sℓ,r of the remaining instances. The motif with the highest total ALLR score for the cluster is the one with endpoints argmaxℓ,r sℓr in profile Pq. Our enumerative algorithm requires time Θ(m2n), where n is the number of HSPs in the cluster and m = R - L + 1. However, the ALLR scores for each column of the alignment between each Pj and Pq can be precomputed and stored in total time Θ(mn). Hence, the constant factor associated with the quadratic cost in m is small in practice, consisting mostly of addition and table lookup. We also note that when the goal is instead to minimize the statistical p-value defined in (Wang and Stormo, 2005) for the motif, the motif with best p-value for a cluster can still be found in time Θ(m2 n log n).
Reducing redundant motifs
The motifs obtained by HSP finding and clustering may contain many overlapping, partially redundant motifs. The major source of redundancy is the re-use of overlapping profiles in construction of multiple motifs. Since we know the genomic coordinates of all the exemplar sites that were used to construct every motif, we can re-describe this problem as an NP-complete set-covering problem (Karp, 1972; Vazirani, 2001). Given a universe U of exemplar contigs (i.e., contiguous regions built from overlapping exemplar sites) and a collection of motifs S, each of which covers a subset of U, a cover is a subset C of S whose union of exemplar sites covers all of U.
We implement a fast greedy approximation for the set-covering problem to significantly reduce the motif redundancy in the final output. Greedy algorithms for minimum set-covering achieve a log n approximation, where n is the size of the largest set (Chvatal, 1979). This means we use at most log n times the minimum number of motifs needed to cover all instances. Our implementation is similar to other set-covering solutions but with some slight modifications. At each iteration, we define a cover as the set of sites from the most occurring motif (m*), as well as sites from any other motif that overlaps m* sites by at least d sites. Thus, at each iteration, we remove a set of sites u* in U and their associated motifs from the problem. We continue this recursion as long as |u*| ≥ Mu minimum unique sites (a default value of 10 unique sites per motif ). The redundant motifs in each resulting cover are subsequently resolved by iteratively scanning all the sites with each motif (by order of most occurrences) and masking their instances. This continues until there are fewer than Mu sites left in the cover.
3. Results
Magma is a fast genome-wide motif-finder with tractable scaling for higher-order eukaryotes
Magma was designed in part to overcome performance limitations in the earlier PhyloNet motif-finding software. To measure Magma's performance relative to PhyloNet, we ran both programs to discover initial motifs in yeast promoters. On a cluster of 2.4-GHz AMD Opteron processors, we observed a ∼70-fold speedup. Moreover, Magma's ability to use gapped profiles, which better aligns motif instances in different parts of the same profile, allowed it to discover more known motifs than PhyloNet while still including less of the reference sequence in its output. We also examined how Magma scales when applied to more complex eukaryotes (Table 1). Running Magma on D. melanogaster's conserved promoter regions (an approximately ninefold increase in search space) required about 30-fold more time than the yeast experiment. The complete C. elegans conserved regulome from six species (∼40-fold search space) required ∼130-fold more time (∼3.5 h). In practice, we implement Magma such that the set of all queries is distributed across several processors, so that the actual running time for C. elegans was only ∼0.75 h.
Table 1.
Magma (Multiple Aligner of Genomic Multiple Alignments) Scales to Higher-Order Eukaryotes with Practical Runtime
| Organism | Search Space (Mbp) | Magma-DiscoveryTime (cpu secs) |
|---|---|---|
| S. cereviasae | 1.74 | 101 |
| D. melanogaster | 15.36 | 3184 |
| C. elegans | 69.56 | 12915 |
Characteristics of Magma C. elegans motifs
We discovered 2,309 motifs in C. elegans, with lengths of 6–20 bases. These motifs are composed of 65,747 unique, non-coding, conserved exemplar sites covering 566,666 bp (∼0.8% of the C. elegans input sequence). These sites are distributed across all non-coding regions but have the most occurrences in the promoter regions, as would be expected for regulatory sites (Table 2). We make these motifs available on our website as position-specific count matrices (PSCMs).
Table 2.
Distribution of Sites in Different Non-Coding Sequence Classes
| Location | Number of Sites* | Coverage (bp) | Size of input region (bp) | Fraction of input region |
|---|---|---|---|---|
| 2kb 5′ Intergenic | 34,278 | 258,322 | 21,532,733 | 1.20% |
| 5′UTR | 2,596 | 15,411 | 461,624 | 3.34% |
| 1st Intron | 15,514 | 73,904 | 7,918,585 | 0.93% |
| Other Intron | 27,787 | 122,333 | 23,691,626 | 0.52% |
| 3′UTR | 4,436 | 27,111 | 1,934,557 | 1.40% |
Note: Some of these sites overlap different regulatory regions of multiple genes.
Evaluation of Magma C. elegans motifs
We assessed whether Magma's motifs are consistent with the known binding sites for the few characterized factors and with other information about regulatory interactions. Because we do not expect Magma's exemplar sites for each motif to be a comprehensive list of all sites for its associated TF, we scan each non-coding region in our input with the PWM for each motif to determine if it was significantly enriched in instances of the motif. The expected number of motif instances arising by chance is determined by the information content of the motif (Schneider et al., 1986; Hertz and Stormo, 1999), whereas the observed number is the actual number of sites within each dataset whose score exceeds the information content of the motif. The score of a putative motif with respect to a given dataset is the log-likelihood ratio
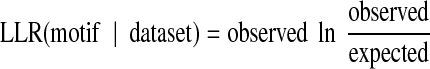 |
.
One of the best-characterized TFs in C. elegans is the Nuclear Factor I (NFI). Whittle et al. (2009) performed ChIP-CHIP for NFI, probing its in vivo targets at 55 regions (∼1500 bp each). Magma finds two motifs that are strongly enriched within those regions, both very similar to the known consensus of TTGGCAN3TGCCAA (Fig. 1).
FIG. 1.

Log likelihood ratio uncovers NPS-like motifs on NPI CNP peaks.
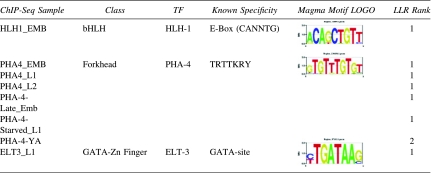
The modENCODE consortium identified regions from ChIP-Seq experiments that bound several TFs (Gerstein et al., 2010). These regions, with average length of 200 bases, were filtered to remove those that overlapped ubiquitous HOT sites, leaving 74,065 regions from 28 samples that bound a total of 23 different TFs (PHA-4 was assayed at six different developmental and environmental conditions). For each sample, we ranked the motifs using the above LLR score. For the three TFs with known motifs, the most significant Magma motif matches the known consensus (Table 3; for the PHA-4-YA set the second-ranked motif matches the consensus). Significant motifs were found for each of the remaining ChIP-Seq datasets, but since the TFs binding these sites have unknown motifs, we could not use them to validate Magma's performance.
Table 3.
Magma (Multiple Aligner of Genomic Multiple Alignments) Motifs in modENCODE ChIP Peaks
 |
We also identified significant motifs for 12 factors with at least 10 promoter binding observations from the EDGE database of Yeast-One Hybrid (Y1H) experiments (Barrasa et al., 2007), though again the correct motifs for these sites are not known a priori. The Oreganno database lists 187 different experimentally tested binding sites and cis-regulatory modules in C. elegans (Montgomery et al., 2006; Griffith et al., 2008), which includes the annotated bound factors for several sites. We find significant matches among our Magma motifs for 185 of these sites, including motifs whose specificity resembles that of TFs matching annotated PHA-4, ELT-2, and DAF-19 sites.
Hunt-Newbury et al. (2007) built promoter/GFP fusion libraries for approximately 2,000 C. elegans genes and cataloged the temporal and spatial expression of the green fluorescent protein. Dupuy et al. (2007) conducted similar studies that monitored the tempo-spatial expression of promoter reporter constructs. Chikina et al. (2009) used support vector machines (SVMs) to predict other genes from C. elegans with similar expression profiles to these experimental results and achieved 90% precision for all of the major tissues (intestine, hypodermis, muscle, neurons, pharynx) except germ-line. Using this SVM predicted dataset, we identified enriched motifs by computing an occupancy score for each motif and each promoter in each tissue-specific gene set (Granek and Clarke, 2005). We recovered several known cis-regulatory elements that regulate or establish tissue expression. For instance, ELT-2 is a zinc finger protein that is known to bind to GATA cis-based elements to regulate transcription in C. elegans intestines (McGhee et al., 2007). Figure 2 shows three GATA motifs and their tissue enrichments (log p-values). Although GATA elements are mostly enriched in the promoters of intestine-expressed genes, we also found it enriched in the introns (especially the first intron) of neuronal and muscle tissue-types such as pharynx, uterus, and vulva, consistent with previous developmental studies highlighting the broad role of GATA factors in development (Spencer et al., 2011). We re-discovered other known cis-acting elements that endow tissue-specific expression, such as PHA-4- and PHA-4-variant-like motifs enriched in the pharynx.
FIG. 2.

Enrichments of three GATA-like motifs in different tissues. All three motifs are enriched in promoter regions of intestinal genes. Alternative motifs are enriched in a few other tissues.
We further analyzed 88 C. elegans ChIP and expression microarray series data sets from the GEO Omnibus database, including 1,362 total samples. Similarly to the previous section, we analyzed the occupancy scores for our discovered motifs to uncover significant enrichments with the differentially regulated genes from each expression sample. We identified significant motifs for 991 different samples. We found that a motif matching the known specificity of DAF-16 (GTTGTTTAC) is significantly enriched in daf-2/daf-16 mutant experiments (McElwee et al., 2004). DAF-16 has also been shown to be involved in starvation response in C. elegans (Henderson and Johnson, 2001), and samples from starvation experiments (Baugh et al., 2009) are significantly enriched for the same motif.
4. Discussion
We have described Magma, a program that identifies motifs that are conserved across species and occur in several locations within the reference genome. In a comparison to the PhyloNet program on the yeast genome, we found slightly higher sensitivity with greatly increased speed, about 70× faster. The entire non-coding conserved genome of C. elegans, about 70 Mbp, can be analyzed in <4 h on a single CPU. We observed that Magma scales sub-quadratically with its input size, due to lower density of strongly conserved regions hence less HSP extensions per seed. Although the lack of extensive knowledge about regulatory motifs in C. elegans hinders a comprehensive evaluation of Magma's specificity, comparison to known motifs from a variety of experimental datasets show that its motifs are generally consistent with existing knowledge. Finally, we posit that these motifs likely represent specificities for TFs involved in various regulatory networks controlling gene expression in different conditions and developmental processes.
Acknowledgments
We acknowledge Ting Wang for helpful comments. Funding was provided by NIH (grant HG00249 to G.D.S.) and NSF (grant ITR-427794 to J.B.).
Disclosure Statement
No competing financial interests exist.
References
- Bailey T.L. Williams N. Misleh C., et al. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–W373. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrasa M.I. Vaglio P. Cavasino F., et al. EDGEdb: a transcription factor–DNA interaction database for the analysis of C. elegans differential gene expression. BMC Genomics. 2007;8:21. doi: 10.1186/1471-2164-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh L.R. Demodena J. Sternberg P.W. RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science. 2009;324:92–94. doi: 10.1126/science.1169628. [DOI] [PubMed] [Google Scholar]
- Chikina M.D. Huttenhower C. Murphy C.T., et al. Global prediction of tissue-specific gene expression and context-dependent gene networks in Caenorhabditis elegans. PLoS Comput. Biol. 2009;5:e1000417. doi: 10.1371/journal.pcbi.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvatal V. A greedy heuristic for the set-covering problem. Math. Operations Res. 1979;4:233–235. [Google Scholar]
- Dupuy D. Bertin N. Hidalgo C.A., et al. Genome-scale analysis of in vivo spatiotemporal promoter activity in Caenorhabditis elegans. Nat. Biotechnol. 2007;25:663–668. doi: 10.1038/nbt1305. [DOI] [PubMed] [Google Scholar]
- Elemento O. Slonim N. Tavazoie S. A universal framework for regulatory element discovery across all genomes and data types. Mol. Cell. 2007;28:337–350. doi: 10.1016/j.molcel.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer K.A. Elnitski L. Church D.M., et al. Cross-species sequence comparisons: a review of methods and available resources. Genome Res. 2003;13:1–12. doi: 10.1101/gr.222003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand M.S. Recognition of regulatory sites by genomic comparison. Res. Microbiol. 1999;150:755–771. doi: 10.1016/s0923-2508(99)00117-5. [DOI] [PubMed] [Google Scholar]
- Gerstein M.B. Lu Z.J. Van Nostrand E.L., et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granek J.A. Clarke N.D. Explicit equilibrium modeling of transcription-factor binding and gene regulation. Genome Biol. 2005;6:R87. doi: 10.1186/gb-2005-6-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith O.L. Montgomery S.B. Bernier B., et al. ORegAnno: an open-access community-driven resource for regulatory annotation. Nucleic Acids Res. 2008;36:D107–D113. doi: 10.1093/nar/gkm967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta U.I. Lee D.T. Leung J.Y.-T. Efficient algorithms for interval graphs and circular arc-graphs. Networks. 1982;12:459–467. [Google Scholar]
- Henderson S.T. Johnson T.E. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Hertz G.Z. Stormo G.D. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- Hunt-Newbury R. Viveiros R. Johnsen R., et al. High-throughput in vivo analysis of gene expression in Caenorhabditis elegans. PLoS Biol. 2007;5:e237. doi: 10.1371/journal.pbio.0050237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen S.T. Shen L. Liu J.S. Combining phylogenetic motif discovery and motif clustering to predict co-regulated genes. Bioinformatics. 2005;21:3832–3839. doi: 10.1093/bioinformatics/bti628. [DOI] [PubMed] [Google Scholar]
- Karp R.M. Reducibility among combinatorial problems, 85–103. In: Miller R.E., editor; Thatcher J.W., editor. Complexity of Computer Computations. Plenum; New York: 1972. [Google Scholar]
- Lawrence C.E. Altschul S.F. Boguski M.S., et al. Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment. Science. 1993;262:208–214. doi: 10.1126/science.8211139. [DOI] [PubMed] [Google Scholar]
- Marchal K. De Keersmaecker S. Monsieurs P., et al. In silico identification and experimental validation of PmrAB targets in Salmonella typhimurium by regulatory motif detection. Genome Biol. 2004;5:R9. doi: 10.1186/gb-2004-5-2-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCue L. Thompson W. Carmack C., et al. Phylogenetic footprinting of transcription factor binding sites in proteobacterial genomes. Nucleic Acids Res. 2001;29:774–782. doi: 10.1093/nar/29.3.774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwee J.J. Schuster E. Blanc E., et al. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J. Biol. Chem. 2004;279:44533–44543. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- McGhee J.D. Sleumer M.C. Bilenky M., et al. The ELT-2 GATA-factor and the global regulation of transcription in the C. elegans intestine. Dev. Biol. 2007;302:627–645. doi: 10.1016/j.ydbio.2006.10.024. [DOI] [PubMed] [Google Scholar]
- McGuire A.M. Hughes J.D. Church G.M. Conservation of DNA regulatory motifs and discovery of new motifs in microbial genomes. Genome Res. 2000;10:744–757. doi: 10.1101/gr.10.6.744. [DOI] [PubMed] [Google Scholar]
- Monsieurs P. Thijs G. Fadda A.A., et al. More robust detection of motifs in coexpressed genes by using phylogenetic information. BMC Bioinform. 2006;7:160. doi: 10.1186/1471-2105-7-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery S.B. Griffith O.L. Sleumer M.C., et al. ORegAnno: an open access database and curation system for literature-derived promoters, transcription factor binding sites and regulatory variation. Bioinformatics. 2006;22:637–640. doi: 10.1093/bioinformatics/btk027. [DOI] [PubMed] [Google Scholar]
- Panina E.M. Mironov A.A. Gelfand M.S. Comparative analysis of FUR regulons in gamma-proteobacteria. Nucleic Acids Res. 2001;29:5195–5206. doi: 10.1093/nar/29.24.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panina E.M. Vitreschak A.G. Mironov A.A., et al. Regulation of biosynthesis and transport of aromatic amino acids in low-GC Gram-positive bacteria. FEMS Microbiol. Lett. 2003;222:211–220. doi: 10.1016/S0378-1097(03)00303-3. [DOI] [PubMed] [Google Scholar]
- Qin Z.S. McCue L.A. Thompson W., et al. Identification of co-regulated genes through Bayesian clustering of predicted regulatory binding sites. Nat. Biotechnol. 2003;21:435–439. doi: 10.1038/nbt802. [DOI] [PubMed] [Google Scholar]
- Rajewsky N. Socci N.D. Zapotocky M., et al. The evolution of DNA regulatory regions for proteo-gamma bacteria by interspecies comparisons. Genome Res. 2002;12:298–308. doi: 10.1101/gr.207502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T.D. Stormo G.D. Gold L., et al. Information content of binding sites on nucleotide sequences. J. Mol. Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- Spencer W.C. Zeller G. Watson J.D., et al. A spatial and temporal map of C. elegans gene expression. Genome Res. 2011;21:325–341. doi: 10.1101/gr.114595.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stormo G.D. Hartzell G.W., 3rd Identifying protein-binding sites from unaligned DNA fragments. Proc. Natl. Acad. Sci. USA. 1989;86:1183–1187. doi: 10.1073/pnas.86.4.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazirani V.V. Approximation Algorithms. Springer; New York: 2001. [Google Scholar]
- Wang T. Stormo G.D. Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics. 2003;19:2369–2380. doi: 10.1093/bioinformatics/btg329. [DOI] [PubMed] [Google Scholar]
- Wang T. Stormo G.D. Identifying the conserved network of cis-regulatory sites of a eukaryotic genome. Proc. Natl. Acad. Sci. USA. 2005;102:17400–17405. doi: 10.1073/pnas.0505147102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle C.M. Lazakovitch E. Gronostajski R.M., et al. DNA-binding specificity and in vivo targets of Caenorhabditis elegans nuclear factor I. Proc. Natl. Acad. Sci. USA. 2009;106:12049–12054. doi: 10.1073/pnas.0812894106. [DOI] [PMC free article] [PubMed] [Google Scholar]