

Abstract
Legumain or asparaginly endopeptidase (AEP) is a lysosomal cysteine protease with a high level of specificity for cleavage of protein substrates after an asparagine residue. It is also capable of cleaving after aspartic acids sites when in the acidic environment of the lysosome. Legumain expression and activity is linked to a number of pathological conditions including cancer, atherosclerosis and inflammation, yet its biological role in these pathologies is not well-understood. Highly potent and selective inhibitors of legumain would not only be valuable for studying the functional roles of legumain in these conditions, but may have therapeutic potential as well. We describe here the design, synthesis and in vitro evaluation of selective legumain inhibitors based on the aza-asparaginyl scaffold. We synthesized a library of aza-peptidyl inhibitors with various non-natural amino acids and different electrophilic warheads, and characterized the kinetic properties of inactivation of legumain. We also synthesized fluorescently labeled inhibitors to investigate cell permeability and selectivity of the compounds. The inhibitors have second order rate constants of up to 5×104 M−1s−1 and IC50 values as low as 4 nM against recombinant mouse legumain. In addition, the inhibitors are highly selective toward legumain and have little or no cross-reactivity with cathepsins. Overall, we have identified several valuable new inhibitors of legumain that can be used to study legumain function in multiple disease models.
Keywords: Legumain, Protease inhibitors, Covalent inhibitors, Cysteine protease, Asparaginyl endopeptidase, Activity-based probes
Legumain is a lysosomal cysteine protease that is conserved in diverse cell types including, plants, invertebrate parasites and mammals. It has a high propensity to cleave protein substrates on the C-terminal side of asparagine residues1. Mammalian legumain is known to have a role in antigen processing2, 3, albumin maturation4 and matrix degradation5, 6 and it is also implicated in various pathological conditions including parasitic infection7, 8, atherosclerosis9, inflammation10 and tumorigenesis11, 12. In addition, legumain is found to be over-expressed in the majority of human solid tumors such as carcinomas of the breast, colon and prostate11, and knock-down of legumain in mouse models of cancer results in a marked decrease in tumor growth and metastasis12. Based on these recent findings, legumain may be a therapeutically important enzyme, especially in tumor progression and metastasis. Therefore, highly selective and potent legumain inhibitors will be valuable for studying the roles of legumain in diseases but also potentially useful for the treatment of these diseases. A number of legumain inhibitors have been developed and tested in vitro13–16. Notably, Powers and coworkers have developed highly selective and potent inhibitors of legumain in blood flukes and hard ticks17. However, only a few of these inhibitors have been tested in mammalian cells or disease models that have therapeutic impact18.
Previously, we have reported the development of irreversible inhibitors and active site probes of legumain in intact cells and mice19. A unique feature of these inhibitors is that they contain both an aza-asparagine and a proline moiety in the P2 position that minimizes cross-reactivity towards other cysteine proteases such as the cathepsins. In an effort to optimize selectivity and potency of these inhibitors, we decided to extend our dipeptidyl aza-asparaginyl scaffold by adding various non-natural amino acids in the P3 position. We also wanted to test several different electrophilic reactive groups (Fig. 1 and Table 1). Based on the previously published study from our group20, 21, we selected twelve non-natural amino acids that were found to prefer legumain over the caspases in a P3 positional scan of a caspase inhibitor library. We initially synthesized a group of compounds containing the core aza-asparagine and proline from our previously reported lead compound19 linked to each of the selected non-natural amino acids in the P3 position and an aza Michael acceptor electrophile. In addition, we also prepared original dipeptidyl scaffold found in our original lead compound to determine the effects of using this electrophile in place of the original epoxide functional group. We synthesized all compounds using the solid phase synthetic methods previously reported by our laboratory22 with a slight modification in order to attach the third amino acid (Scheme 1 and supplementary data).
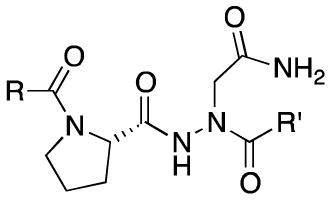
Figure 1.

Structures of the aza-peptidyl inhibitors of legumain.
Table 1.
IC50 values and second-order rate constants for inhibition of legumain by aza-peptidyl inhibitorsa
| ||||
|---|---|---|---|---|
| Inhibitor | Legumain | CathepsinL | ||
| R | R′ | IC50 (nM) | kobs/[I] (M−1s−1) | IC50 (μM) |
| NN1 | Michael Acceptor | 6.5 | 36169 | 964 |
| NN2 | Michael Acceptor | 141 | 3450 | ND |
| NN3b | Michael Acceptor | 148 | 4994 | ND |
| NN4 | Michael Acceptor | 9.3 | 24412 | >1mM |
| NN5 | Michael Acceptor | 198 | 3622 | ND |
| NN6 | Michael Acceptor | 17 | 14480 | 855 |
| NN7b | Michael Acceptor | 638 | 1757 | ND |
| NN8 | Michael Acceptor | 167 | 5376 | ND |
| NN9 | Michael Acceptor | >1000 | ND | ND |
| NN10 | Michael Acceptor | >1000 | ND | ND |
| NN11 | Michael Acceptor | 267 | 3621 | ND |
| NN12b | Michael Acceptor | >1000 | ND | ND |
| Ac | Michael Acceptor | 8.3 | 10802 | >1mM |
| Ac | Epoxide | 9.3 | 27397 | 370 |
| NN1 | Epoxide | 8.1 | 53674 | 890 |
| NN4 | Epoxide | 4.4 | 46467 | 106 |
| NN6 | Epoxide | 8.6 | 25713 | 358 |
Inhibition assays for legumain were performed in 0.1 M citrate-phosphate, 4 mM DTT, pH 5.8, <0.1% DMSO with a final concentration of 10 μM Cbz-Ala-Ala-Asn-AMC as substrate. Second-order rate constants were determined by linear or nonlinear regression analysis as described in the supplementary data. All measurements were triplicated and the average values were reported. Detailed procedures for the enzyme assays can be found in the supplementary data.
These compounds (NN3, NN7 and NN12 with Michael acceptor) were isolated and tested as hydrolized fumaric acid.
Scheme 1.

Synthesis of legumain inhibitors (a) DIC, NMP, 1h (b) Fmoc-hydrazide, DIEA, NMP;(c) Allyl chloroformate, DIEA, DMF; (d) 20% piperidine in DMF then Fmoc-Pro-OH, DIC, HOBT, DMF; (e) 20% piperidine in DMF then Fmoc-NN1~12-OH, HATU, DIEA, DMF; (f) 20% piperidine in DMF then Acetic anhydride, DIEA, DMF; (g) Phenylsilane, Pd(PPh3)4, DCM; (h) Fumaric acid monoethyl ester (for Michael acceptor series) or ethyl L- trans-epoxysuccinic acid (for epoxide series), HBTU, DIEA, DMF, overnight then 95%TFA.
We carried out a simple IC50 determination of each compound against recombinant mouse legumain and the results are shown in Table 1. Within this compound series, we found that when the P3 side chains were small alkyl groups (i.e R = NN1, NN4, NN6), these compounds showed excellent inhibitory effect. If these R groups were relatively bulky groups such as aromatic or piperazine groups (R = NN7, NN9, NN10, NN12), these compounds showed a substantial drop in activity. It should be noted that all the previous work done by other groups mainly focused on modifications of the P1’ site17, however our results suggest that additional specificity and potency may be achieved by modifications at sites distal to the active site cysteine.
Next, we chose three Michael acceptor inhibitors (R = NN1, NN4, NN6) with the most optimal IC50 values and replaced the Michael acceptor electrophile with the epoxide group from our initial lead compound. All six compounds, both with Michael acceptor and epoxide electrophiles had similar potency with IC50 values in the low nano molar range. Interestingly, the second-order inhibition rate constants (kobs/[I]), indicated that the epoxide-containing compounds were approximately 1.5~2-fold more potent than their Michael acceptor counterparts. These data suggest that the epoxide electrophile reacts with the active site cysteine faster than the Michael acceptor electrophile. However, this faster reactivity may lead to an increase in overall off-target modification. In agreement with this idea, we found that while all the compounds had weak inhibition of cathepsin L (> 100 μM), all inhibitors containing the epoxide electrophile showed slightly higher reactivity toward cathepsin compared to the Michael acceptor compounds.
In order to further assess the potential cross reactivity of the compounds, we generated activity based probe of the most potent compounds for use in cells. This was accomplished by attachment of a Cy5 fluorophore followed by labeling of RAW264.7 macrophages. We chose to focus on the compounds containing NN1 with the Michael acceptor electrophile and NN4 with the epoxide electrophile since these two inhibitors showed the highest potency in each series. The results of the cellular labeling study are shown in Figure 2. As expected, all four probes showed highly selective labeling of legumain at nanomolar concentrations. However, the epoxide containing probes (LP-1, LP-4) labeled cathepsins at high probe concentrations indicating that higher reactivity probably contributed to this off-target labeling. We did not observe significant differences in labeling intensity for all four probes, although it seems that epoxide probes were slightly more sensitive compared to the Michael acceptor probes (LP-2, LP-3). Regardless, all the synthesized probes were cell permeable and highly selective toward legumain.
Figure 2.

Intact cell labeling of legumain with fluorescently labeled inhibitors. (a) Structures of Cy5-labeled legumain probes (b) RAW 264.7 macrophages were treated with Cy5-labeled legumain inhibitors for 60 min at different concentrations. After incubation, cells were lysed under hypotonic conditions and all labeled proteins were separated by 12.5% SDS-PAGE and analyzed by scanning the gel with a Typhoon flatbed scanner.
In conclusion, we have designed and synthesized aza epoxide and aza Michael acceptor inhibitors for legumain. We incorporated non-natural amino acids to improve potency and selectivity of the previously developed dipeptidyl scaffold. We found that inhibitors with small alkyl groups in the P3 position demonstrated slightly enhanced inhibitory effect compared to the inhibitor without a P3 amino acid, whereas inhibitors with bulkier aromatic groups had significantly reduced activity. In addition, inhibitors containing the aza-epoxide electrophile have faster inhibition kinetics compared to the Michael acceptor compounds, but also showed more cross-reactivity toward cathepsins. We also attached a Cy5 fluorophore to the selected inhibitors to test specificity and reactivity inside living cells and all the synthesized probes selectively labeled legumain in intact RAW264.7 macrophages. Currently we are moving forward to scale up some of these inhibitors to investigate the therapeutic applications of legumain inhibitors in various mouse models of inflammation and cancer.
Supplementary Material
Acknowledgments
We thank Andrew Guzzetta and Juan Valderramos for HR-MS analysis and Drs. Victoria Albrow and Fangfang Yin for helpful discussions about the project.
Footnotes
Supplementary material includes the synthetic procedures and characterization data of all compounds and the experimental protocols for enzyme assays and cellular labeling experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Ishii S. Methods Enzymol. 1994;244:604. doi: 10.1016/0076-6879(94)44044-1. [DOI] [PubMed] [Google Scholar]
- 2.Manoury B, Mazzeo D, Li DN, Billson J, Loak K, Benaroch P, Watts C. Immunity. 2003;18:489. doi: 10.1016/s1074-7613(03)00085-2. [DOI] [PubMed] [Google Scholar]
- 3.Maehr R, Hang HC, Mintern JD, Kim YM, Cuvillier A, Nishimura M, Yamada K, Shirahama-Noda K, Hara-Nishimura I, Ploegh HL. J Immunol. 2005;174:7066. doi: 10.4049/jimmunol.174.11.7066. [DOI] [PubMed] [Google Scholar]
- 4.Mylne J, Colgrave M, Daly N, Chanson A, Elliott A, McCallum E, Jones A, Craik D. Nat Chem Biol. 2011;7:257. doi: 10.1038/nchembio.542. [DOI] [PubMed] [Google Scholar]
- 5.Morita Y, Araki H, Sugimoto T, Takeuchi K, Yamane T, Maeda T, Yamamoto Y, Nishi K, Asano M, Shirahama-Noda K, Nishimura M, Uzu T, Hara-Nishimura I, Koya D, Kashiwagi A, Ohkubo I. FEBS Lett. 2007;581:1417. doi: 10.1016/j.febslet.2007.02.064. [DOI] [PubMed] [Google Scholar]
- 6.Sottile J, Hocking DC. Mol Biol Cell. 2002;13:3546. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gotz MG, James KE, Hansell E, Dvorak J, Seshaadri A, Sojka D, Kopacek P, McKerrow JH, Caffrey CR, Powers JC. J Med Chem. 2008;51:2816. doi: 10.1021/jm701311r. [DOI] [PubMed] [Google Scholar]
- 8.James KE, Gotz MG, Caffrey CR, Hansell E, Carter W, Barrett AJ, McKerrow JH, Powers JC. Biol Chem. 2003;384:1613. doi: 10.1515/BC.2003.179. [DOI] [PubMed] [Google Scholar]
- 9.Clerin V, Shih HH, Deng N, Hebert G, Resmini C, Shields KM, Feldman JL, Winkler A, Albert L, Maganti V, Wong A, Paulsen JE, Keith JC, Jr, Vlasuk GP, Pittman DD. Atherosclerosis. 2008 doi: 10.1016/j.atherosclerosis.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 10.Chan CB, Abe M, Hashimoto N, Hao C, Williams IR, Liu X, Nakao S, Yamamoto A, Zheng C, Henter JI, Meeths M, Nordenskjold M, Li SY, Hara-Nishimura I, Asano M, Ye K. Proc Natl Acad Sci U S A. 2009;106:468. doi: 10.1073/pnas.0809824105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu C, Sun C, Huang H, Janda K, Edgington T. Cancer Res. 2003;63:2957. [PubMed] [Google Scholar]
- 12.Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, Markowitz D, Wu W, Liu C, Reisfeld RA, Xiang R. J Clin Invest. 2006;116:2132. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loak K, Li DN, Manoury B, Billson J, Morton F, Hewitt E, Watts C. Biological chemistry. 2003;384:1239. doi: 10.1515/BC.2003.136. [DOI] [PubMed] [Google Scholar]
- 14.Niestroj AJ, Feussner K, Heiser U, Dando PM, Barrett A, Gerhartz B, Demuth HU. Biological chemistry. 2002;383:1205. doi: 10.1515/BC.2002.133. [DOI] [PubMed] [Google Scholar]
- 15.Gotz MG, James KE, Hansell E, Dvorak J, Seshaadri A, Sojka D, Kopacek P, McKerrow JH, Caffrey CR, Powers JC. Journal of medicinal chemistry. 2008;51:2816. doi: 10.1021/jm701311r. [DOI] [PubMed] [Google Scholar]
- 16.James KE, Gotz MG, Caffrey CR, Hansell E, Carter W, Barrett AJ, McKerrow JH, Powers JC. Biological chemistry. 2003;384:1613. doi: 10.1515/BC.2003.179. [DOI] [PubMed] [Google Scholar]
- 17.Ovat A, Muindi F, Fagan C, Brouner M, Hansell E, Dvorak J, Sojka D, Kopacek P, McKerrow JH, Caffrey CR, Powers JC. Journal of medicinal chemistry. 2009;52:7192. doi: 10.1021/jm900849h. [DOI] [PubMed] [Google Scholar]
- 18.Stern L, Perry R, Ofek P, Many A, Shabat D, Satchi-Fainaro R. Bioconjugate chemistry. 2009;20:500. doi: 10.1021/bc800448u. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Bogyo M. ACS Chem Biol. 2010;5:233. doi: 10.1021/cb900232a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger AB, Witte MD, Denault JB, Sadaghiani AM, Sexton KM, Salvesen GS, Bogyo M. Mol Cell. 2006;23:509. doi: 10.1016/j.molcel.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 21.Edgington LE, Berger AB, Blum G, Albrow VE, Paulick MG, Lineberry N, Bogyo M. Nat Med. 2009;15:967. doi: 10.1038/nm.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kato D, Verhelst SH, Sexton KB, Bogyo M. Org Lett. 2005;7:5649. doi: 10.1021/ol052275v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.