

Abstract
Purpose
The goal of the current studies was to elucidate the role of the principal poly(ADP-ribose)polymerase isoform, PARP1 in the regulation of cellular energetics in endothelial cells under resting conditions and during oxidative stress.
Methods
We utilized bEnd.3 endothelial cells and A549 human transformed epithelial cells. PARP1 was inhibited either by pharmacological inhibitors or by siRNA silencing. The Seahorse XF24 Extracellular Flux Analyzer was used to measure indices of mitochondrial respiration (oxygen consumption rate) and of glycolysis (extracellular acidification rate). Cell viability, cellular and mitochondrial NAD+ levels and mitochondrial biogenesis were also measured.
Results
Silencing of PARP1 increased basal cellular parameters of oxidative phosphorylation, providing direct evidence that PARP1 is a regulator of mitochondrial function in resting cells. Pharmacological inhibitors of PARP1 and siRNA silencing of PARP1 protected against the development of mitochondrial dysfunction and elevated the respiratory reserve capacity in endothelial cells exposed to oxidative stress. The observed effects were unrelated to an effect on mitochondrial biogenesis. Isolated mitochondria of A549 human transformed epithelial cells exhibited an improved resting bioenergetic status after stable lentiviral silencing of PARP1; these effects were associated with elevated resting mitochondrial NAD+ levels in PARP1 silenced cells.
Conclusions
PARP1 is a regulator of basal cellular energetics in resting endothelial and epithelial cells. Furthermore, endothelial cells respond with a decrease in their mitochondrial reserve capacity during low-level oxidative stress, an effect, which is attenuated by PARP1 inhibition. While PARP1 is a regulator of oxidative phosphorylation in resting and oxidatively stressed cells, it only exerts a minor effect on glycolysis.
Keywords: Oxidative stress, poly(ADP)ribose polymerase, mitochondrial bioenergetics, oxidative phosphorylation, intracellular NAD+ content, respiratory reserve capacity
1. Introduction
One of the common features of cardiovascular diseases (hypertension, atherosclerosis, reperfusion injury, diabetes) and of critical illness (sepsis, shock, hemorrhage, trauma, burns) is endothelial dysfunction, characterized by reduced vasorelaxant responses to endothelium-dependent relaxant agents [1–7]. The pathogenesis of endothelial dysfunction is attributed, at least in part, to the formation of oxygen- and nitrogen-derived reactive species, which induce endothelial dysfunction via multiple interacting cellular actions [1–7].
The mitochondria of endothelial cells are important in the regulation of endothelial function both in health and disease [8–10]. When cells are subjected to stress, mitochondria are capable of drawing upon a ‘reserve capacity’, which is available to serve the increased energy demands for maintenance of organ function, cellular repair or detoxification of reactive species [11,12]. Impairment or depletion of this reserve capacity ultimately leads to excessive protein damage and cell death. Exhaustion of the reserve capacity and subsequent loss of bioenergetic control by exposure to reactive oxygen and nitrogen species results in mitochondrial protein modifications, followed by inhibition of mitochondrial respiration, processes that may ultimately result in cell death [11,12].
The nuclear enzyme poly(ADP-ribose) polymerase 1 (PARP1, EC 2.4.2.30) is the most abundant isoform of the PARP enzyme family [3, 13–16]. PARP1 functions as a DNA damage sensor and signaling molecule. PARP1 has an important role in the cellular repair mechanism of single-stranded DNA breaks. Upon recognizing breaks in the DNA strands, PARP1 forms homodimers and catalyzes the cleavage of NAD+ into nicotinamide and ADP-ribose to form long branches of ADP-ribose polymers on a number of target proteins including histones, DNA polymerase and PARP1 itself. Poly(ADP-ribosylation) confers negative charge on histones leading to electrostatic repulsion among histones and DNA, a process implicated in chromatin remodeling, DNA repair and transcriptional machinery. Poly(ADP-ribosyl)action (PARylation) is a fast dynamic process, which is also indicated by the short half-life of the polymer, and determined by two catabolic enzymes, poly(ADP-ribose) glycohydrolase (PARG) and ADP-ribosyl protein lyase [13–16]. The cellular overactivation of PARP1 has been linked to cell necrosis; pharmacological inhibition or genetic depletion of PARP1 affords a protective phenotype in a variety of cardiovascular, inflammatory and neurological disorders [13–16].
A number of prior studies investigated the role of PARP1 in the regulation of cellular energetics during oxidative and nitrosative stress [3,13–16]. The majority of these studies relied on indirect end-point measurements for cellular energetics, and utilized pharmacological inhibitors of PARP, as opposed to genetic inactivation. Although these studies demonstrated a significant role of PARP in the regulation of cellular energetics in cells subjected to conditions of oxidative stress, the role of PARP1 in resting cellular energetics have not yet been delineated. Our goal was to define the role of PARP1 in the regulation of cellular energetics in resting endothelial cells, as well as under conditions of mild oxidative stress, by a non-invasive continuous measurement of oxidative phosphorylation and glycolysis, utilizing extracellular flux analysis technology [11,12,17–22].
2. Materials and methods
2.1. Materials
Adenosine diphosphate, alcohol dehydrogenase, antimycin A, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), 3-(4,5-dimethyl-2- thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT), Gly-Gly buffer, 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride (INT), lactic acid, oligomycin, nicotinamide adenine dinucleotide (NAD+), N-methylphenazonium methyl sulfate (PMS), 1% Non Essential Amino Acid, and PARP inhibitors, the PJ34 (23) and 5-AIQ (24) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum, glutamine, penicillin and streptomycin were procured from Invitrogen (Carlsbad, CA, USA). Small interfering RNA (siRNA) for PARP1 was purchased from Applied Biosystems/Ambion (Austin, TX, USA). The lipofectamineTM 2000 transfection reagent and the Mitotracker green FM fluorescent stain were purchased from Invitrogen (Carlsbad, CA, USA). Anti-PARP1 and anti-actin HRP linked antibodies for Western blotting were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA).
2.2. Cell culture
The bEnd.3 cerebral vascular endothelial cell line was purchased from the European Collection of Cell Cultures (ECACC, Salisbury, UK), and cultured in a 37 °C, 5% CO2 atmosphere with 1 g/l glucose (5.5 mmol/l) containing Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA), 2 mmol/l glutamine, 100 IU/ml penicillin and 100 µg/ml streptomycin and 1% Non Essential Amino Acid (Sigma, St. Louis, MO, USA). 60.000 cells per well were plated into 96-well cell culture plates (Corning Glass, Corning, NY, USA) or into XF24 V7 Seahorse cell culture plates. bEnd.3 cells from passage numbers 21–35 were used for subsequent assays. In an additional set of experiments, we also utilized the A549 human transformed pulmonary epithelial cell line (control and stable lentiviral silencing of PARP1), obtained from the European Collection of Cell Cultures (ECACC, Salisbury, UK) [25].
2.3. MTT cell viability assay
To estimate the number of viable cells, 3-(4,5-dimethyl-2- thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) was added to the cells at a final concentration of 0.5 mg/ml, and cultured at 37 °C, 5% CO2 atmosphere for 1 hour (26,27). Cells were washed with PBS and the converted formazan dye was dissolved in isopropanol and measured at 570 nm with background measurement at 690 nm on a SpectraMax M2 reader (Molecular Devices Corp., Sunnyvale, CA, USA).
2.4. LDH cytotoxicity assay
Cell culture supernatants (30 µl) were mixed with 100 ml freshly prepared lactate dehydrogenase assay reagent to reach final concentrations of 85 mM lactic acid, 1040 mM nicotinamide adenine dinucleotide (NAD), 224 mM N-methylphenazonium methyl sulfate (PMS), 528 mM 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride (INT) and 200 mM Tris (pH 8.2). The changes in absorbance were read kinetically at 492 nm for 15 min. LDH activity values are shown as Vmax for kinetic assays in mOD/min [27].
2.5. Measurement of cellular or mitochondrial NAD+ levels
Total cellular and mitochondrial NAD+ content was determined using a previosly described method [26] with minor modifications. Cells or mitochondrial preparations were subjected to 0.5 N HClO4, then neutralized with 3 M KOH/125 mM Gly-Gly buffer (pH 7.4). After gently mixing, the samples were centrifuged at 10,000 × g for 5 min at 4 °C. The supernatants were added to 61 mM Gly-Gly buffer (pH 7.4) containing final concentrations of 0.1 mM MTT, 0.9 mM phenazine methosulfate, 0.3 U/ml alcohol dehydrogenase, 100 mM nicotinamide and 5.7% ethanol. The NAD+ assay is based on an alchohol dehydrogenase cyclic reaction, in which the intensity of reduced terazolium dye (MTT) is measured kinetically at 560 nm for 1 h using a SpectraMax M2 reader (Molecular Devices Corp., Sunnyvale, CA, USA). NAD+ values were calculated as nmoles per mg protein of total cell homogenate or mitochondrial homogenate, respectively.
2.6. PARP-1 silencing by siRNA
The small interfering RNA (siRNA) oligonucleotides, which we used to silence PARP1 in bEnd.3 cells, were pre-designed by Applied Biosystems/Ambion (Austin, TX, USA). Cells (100.000 per well) were seeded into 6-well tissue culture plates and cultured in normal culture medium to reach approximately 50% confluence in 24-hours. At this point, the growth medium was replaced with Opti-Mem medium containing 10% fetal bovine serum (FBS) lacking antibiotics, and then cells were transfected with siRNA fragments that were allowed to form complexes with 10 µg/ml Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) for 20 min at room temperature in Opti-Mem medium lacking fetal calf serum and antibiotics. siRNA was used at a final concentration of 30 nM. To identify potential nonspecific effects, cells were transfected in parallel with scrambled siRNA (Ambion, Silencer® Negative Control#1). After 24-hours, cells were harvested from each transfected groups and 60,000 cells/well/0.32 cm2 were plated into 24-well XF24 V7 Seahorse cell culture plates. On the following day, Seahorse experiments were conducted to observe the effect of PARP1 knockdown on mitochondrial bioenergetics.
2.7. Bioenergetic analysis in intact cells
The XF24 Analyzer (Seahorse Biosciences, North Billerica, MA, USA) was used to measure bioenergetic function in intact bEnd.3 cells as described [11,12,17–22]. The XF24 creates a transient 7-µl chamber in specialized microplates that allows for OCR (oxygen consumption rate) and ECAR (extracellular acidification rate) or PPR (proton production rate, that is calculated from ECAR) to be monitored in real time over 3 hours. The changes of oxygen and proton concentrations are performed in real-time measurements via specific fluorescent dyes incorporated in Seahorse Flux Pak cartridges. Cells were subjected to hydrogen peroxide insults to create oxidative stress in bEnd.3 cells. Prior to the bioenergetic measurements, the culture medium was changed to unbuffered 5 mM glucose containing Dulbecco’s modified Eagle’s medium (DMEM, pH 7.4) supplemented with 2 mM L-glutamine and 1 mM sodium pyruvate. In preliminary studies, the optimum number of cells per well was determined as 60,000/0.32 cm2. This cell number allows the appropriate detection of changes in OCR, ECAR and PPR values for the subsequent experiments. Next, a protocol was implemented to measure indices of mitochondrial function. Oligomycin, FCCP and antimycin A were injected sequentially through ports of the Seahorse Flux Pak cartridges to reach final concentrations of 1 µg/ml, 0.3 µM and 2 µg/ml respectively. Using these agents, we determined the basal level of oxygen consumption, the amount of oxygen consumption linked to ATP production, the level of non-ATP-linked oxygen consumption (proton leak), the maximal respiration capacity and the non-mitochondrial oxygen consumption. Four basal OCR were recorded prior to injection of appropriate concentration of hydrogen peroxide, which oxidatively damaged bEnd.3 endothelial cells. After recording the H2O2-induced changes in the OCR and ECAR, oligomycin and FCCP were injected and further OCR-ECAR measurements were conducted. The OCR value measured after FCCP injection represents the maximal mitochondrial respiratory capacity of the cells. Finally, antimycin A was injected to inhibit the flux of electrons through complex III, and thus no oxygen is further consumed at cytochrome c oxidase. The remaining OCR after this treatment is primarily non-mitochondrial and is considered to be due to cytosolic oxidase enzymes.
2.8. Bioenergetic analysis in isolated mitochondria
Mitochondria were isolated and subjected to extracellular flux analysis as previously described [28]. We used a detailed method with minor modifications for mitochondria isolation from cell cultures developed by Christian Frezza and his colleagues [29]. Total protein (mg/ml) was determined using Pierce BCA Protein Assay reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA). Bioenergetic measurements commenced immediately after the isolation of the mitochondria. Mitochondrial assay solution (MAS, 1X) comprises 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA and 0.2% (w/v) fatty acid-free BSA, pH 7.2 at 37°C. The substrates, ADP and respiration reagents A were diluted in MAS, 1X. Respiration by the mitochondria (20 µg/well) was sequentially measured in a coupled state with substrate present (basal respiration), followed by State 3 (phosphorylating respiration, in the presence of ADP and substrate), State 4 (non-phosphorylating or resting respiration) following conversion of ADP to ATP, State 4o induced with the addition of oligomycin, and then maximal uncoupler-stimulated respiration (State 3u). This allowed the assessment of respiratory control ratios (RCR; State 3/State 4 and/or State 3u/State 4o).
2.9. Western blotting
Cells were washed with ice-cold phosphate-buffered saline (PBS, pH 7.4) and were lysed in 110 µl/well denaturing loading buffer (20 mM Tris, 2% SDS, 10% glycerol, 6 M urea, 100 µg/ml bromophenol blue, 200 mM β-mercaptoethanol), sonicated and boiled. Lysates (20 µl) were resolved on 4–12% NuPage Bis-Tris acrylamide gels (Invitrogen Carlsbad, CA, USA) and transferred to PVDF membranes. Membranes were blocked in 10% non-fat dried milk or Starting Block™ T20 (TBS), a commercially available blocking buffer solution (Fischer Scientific, Pittsburgh, PA, USA). Then membranes were probed overnight with anti-poly(ADP-ribose) enzyme 1 antibody (anti-PARP1, 1:1000). On the following day, anti-rabbit-horseradish peroxidase conjugate antibody (HRP, 1:2000, Cell Signaling or HRP, 1:3000, Abcam) was applied and enhanced chemiluminescent substrate (ECL, Pierce) were used to detect the signal in a CCD-camera-based chemiluminescence detection system (G:Box chemi XT, Syngene, Frederick, MD, USA). To normalize signals, membranes were re-probed with a HRP conjugated antibody against actin (anti-actin HRP linked, 1:2000, Santa Cruz Biotechnology, Inc.). PARP1 was detected at 116 kDa. Actin was determined at 43 kDa. The intensity of Western blot signals was quantified by densitometry using Image analysis software (GeneSnap, Syngene, Frederick, MD, USA). The ratios of the signals were expressed as normalized densitometry units.
2.10. Analysis of mitochondrial biogenesis
To estimate the cellular mitochondrial content, a specific green-fluorescent mitochondrial stain, the MitoTracker Green FM (Ex. 490 nm, Em 516 nm) (Invitrogen, Carlsbad, CA, USA) was applied, which localizes to mitochondria regardless of mitochondrial membrane potential. MitoTracker fluorescence signal was normalized to the cellular DNA content using a cell-permeant nuclear counterstain, Hoechst 33342 (Ex. 361 nm, Em. 486 nm) (Invitrogen, Carlsbad, CA, USA), which emits blue fluorescence upon binding to dsDNA [30].
2.11. Statistical analysis
Data are shown as means ± SEM. One-way and two-way ANOVA was used to detect differences between groups, as appropriate. Post-hoc comparisons were made using Tukey's test. The value of p < 0.05 was considered statistically significant. All statistical calculations were performed using Graphpad Prism 5 analysis software. The experiments were repeated independently at least three times performed on 3 different days.
3. Results
3.1. Characterization of the bioenergetic profile of bEnd.3 endothelial cells
To characterize the cellular bioenergetics of intact endothelial cells, extracellular flux analysis was used to determine rates of O2 consumption and glycolysis. In the first series of experiments, the optimal number of bEnd.3 cells per well was determined applying different cell number per well with various concentrations of FCCP. Both extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) showed a proportional response with cell number (data not shown). 60,000 cells/well/0.32 cm2 obtained measurable OCR and ECAR values. Accordingly for the subsequent experiments a seeding density of 60,000 cells/well/0.32 cm2 was selected to allow for optimal detection of changes in OCR and ECAR due to exposure to hydrogen peroxide. Mitochondrial function of the cells was determined by sequentially adding pharmacological inhibitors to probe the function of individual components of the respiratory chain, similar to previous approaches [11,12,17–22] (Fig. 1). According to our bioenergetic measurements, bEnd.3 cells have a basal O2 consumption rate of 213±40 pmoles/min, representing the mean and SEM of n=9 independent experiments), which amounts to approximately 60% of the maximal oxygen consumption achievable using the uncoupling agent FCCP. This finding indicates the presence of a significant reserve or spare respiratory capacity that is available for the cells to call upon when bioenergetic demand is increased (Fig. 1).
Fig. 1. Mitochondrial profile of cultured endothelial cells in response to sequential administration of pharmacological modulators of mitochondrial electron transport.

Elevated cell respiratory control ratio (RCR) represents adequate mitochondrial function, which provides sufficient spare respiratory capacity against cellular insults. During mitochondrial dysfunction the respiratory reserve capacity decreases, as evidenced by a reduced RCR value. Data are shown as means ± SEM.
Using the extracellular flux analysis methodology, mitochondrial function of the cells was determined by sequentially adding pharmacological inhibitors to probe the function of individual components of the respiratory chain, similar to previous approaches [11,12,17–22] (Fig. 1). First, the basal OCR was measured in adherent endothelial cells. To estimate the proportion of the basal OCR coupled to ATP synthesis, oligomycin (1 µg/ml) is applied to inhibit the ATP synthase (Complex V). The OCR decreases in response to oligomycin to the extent to which the cells are using mitochondria to generate ATP, while the remaining OCR can be ascribed to both proton leak across the mitochondrial anion carriers or through the lipid bilayer, and non-mitochondrial oxygen consumption [11,12]. This measurement is useful to compare the ATP-linked OCR among various experimental groups of cells and yields novel information regarding mitochondrial integrity. Furthermore, extended proton leak is implicated in mitochondrial dysfunction as an indicator of mitochondrial inner membrane damage. To determine the maximal OCR that the cells can sustain, the mitochondrial uncoupling agent FCCP (0.3 µM) was used. This agent results in stimulation of OCR, which occurs as the mitochondrial inner membrane becomes permeable to protons and electron transfer is no longer constrained by the proton gradient across the mitochondrial inner membrane. The more is the increase of the proton concentration in the mitochondrial matrix, the more elevated is the OCR response after FCCP administration. Subtraction of the basal rate from the maximal rate yields the mitochondrial reserve capacity [11,12,17–22], a relevant parameter of mitochondrial bioenergetics. The last component of our experimental protocol was the application of the Complex V inhibitor antimycin A (2 µg/ml) to inhibit the electron flow through Complex III. This agent causes a dramatic suppression of the OCR, such that the small amount of residual OCR is attributable to O2 consumption due to the formation of reactive oxygen species and non-mitochondrial sources (i.e. extramitochondrial enzymes which have the ability to convert oxygen).
Fig. 2. shows the results of a hydrogen peroxide (H2O2) concentration-response experiment on cell viability (MTT) and cytotoxicity (LDH release into the medium) in cultured bEnd.3 endothelial cells, showing a concentration-dependent decrease in MTT conversion, coupled with a concentration-dependent release of LDH release to the medium. The decrease in cell viability was slight at 900 µM, became prominent at 1300 µM, but was not associated with a significant increase in LDH release up to 3 mM H2O2 concentration. From these experiments, we selected concentration of 500–900 µM H2O2 as a stimulus that can induce a slight, but detectable cellular dysfunction, which, however, is not yet associated with overt cytotoxicity or the breakdown of the cell membrane integrity.
Fig. 2. Hydrogen peroxide decreases cell viability in cultured endothelial cells in a concentration-dependent fashion.
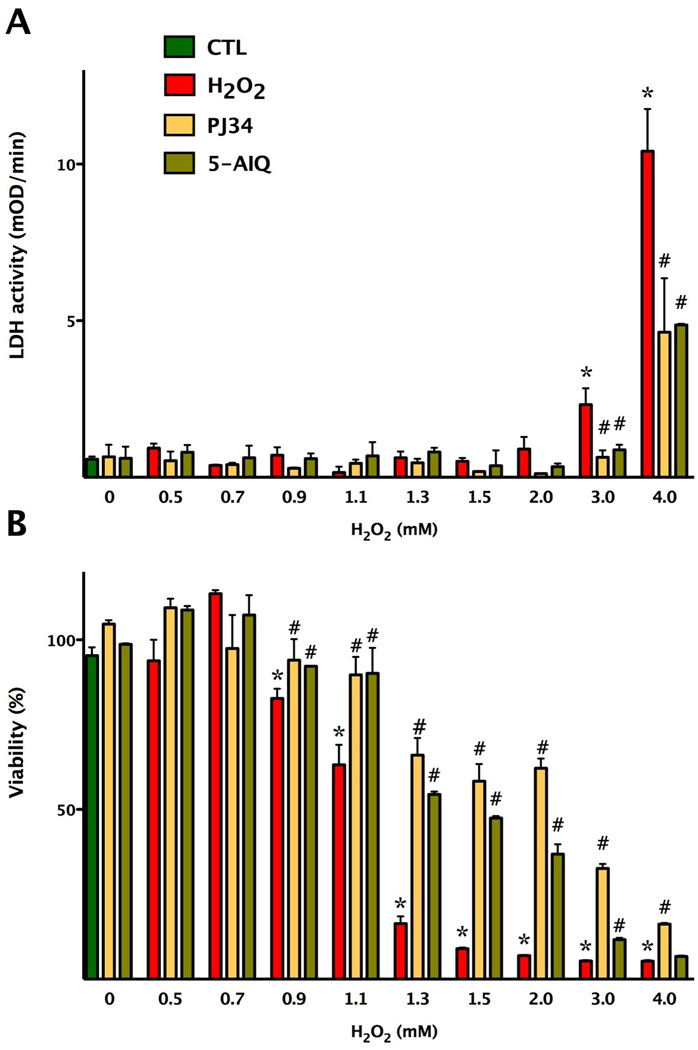
Confluent bEnd.3 cultures were subjected to different concentrations of hydrogen peroxide for 3 hours. Viability was determined by the MTT assay (A) and by measurement of LDH activity in the supernatant (B). Cells were pretreated with the PARP inhibitors PJ34 and 5-AIQ at 3 µM and at 10 µM concentrations, respectively, prior to H2O2. Data are shown as means ± SEM (n=4). *p<0.05 compared to the control group (CTL), #p<0.05 compared to hydrogen peroxide treated group.
Next, we examined the effects of acute oxidative stress on mitochondrial bioenergetics in bEnd.3 cells by exposing them to various concentrations (500 µM, 700 µM, 900 µM and 1.1 mM) H2O2. H2O2 diminished the FCCP mediated oxygen consumption rate in a concentration-dependent manner, and this effect correlated well with the area under the curve (AUC) analysis of oxygen consumption rates after the administration of different concentrations of hydrogen peroxide (Fig. 3). Furthermore, after the administration of H2O2, the oxygen consumption rate was lowered throughout the experimental period. Importantly, the reduced respiratory reserve capacity was already apparent at 500 and 700 µM H2O2, i.e. concentrations where no change in overall cell viability was noted by the MTT and LDH method (Figs 2–3). The decline of OCR values in response to oligomycin administration was completely abolished in H2O2 treated groups compared to the control group (Fig. 3), indicative of an extended proton leak. In parallel with the decrease in mitochondrial reserve capacity, the cells exhibited an elevated glycolytic activity during oxidative stress (Fig. 4).
Fig. 3. Hydrogen peroxide reduces the respiratory reserve capacity of oxidatively stressed endothelial cells in a concentration-dependent fashion.
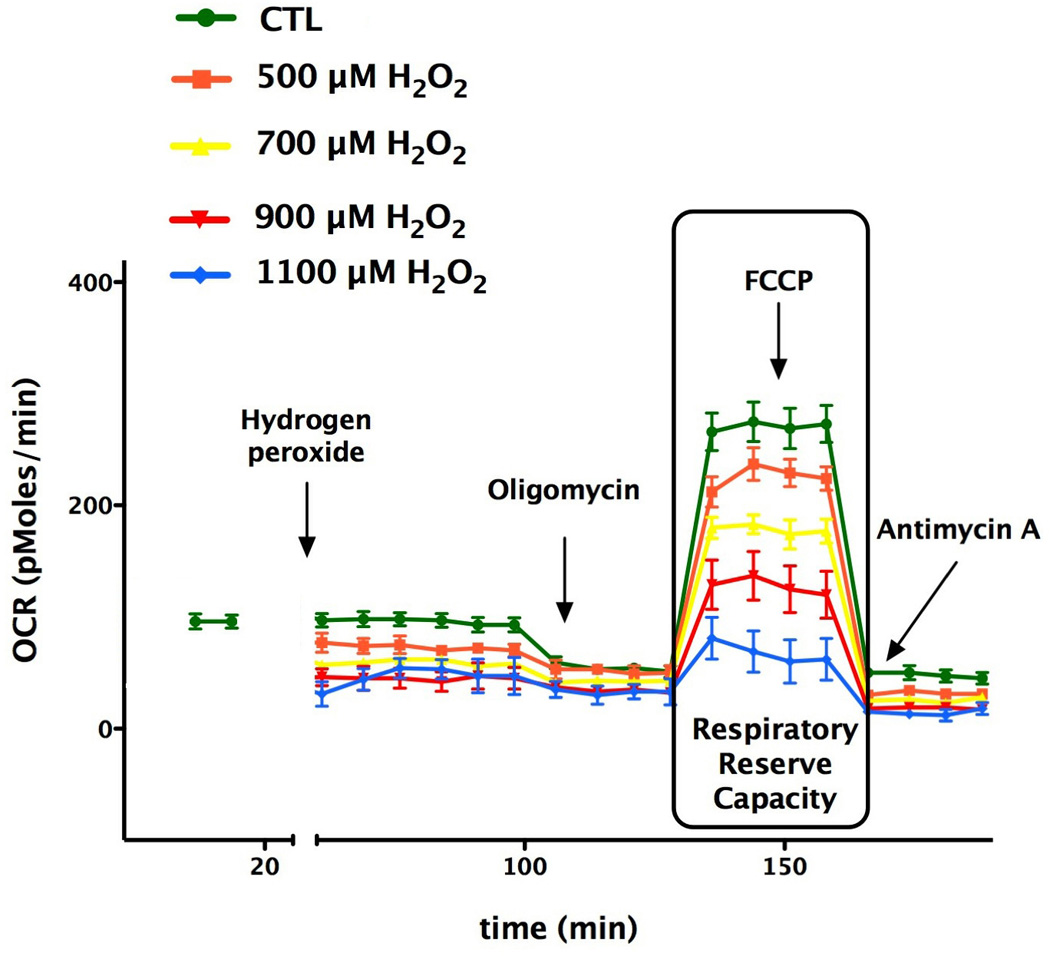
Bioenergetic analysis of the cells was performed using extracellular flux analysis. In the figure a time course for measurement of OCR for 60,000 cells/well/0.32 cm2 under the basal condition followed by the administration of different concentration of hydrogen peroxide (500–1100 µM) and the sequential addition of oligomycin (1 µg/ml), FCCP (0.3 µM), and antimycin A (2 µg/ml) are shown. H2O2 induced in a concentration-dependent manner the suppression of oxidative phosphorylation and respiratory reserve capacity as compared with control cell groups. Data are shown as means ± SEM of n=9 wells (each concentration of H2O2-treated groups) n= 21 wells (control group) collected from n=3 experiments performed on 3 different days.
Fig. 4. Hydrogen peroxide reduces the respiratory reserve capacity of oxidatively stressed endothelial cells, while elevating glycolytic activity.
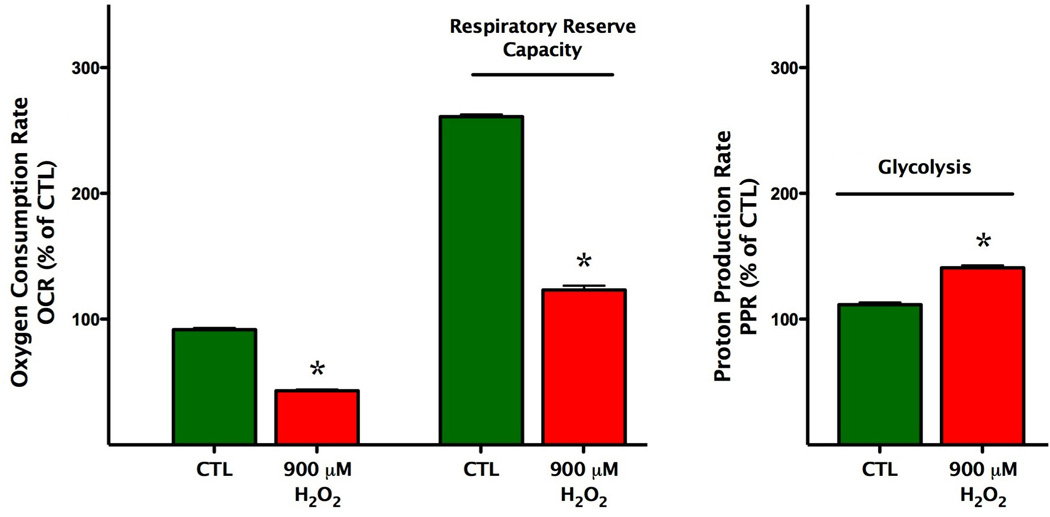
Bioenergetic analysis of the cells was performed using extracellular flux analysis. Data are shown for basal conditions and following by the administration of hydrogen peroxide (900 µM). H2O2 induced a suppression of oxidative phosphorylation and respiratory reserve capacity of the cells as compared with control cell groups (left panel), and elevated the glycolytic activity of the cells (right panel) (*p<0.05). Data are shown as means ± SEM of n=9 wells (each concentration of H2O2-treated groups) n=21 wells (control group) collected from n=3 experiments performed on 3 different days.
3.2. Effect of PARP1 silencing or PARP inhibition on mitochondrial function and cellular energetics
We selected the 900 µM concentration of H2O2 for the subsequent experiments to induce a reversible impairment in the mitochondrial respiratory reserve capacity of endothelial cultures, without eliciting overt cytotoxicity. PARP1 siRNA knockdown, resulting in approximately 50% suppression of PARP1 protein levels (Fig. 5), markedly enhanced baseline mitochondrial respiration and baseline mitochondrial reserve capacity in cultured bEnd.3 endothelial cells (Figs. 6–7).
Fig. 5. Effect of PARP1 siRNA silencing on the cellular expression of PARP1, as determined by Western blotting.
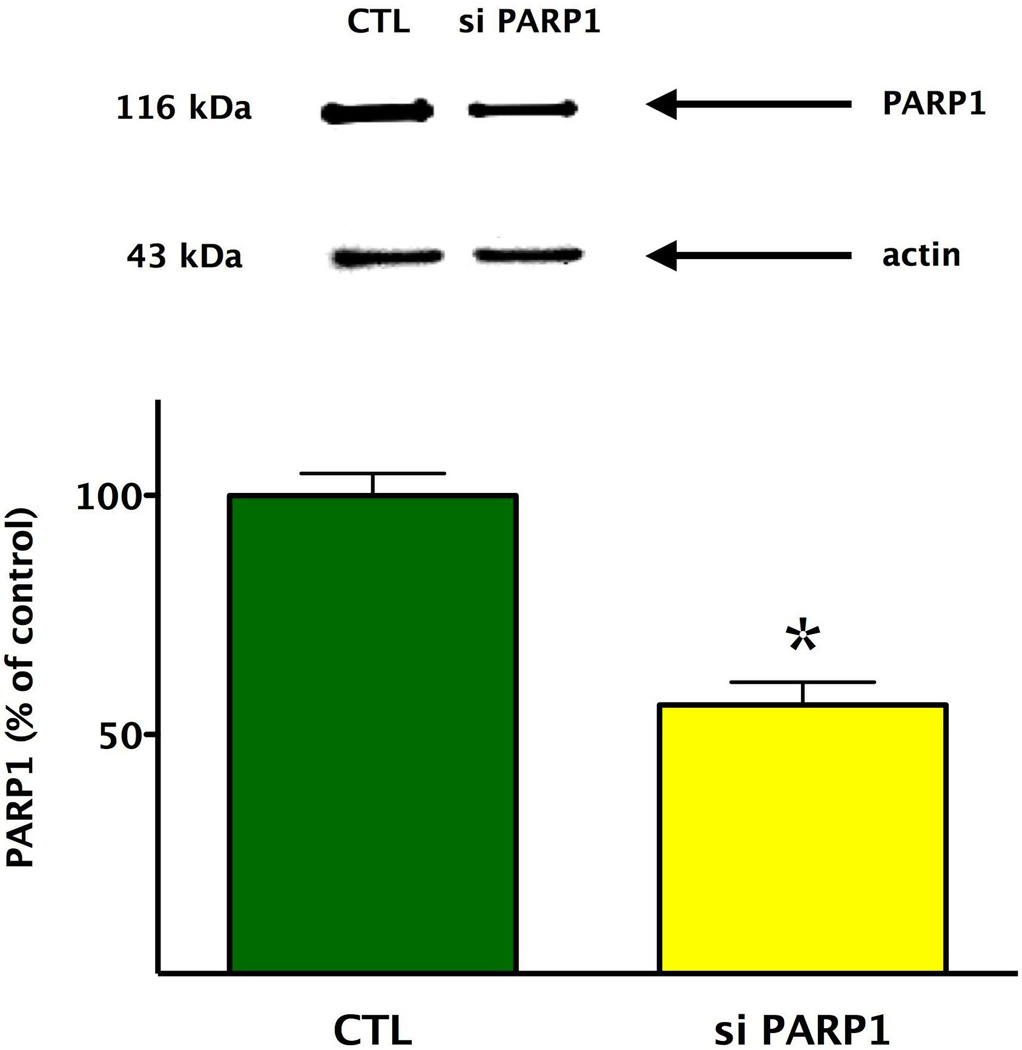
The efficiency of the PARP1 siRNA transfection method in oxidatively stressed endothelial cells was confirmed by western blot analysis, showing a significant (*p<0.05) decrease in PARP1 expression. Data are shown as means ± SEM of n=15 wells (each group) collected from n=3 experiments performed on 3 different days.
Fig. 6. SiRNA knock-down of PARP1 increases the basal mitochondrial reserve capacity and protects from oxidative stress.

Bioenergetic analysis of the endothelial cells was obtained by using extracellular flux analysis. In the figure a time course for measurement of OCR (A) and ECAR (B) for 60,000 cells/well/0.32 cm2 under basal condition followed by the sequential addition of hydrogen peroxide (900 µM), oligomycin (1 µg/ml), FCCP (0.3 µM), and antimycin A (2 µg/ml) as shown. The PARP1 knockdown cells show significantly elevated OCR values during basal and uncoupled respiration, and are protected from H2O2 challenge. Data are shown as means ± SEM of n=15 wells (each group) collected from n = 3 experiments performed on 3 different days.
Fig. 7. SiRNA knock-down of PARP1 increases the basal mitochondrial reserve capacity and protects from oxidative stress.
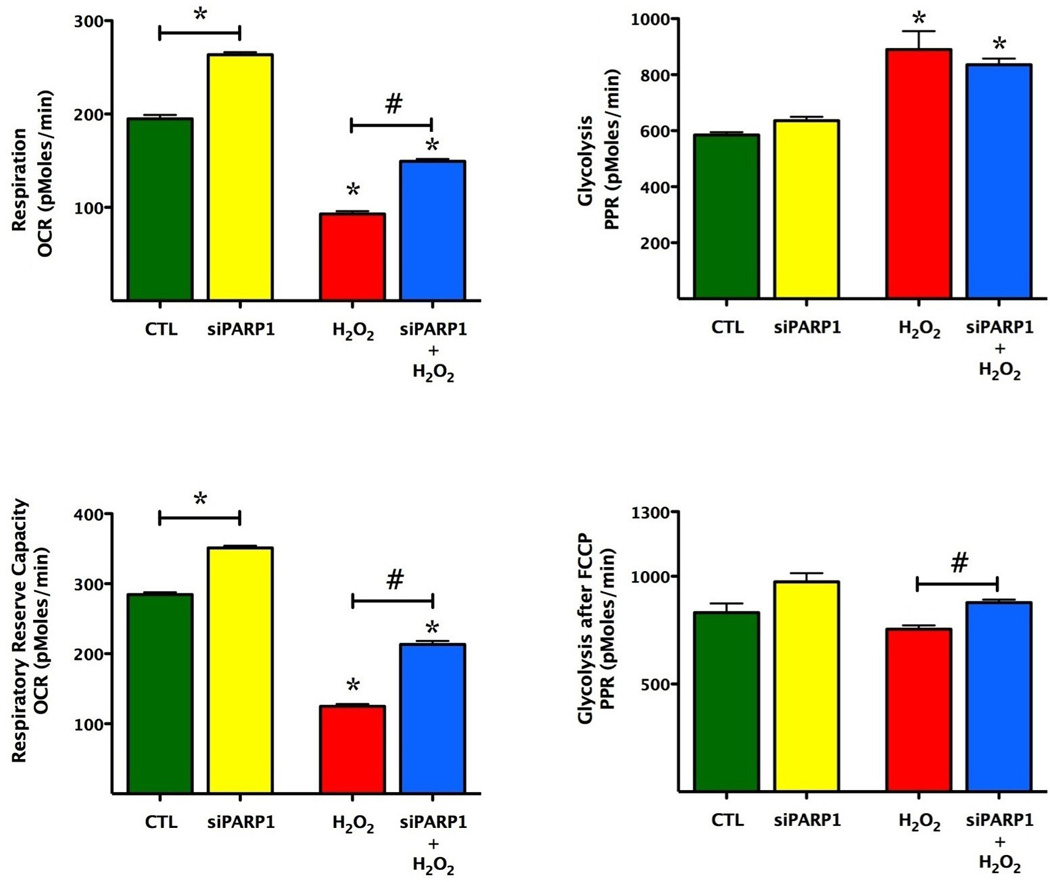
Bioenergetic analysis of the endothelial cells was obtained by using extracellular flux analysis. Relevant bioenergetic parameters are shown in resting cells and in cells after siRNA silencing of PARP1 under basal conditions and after the addition of hydrogen peroxide (900 µM). Please note the increase in basal bioenergetic status after PARP1 silencing, and the protection against hydrogen peroxideinduced suppression of mitochondrial respiration. PARP1 silencing also exerted a slight but significant effect on glycolytic parameters. Significant differences are shown between control and H2O2 treated cells (*p<0.05), and between groups as indicated (#p<0.05). Data are shown as means ± SEM of n = 15 wells (each group) collected from n=3 experiments performed on 3 different days.
The absolute values of total cellular NAD+ in unstimulated, resting bEnd.3 cells amounted to 9.8±0.6 nmol/mg protein (n=4). PARP1 siRNA knockdown failed to affect resting cellular NAD+ levels: NAD+ levels in PARP1 silenced cells amounted to 102±2% of the value in sham-silenced cells (n=4). PARP1 silencing prevented the H2O2-induced loss of mitochondrial reserve capacity, and maintained cellular NAD+ levels. Cellular NAD+ levels in sham-silenced cells decreased by 36±3% (p<0.01 compared to controls not subjected to hydrogen peroxide), while in PARP1 silenced cells decreased by a markedly smaller degree (p<0.05) by 11±1% (n=4).
The measurement of extracellular acidification rate (ECAR) reflects lactate production and is used as an index of glycolysis [11,12]. Our results indicate that oxidative stress enhances glycolysis (indicative of a possible compensatory mechanism), and PARP1 silencing slightly, but significantly further enhances this response.
Treatment of resting endothelial cells with the pharmacological PARP inhibitors PJ34 and 5-AIQ attenuated the decrease in mitochondrial function in oxidatively stressed endothelial cells (Fig. 8), which was also associated with maintenance of cellular NAD+ content. Cellular NAD+ levels in sham-silenced cells decreased by 36±3% (p<0.01 compared to controls not subjected to hydrogen peroxide), while in cells pretreated by PJ34 or 5-AIQ decreased by a markedly smaller degree (p<0.05) by 6±1% or 12±2%, respectively (n=4).
Fig. 8. Effect of pharmacological PARP inhibition on the bioenergetic parameters of oxidatively stressed cells.
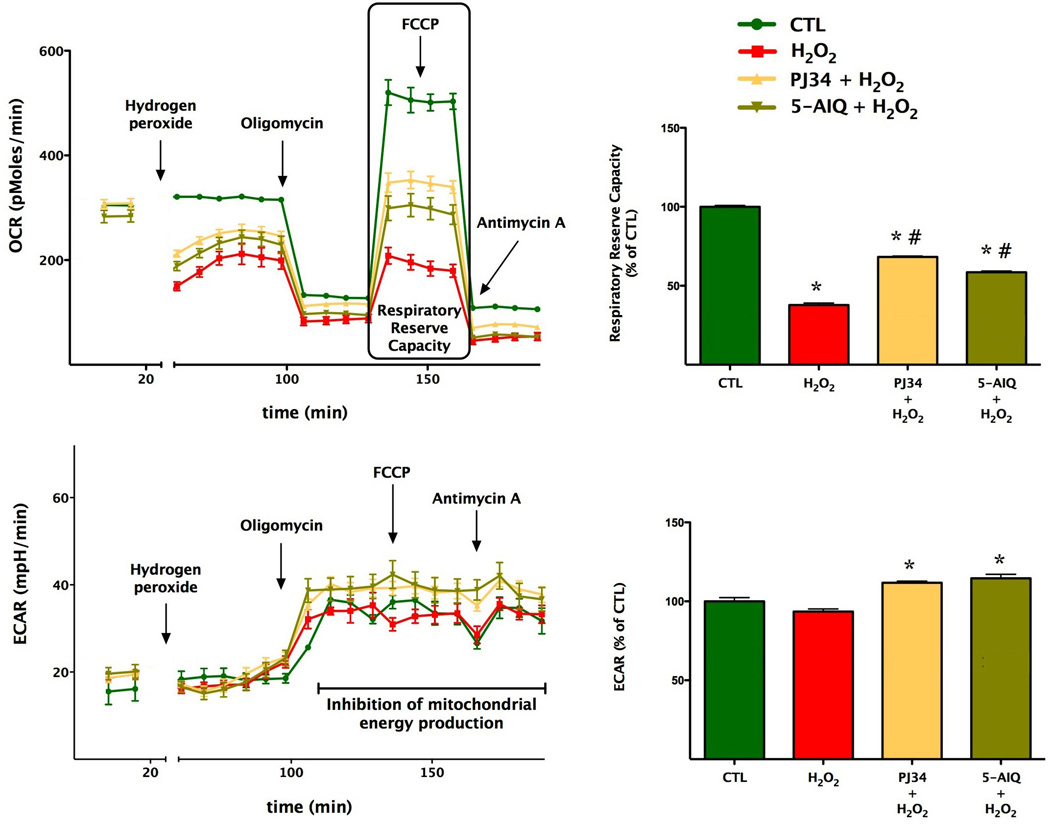
Bioenergetic analysis of the endothelial cells was determined using extracellular flux analysis. Measurement of OCR for 60,000 cells/well/0.32 cm2 is shown under resting conditions, followed by the sequential addition of hydrogen peroxide (900 µM), oligomycin (1 µg/ml), FCCP (0.3 µM), and antimycin A (2 µg/ml). bEnd.3 cultures were pre-treated with the PARP inhibitors PJ34 (3, or 5-AIQ for 48 hours prior to the H2O2 insult). Hydrogen peroxide suppresses mitochondrial parameters (*p<0.05), while PJ34 or 5-AIQ pre-treatment exerts a partial protective effect against the H2O2-induced loss of mitochondrial reserve capacity (#p<0.05). Data are shown as means ± SEM of n=12 wells from n=3 experiments performed on 3 different days.
We have not observed any significant changes in mitochondrial mass with PARP1 silencing (104±11%, n=8) or PARP inhibition (90±7%, PJ34 or 123±10%, 5-AIQ, n=8) in the current studies. Furthermore, we have not observed any changes in basal NAD+ levels between control and PARP1 silenced cells (see above).
3.3. Stable lentiviral PARP1 silencing enhances cellular energetics in human A549 cells and in isolated mitochondria
We next determined whether the marked alterations in basal mitochondrial bioenergetics observed after siRNA silencing of PARP1 were also applicable to other cell types and other methods of PARP silencing. Extracellular flux analysis of A529 epithelial cells in which PARP was stably silenced by a lentiviral approach [25] also exhibited a markedly enhanced cellular metabolic phenotype under baseline conditions, as well as in response to oxidative stress (Fig. 9). Furthermore, mitochondria isolated from PARP1 silenced cells also exhibited a higher metabolic status than mitochondria isolated from wild-type cells, characterized by higher oxygen consumption rate, as well as a doubling of their respiratory reserve capacity (Fig. 10). These bioenergetic changes were not associated with any differences in total cellular NAD+ content, when wild-type and PARP1 silenced cells were compared (Fig. 11). However, there was a significant difference in the basal mitochondrial NAD+ levels between the wild-type and the PARP1 silenced cells; mitochondrial NAD levels in the PARP1 silenced cells amounted to approximately 4-times higher than the corresponding mitochondrial NAD+ levels of the control cells (Fig. 11).
Fig. 9. Stable lentiviral silencing of PARP1 increases the basal mitochondrial reserve capacity in A549 cells.

Bioenergetic analysis of the A549 epithelial cells was performed by using extracellular flux analysis. Relevant bioenergetic parameters are shown in resting cells and in response to hydrogen peroxide (600 µM). Please note the increase in basal bioenergetic status after PARP1 silencing, and the protection against the hydrogen peroxide-induced suppression of cellular bioenergetics in PARP1 deficient cells. Data are shown as means ± SEM of n=15 wells (each group) collected from n=3 experiments performed on 3 different days.
Fig. 10. Stable lentiviral silencing of PARP1 increases the oxygen consumption rate and the respiratory reserve capacity of isolated mitochondria.

Mitochondrial coupling experiments were performed as described in the Methods. The figure shows OCR in response to the sequential administration of ADP, oligomycin, FCCP and Antimycin A to mitochondria from wild-type and stably silenced PARP1 deficient A549 cells. Please note the increase in respiratory reserve capacity (*p<0.05) in the mitochondria prepared from cells with PARP1 silencing, as compared to mitochondria from wild-type cells. Data are shown as means ± SEM of n=15 wells collected from n=3 experiments performed on 3 different days.
Fig. 11. Stable lentiviral silencing of PARP1 increases mitochondrial NAD+ levels.

Total cellular and mitochondrial NAD+ levels are shown from wild-type and PARP1-silenced cells. Although total cellular NAD+ contents were similar in the two cell lines, there was a marked increase in the mitochondrial NAD+ content (*p<0.05) in the cells with PARP1 silencing, as compared to wild-type cells. Data are shown as means ± SEM of n=4 determinations.
4. Discussion
According to our bioenergetic measurements, the basal O2 consumption rate of bEnd.3 endothelial cells amounts to approximately 60% of the maximal oxygen consumption achievable using the uncoupling agent FCCP. Thus, these cells possess a significant reserve or spare respiratory capacity that is available for the cells to call upon when bioenergetic demand is increased. The loss of this reserve respiratory capacity (and also the decrease in maximum respiratory capacity) is a strong indicator of potential mitochondrial dysfunction that may not be particularly apparent under basal conditions, when respiration rate is strongly controlled by ATP turnover, but becomes manifest only under load (for instance in oxidative stress) when ATP demand increases and substrate oxidation is more limited to respiratory electron flow.
The extracellular flux analysis technology allows the determination of not only the respiratory reserve capacity of the mitochondria, but also a calculation of the cellular respiratory control ratio (RCR) [11,12,17–22]. Both the reserve respiratory capacity and the cellular respiratory control ratio are parameters that are useful for the characterization of mitochondrial function in intact cells. During the calculation of RCR, the respiration of state 3 is considered equivalent to the rate measured after addition of FCCP, while state 4 is considered to be the rate measured after addition of the Complex V inhibitor oligomycin. Accordingly, the RCR is estimated as the State 3 rate divided by the State 4 rate. RCR can be used to analyze the respiration of mitochondria in endothelial cells both under resting conditions and in response to various stress conditions (e.g. during oxidative stress) (Fig. 1). Typical values for this ratio vary from 3 to 15 in different cell lines, depending on the passage number of the cell lines and the extent of the mitochondrial damage. After the characterization of the basic bioenergetic parameters of bEnd.3 cells, we determined the sensitivity of the cells to the cytotoxic oxidant hydrogen peroxide. As shown in Fig. 2., a rather steep concentration-response was noted, and a good correlation between cell viability (MTT) and cytotoxicity (LDH release) was seen. Because in the present study we were interested in the role of PARP in the modulation of basal cellular energetics, as well as in changes in cellular energetics during non-cytotoxic conditions of oxidative stress, we selected 900 µM concentration of H2O2 to be used for subsequent experiments. The studies demonstrated that although this oxidant exposure does not result in substantial changes in MTT or LDH readouts, it does induce a substantial decrease in FCCP-mediated oxygen consumption rate (Fig. 3) and a reduced respiratory reserve capacity. The way we interpret these findings is that oxidative stress induces early changes in mitochondrial function that are not yet associated with overall alterations in cell viability. It is well documented that H2O2 can trigger cellular damage via multiple mechanisms, including intracellular and intramitochondrial Ca2+ overload, the opening of the mitochondrial permeability transition pore, activation of PARP1 and other pathways [6–12]. However, it appears that moderate changes in mitochondrial reserve capacity precede overt alterations in cell viability.
At higher concentrations, H2O2 resulted in an increase in proton leak (as determined by the addition of oligomycin), as well as in preventing the effect of FCCP to stimulate the oxygen consumption. Moreover, the decline of OCR values in response to oligomycin administration was completely abolished in H2O2 treated cells compared to controls (Fig. 3), a finding that is most likely due to a reduction in cellular ATP content in the cells exposed to higher concentrations of H2O2. In parallel with the decrease in mitochondrial reserve capacity, the cells exhibited an elevated glycolytic activity during oxidative stress (Fig. 4), consistent with a model whereby the decreased mitochondrial respiration after H2O2 insult forces the cells to switch to glycolysis. It is interesting to note that a similar switch has recently been observed in endothelial cells placed in elevated extracellular glucose, a condition that induces the production of mitochondrial ROS [20].
The role of PARP in the regulation of resting cellular bioenergetics, and in the changes in bioenergetics under non-cytotoxic levels of oxidative stress, was next evaluated by a combination of a pharmacological and PARP1 gene silencing-based approaches. The results showed that PARP1 siRNA knockdown, which resulted in approximately 50% suppression of PARP1 protein levels, markedly enhanced baseline mitochondrial respiration and baseline mitochondrial reserve capacity in cultured bEnd.3 endothelial cells. Based on these findings we conclude that PARP1 acts as an important regulator of mitochondrial function and cellular bioenergetics in resting endothelial cells. This finding is in stark contrast to most prior work, which considered PARP1 strictly in the context of damage-response, and not in the context of the regulation of physiological cellular energetic homeostasis [13–16]. Under our current experimental conditions, the regulation by PARP1 of resting bioenergetic parameters was not related to changes in global cellular NAD+ levels or to potential PARP1-mediated regulation [31] of mitochondrial biogenesis [31].
There are several different pools of NAD+ in the cell [32,33] and the mitochondrial membrane is impermeable to the nicotinamide adenine dinucleotides NAD+, NADH, NADP+, NADPH. Nevertheless, the NADH generated in the cytoplasm by glycolysis is oxidized by the respiratory chain. This is achieved by two main pathways, the glutamate/aspartate shuttle, and dihydroxyacetone phosphate shuttle [32,33]. The present study demonstrates that PARP1 deficiency results in a marked and selective increase in the mitochondrial NAD+ pool. We hypothesize that prolonged PARP inhibition/PARP1 silencing may produce an improved mitochondrial function by increasing the rate of electron flow (by converting NAD+ to NADH), thereby increasing mitochondrial potential and elevating mitochondrial respiratory reserve capacity. Additional mechanisms may also be conceivable. For instance, poly(ADP-ribosyl)ation of mitochondrial and/or nuclear enzymes may also modulate mitochondrial electron transport chain activities, which may also contribute to the observed alterations in resting cellular bioenergetics in cells subjected to PARP1 silencing. Another question remains whether it is the enzymatic activity of PARP, or the physical presence of PARP that (via its molecular scaffolding functions) is the key determinant in the regulation of mitochondrial function. The effects of the pharmacological PARP inhibitors observed in the current study suggest that the catalytic activity of PARP is an important (although possibly not an exclusive) factor in this process.
Consistent with the previously well-recognized role of PARP1 in oxidatively stressed cells [13–16], PARP1 silencing or pharmacological PARP inhibition prevented the H2O2-induced loss of mitochondrial reserve capacity, and maintained cellular NAD+ levels (Fig. 12). The two PARP inhibitors used in the current study are from different structural classes, but both of them bind to the NAD+-binding site of the enzyme and prevent the conversion of NAD+ to poly(ADP-ribose) [23,24]. The current findings indicate that PARP enzymatic activity (as opposed to the presence of the enzyme, i.e. its scaffolding functions) is a regulator of mitochondrial function and cellular bioenergetics in endothelial cells. On the other hand, the measurement of extracellular acidification rate (ECAR, reflecting lactate production and used as an index of glycolysis [11,12]) showed that oxidative stress mediated enhancement of cellular glycolysis (most likely a compensatory energetic response of the cell) is affected by PARP inhibition or PARP1 silencing by a relatively lower degree.
Fig. 12. Graphical abstract: a schematic representation of the role of PARP1 in the regulation of mitochondrial function under resting condition and during oxidative stress.
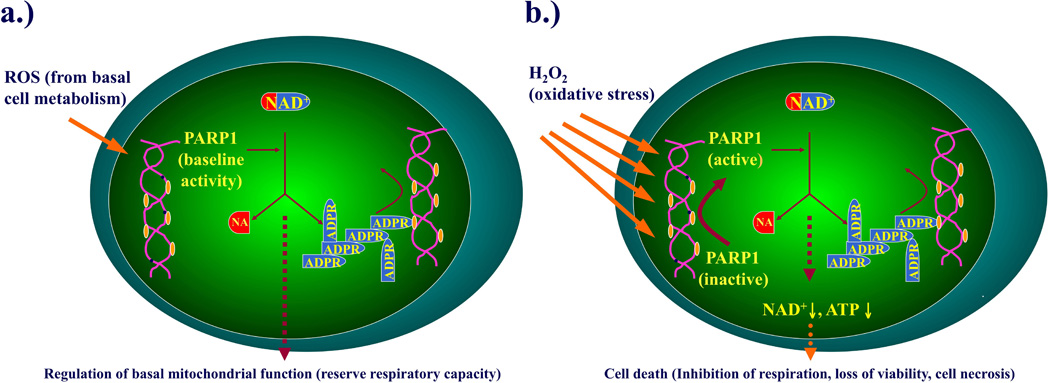
The goal of the current study was to delineate the regulatory effect of PARP1 on resting cellular bioenergetics, and on the bioenergetic alterations induced by oxidative stress (hydrogen peroxide). During resting conditions (a), PARP1 regulates cellular bioenergetics, as evidenced by an enhanced reserve respiratory capacity in cells where PARP1 was silenced. During conditions of oxidative stress (b), PARP1 becomes activated due to DNA strand breakage, and catalyzes the cleavage of NAD+ into nicotinamide and ADP-ribose to form long branches of ADP-ribose polymers on a number of target proteins including histones, DNA polymerase, DNA ligase and other proteins. A result of this process is a reduction in cellular NAD+ and ATP pools, leading to a deterioration of cellular energetics, cell dysfunction, and, in case of severe oxidative stress, cell necrosis.
In the final part of our study, we determined whether the alterations in basal mitochondrial bioenergetics obtained after siRNA silencing of PARP1 in endothelial cells can also observed in epithelial cells, and whether these differences also persist in isolated mitochondrial preparations. Extracellular flux analysis of A529 epithelial cells in which PARP was stably silenced by a lentiviral approach [25] also exhibited a markedly enhanced cellular metabolic phenotype under baseline conditions. Interestingly, mitochondria isolated from PARP1 silenced cells exhibited a higher metabolic status than mitochondria isolated from wild-type cells, characterized by higher oxygen consumption rate, as well as an increased respiratory reserve capacity. These findings, coupled with our data showing significantly higher intramitochondrial NAD+ levels in the PARP1 silenced cells, further support the hypothesis that PARP1 serves as a physiological regulator of an intramitochondrial bioenergetic pathway in resting cells. This conclusion is in agreement with prior studies suggesting the potential importance of mitochondrial PARP1 in the regulation of cellular functions in health and disease [34–36].
In conclusion, the current study is the first one to implicate PARP1 in the regulation of physiological energetic homeostasis in resting cells (Fig. 12). In endothelial cells (transiently silenced by siRNA) or in epithelial cells (stably silenced by a lentiviral approach) there was clear evidence for an increased basal respiratory reserve capacity, indicative of a fundamental role of PARP1 in the regulation of normal cellular energetic homeostasis. These changes occurred in the absence of alterations in mitochondrial biogenesis and were seen both in intact cells as well as in isolated mitochondria. The present data (including the higher mitochondrial NAD+ levels detected in the PARP1 silenced A549 cells) are consistent with the hypothesis that basal PARP activity in resting mitochondria leads to a physiological decrease in mitochondrial capacity for substrate oxidation and ATP turnover. This response, coupled with an increase in the proton leak, possibly through attenuation of the proton motive force between the intermembrane and mitochondrial matrix, may exert a 'physiological dampening' effect on respiratory spare capacity. This phenomenon may be related to the fact that all cells, even under resting conditions, produce small amounts of reactive oxygen and nitrogen species as part of their normal cellular respiration [37–40]. It is, therefore, conceivable that this basal ROS/RNS production is involved in the maintenance of a low baseline level of PARP1 activity, leading to the effects on cellular bioenergetics reported here.
Acknowledgements
This work was supported by a grant from the National Institutes of Health to C.S. (R01GM60915) and by a McLaughlin Fellowship of the University of Texas to Katalin Módis.
Abbreviations
- 5-AIQ
5-aminoisoquinolinone hydrochloride
- DMEM
Dulbecco’s Eagle Modified Medium
- ECAR
extracellular acidification rate
- ECL
enhanced chemiluminescence
- FBS
fetal bovine serum
- FCCP
carbonyl cyanide p-[trifluoromethoxy]-phenyl-hydrazone
- H2O2
hydrogen peroxide
- INT
2-(4-iodophenyl)-3-(4-nitrophenyl)-5-phenyl-2H-tetrazolium chloride
- LDH
lactate dehydrogenase
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAD+
nicotinamide adenine dinucleotide
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- PARP1
poly(ADP-ribose) polymerase 1
- PBS
phosphate buffered saline
- PJ34
N-(-oxo-5,6-dihydro-phenanthridin-2-yl)-N,N-dimethylacetamide HCl
- PMS
N-methylphenazonium methyl sulfate
- PVDF
polyvinylidene fluoride
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SDS
sodium dodecyl sulfate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thomas SR, Witting PK, Drummond GR. Redox control of endothelial function and dysfunction: molecular mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2008;10:1713–1765. doi: 10.1089/ars.2008.2027. [DOI] [PubMed] [Google Scholar]
- 2.Vanhoutte PM, Shimokawa H, Tang EH, Feletou M. Endothelial dysfunction and vascular disease. Acta Physiol (Oxf) 2009;196:193–222. doi: 10.1111/j.1748-1716.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 3.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Szabo C. Role of nitrosative stress in the pathogenesis of diabetic vascular dysfunction. Br J Pharmacol. 2009;156:13–27. doi: 10.1111/j.1476-5381.2008.00086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabo C, Modis K. Pathophysiological roles of peroxynitrite in circulatory shock. Shock. 2010;34 Suppl 1:4–14. doi: 10.1097/SHK.0b013e3181e7e9ba. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tousoulis D, Briasoulis A, Papageorgiou N, Tsioufis C, Tsiamis E, Toutouzas K, et al. Oxidative stress and endothelial function: therapeutic interventions. Recent Pat Cardiovasc Drug Discov. 2011;6:103–114. doi: 10.2174/157489011795933819. [DOI] [PubMed] [Google Scholar]
- 7.Gielis JF, Lin JY, Wingler K, Van Schil PE, Schmidt HH Moens AL. Pathogenetic role of eNOS uncoupling in cardiopulmonary disorders. Free Radic Biol Med. 2011;50:765–776. doi: 10.1016/j.freeradbiomed.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Förstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010;459:923–939. doi: 10.1007/s00424-010-0808-2. [DOI] [PubMed] [Google Scholar]
- 9.Davidson SM. Endothelial mitochondria and heart disease. Cardiovasc Res. 2010;88:58–66. doi: 10.1093/cvr/cvq195. [DOI] [PubMed] [Google Scholar]
- 10.Shen GX. Oxidative stress and diabetic cardiovascular disorders: roles of mitochondria and NADPH oxidase. Can J Physiol Pharmacol. 2010;88:241–248. doi: 10.1139/Y10-018. [DOI] [PubMed] [Google Scholar]
- 11.Dranka BP, Hill BG, Darley-Usmar VM. Mitochondrial reserve capacity in endothelial cells: The impact of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2010;48:905–914. doi: 10.1016/j.freeradbiomed.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sansbury BE, Jones SP, Riggs DW, Darley-Usmar VM, Hill BG. Bioenergetic function in cardiovascular cells: The importance of the reserve capacity and its biological regulation. Chem Biol Interact. 2011;191:288–295. doi: 10.1016/j.cbi.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 14.Peralta-Leal A, Rodríguez-Vargas JM, Aguilar-Quesada R, Rodríguez MI, Linares JL, de Almodóvar MR, et al. PARP inhibitors: new partners in the therapy of cancer and inflammatory diseases. Free Radic Biol Med. 2009;47:13–26. doi: 10.1016/j.freeradbiomed.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 15.Giansanti V, Donà F, Tillhon M, Scovassi AI. PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol. 2010;80:1869–1877. doi: 10.1016/j.bcp.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 16.Sodhi RK, Singh N, Jaggi AS. Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic implications. Vascul Pharmacol. 2010;53:77–87. doi: 10.1016/j.vph.2010.06.003. [DOI] [PubMed] [Google Scholar]
- 17.Trudeau K, Molina AJ, Guo W, Roy S. High glucose disrupts mitochondrial morphology in retinal endothelial cells: implications for diabetic retinopathy. Am J Pathol. 2010;177:447–455. doi: 10.2353/ajpath.2010.091029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varum S, Rodrigues AS, Moura MB, Momcilovic O, Easley CAt, Ramalho-Santos J, et al. Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One. 2011;6:e20914. doi: 10.1371/journal.pone.0020914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oláh G, Módis K, Gero D, Suzuki K, Dewitt D, Traber DL, et al. Cytoprotective effect of gamma-tocopherol against tumor necrosis factor alpha induced cell dysfunction in L929 cells. Int J Mol Med. 2011;28:711–720. doi: 10.3892/ijmm.2011.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gero, et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:13829–13834. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today. 2008;13:268–274. doi: 10.1016/j.drudis.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 22.Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 23.Jagtap P, Soriano FG, Virág L, Liaudet L, Mabley J, Szabó E, et al. Novel phenanthridinone inhibitors of poly (adenosine 5'-diphosphate-ribose) synthetase: potent cytoprotective and antishock agents. Crit Care Med. 2002;30:1071–1082. doi: 10.1097/00003246-200205000-00019. [DOI] [PubMed] [Google Scholar]
- 24.McDonald MC, Mota-Filipe H, Wright JA, Abdelrahman M, Threadgill MD, Thompson AS, et al. Effects of 5-aminoisoquinolinone, a water-soluble, potent inhibitor of the activity of poly (ADP-ribose) polymerase on the organ injury and dysfunction caused by haemorrhagic shock. Br J Pharmacol. 2000;130:843–850. doi: 10.1038/sj.bjp.0703391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Erdélyi K, Bai P, Kovács I, Szabó E, Mocsár G, Kakuk A, et al. Dual role of poly(ADP-ribose) glycohydrolase in the regulation of cell death in oxidatively stressed A549 cells. FASEB J. 2009;23:3553–3563. doi: 10.1096/fj.09-133264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szabo C, Zingarelli B, O'Connor M, Salzman AL. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc Natl Acad Sci U S A. 1996;93:1753–1758. doi: 10.1073/pnas.93.5.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerö D, Módis K, Nagy N, Szoleczky P, Tóth ZD, Dormán G, et al. Oxidant-induced cardiomyocyte injury: identification of the cytoprotective effect of a dopamine 1 receptor agonist using a cell-based high-throughput assay. Int J Mol Med. 2007;20:749–761. [PubMed] [Google Scholar]
- 28.Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, et al. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6:e21746. doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2(2):287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 30.Addabbo F, Ratliff B, Park HC, Kuo MC, Ungvari Z, Csiszar A, et al. The Krebs cycle and mitochondrial mass are early victims of endothelial dysfunction: proteomic approach. Am J Pathol. 2009;174:34–43. doi: 10.2353/ajpath.2009.080650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rongvaux A, Andris F, Van Gool F, Leo O. Reconstructing eukaryotic NAD metabolism. Bioessays. 2003;25:683–690. doi: 10.1002/bies.10297. [DOI] [PubMed] [Google Scholar]
- 33.Magni G, Amici A, Emanuelli M, Orsomando G, Raffaelli N, Ruggieri S. Enzymology of NAD+ homeostasis in man. Cell Mol Life Sci. 2004;61:19–34. doi: 10.1007/s00018-003-3161-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, et al. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- 35.Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, et al. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem. 2008;104:1700–1711. doi: 10.1111/j.1471-4159.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- 36.Rossi MN, Carbone M, Mostocotto C, Mancone C, Tripodi M, Maione R, et al. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J Biol Chem. 2009;284:31616–31624. doi: 10.1074/jbc.M109.025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brookes PS. Mitochondrial H(+) leak and ROS generation: an odd couple. Free Radic Biol Med. 2005;38:12–23. doi: 10.1016/j.freeradbiomed.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 38.Ungvari Z, Buffenstein R, Austad SN, Podlutsky A, Kaley G, Csiszar A. Oxidative stress in vascular senescence: lessons from successfully aging species. Front Biosci. 2008;13:5056–5070. doi: 10.2741/3064. 2008. [DOI] [PubMed] [Google Scholar]
- 39.Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA, et al. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal. 2010;12:1431–1470. doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- 40.Hwang AB, Lee SJ. Regulation of life span by mitochondrial respiration: the HIF-1 and ROS connection. Aging (Albany NY) 2011;3:304–310. doi: 10.18632/aging.100292. [DOI] [PMC free article] [PubMed] [Google Scholar]