

Abstract
We investigated the relevance of gene expression heterogeneity to virulence properties of a major fungal pathogen, Candida glabrata. The organism's key virulence-associated factors include glycosylphosphatidylinositol-anchored adhesins, encoded subtelomerically by the EPA gene family. Individual-cell analyses of expression revealed very striking heterogeneity for Epa1, an adhesin that mediates ∼95% of adherence to epithelial cells in vitro. The heterogeneity in Epa1 was markedly greater than that known for other yeast genes. Sorted cells expressing high or low levels of Epa1 exhibited high and low adherence to epithelial cells, indicating a link between gene expression noise and potential virulence. The phenotypes of sorted subpopulations reverted to mixed phenotypes within a few generations. Variation in single-cell Epa1 protein and mRNA levels was correlated, consistent with transcriptional regulation of heterogeneity. Sir-dependent transcriptional silencing was the primary mechanism driving heterogeneous Epa1 expression in C. glabrata BG2, but not in CBS138 (ATCC 2001). Inefficient silencing in the latter strain was not due to a difference in EPA1 sequence or (sub)telomere length and was overcome by ectopic SIR3 expression. Moreover, differences between strains in the silencing dependence of EPA1 expression were evident across a range of clinical isolates, with heterogeneity being the greatest in strains where EPA1 was subject to silencing. The study shows how heterogeneity can impact the virulence-related properties of C. glabrata cell populations, with potential implications for microbial pathogenesis more broadly.
INTRODUCTION
Candida glabrata has risen to being second only to C. albicans as the most prevalent yeast pathogen in humans, being responsible for approximately 26% of Candida bloodstream infections in the United States (22). Mortality rates up to 50% are commonly associated with C. glabrata infection and are higher than those associated with C. albicans. A likely contributor to this is increased resistance to antifungal agents, specifically, azoles, in C. glabrata (15). Another major virulence-associated factor of C. glabrata is the expression of adhesin proteins encoded by the subtelomeric EPA gene family. Approximately 67 genes encoding adhesin-like glycosylphosphatidylinositol (GPI)-anchored proteins reside within the C. glabrata genome, and at least 17 or 23 (depending on the strain) of these proteins can be allocated to the Epa family (12, 24). Several of the Epa proteins are important for adherence and virulence. Kidney infections in mice are attenuated between three- and fivefold by deletion of the EPA1 gene cluster (HYR/EPA1-EPA2-EPA3) (13). EPA6 and EPA7 have also been implicated in colonization of the kidney (9), while in vitro adhesion to epithelial cells is ∼95% mediated by EPA1 (11). The different Epa proteins have different specificities for glycan-containing ligands (57), and deletion of EPA1 alone produced no significant virulence phenotype in murine models of systemic or vaginal candidiasis (11). It has been postulated that the EPA genes could be differentially regulated in order to maximize adherence to different host cell types during infections (9, 12–14, 57). The limited availability of nicotinic acid (NA) stimulates EPA6 expression during urinary tract infection. EPA1 and EPA7 expression is also increased in response to this signal (14). Nicotinic acid limitation is thought to reduce the activity of the NAD+-dependent histone deacetylase Sir2p, leading to loss of silencing of the EPA genes, which are subject to the telomere position effect due to their subtelomeric location. Deletion of SIR3 results in EPA gene derepression (9, 13).
A contribution of gene silencing to regulation is also evident in the FLO gene family of Saccharomyces cerevisiae, which is functionally related to the EPA gene family. Studies on FLO gene silencing have indicated the potential for individual FLO genes to be differentially expressed between individual cells (20). Heterogeneity of this type is masked in conventional population-wide analyses of gene expression but may have profound implications for phenotypes like virulence where initiation of an infection could require just a few variant virulent cells within a larger avirulent population.
Research on gene expression noise during the last decade has revealed multiple underlying sources of such cell-to-cell heterogeneity, including contributions from stochasticity, the cell cycle, and epigenetic regulation (3, 48). In turn, a diverse range of affected phenotypes may vary between individual cells of a genetically uniform population. Evidence from laboratory and modeling studies indicates that this nongenotypic heterogeneity confers advantages under certain conditions, by offering to cell subpopulations alternative adaptive strategies which may be exploited during changing conditions (1, 7, 18, 47). Such advantages seem likely to extend to pathogenic microorganisms during colonization of alternative host niches, similar to the genome rearrangement-driven variation in virulence gene expression described in certain protozoal and bacterial pathogens (5, 44). To date, the relevance of gene expression noise to virulence of Candida spp. has not been examined, although it has been proposed that an ability to produce phenotypic variants could be crucial for optimal host interaction (26).
In this study, expression of the major adhesion Epa1 of C. glabrata was found to be more heterogeneous than that determined previously for any other yeast protein. Epa1 expression level was correlated with adherence properties of individual cells and was driven by Sir-mediated silencing in some but not all tested strains, revealing additional strain-to-strain heterogeneity.
MATERIALS AND METHODS
Strains and plasmids.
Candida glabrata BG2 (11) and the type strain CBS138 were the backgrounds from which other strains were derived. C. glabrata BG2 and derivatives ura3Δ (BG14), epa1Δ (BG64), and BG198, an EPA1-green fluorescent protein (GFP) transcriptional fusion strain, were gifts from Brendan Cormack (Johns Hopkins University). Clinical isolates of C. glabrata from the collection at the Department of Medicine, Imperial College London, were provided by Michael Petrou. C. glabrata NCYC388 was from the NCYC, Norwich, United Kingdom.
To construct strains expressing hemagglutinin (HA)-tagged EPA1, a 1-kb fragment immediately upstream of the EPA1 open reading frame (ORF) encompassing the EPA1 promoter region was amplified with PCR (using Phusion high-fidelity polymerase [Finzymes]; all primer sequences are available on request) from C. glabrata BG2 genomic DNA. Ligation at the XbaI site of pBC214 (from Brendan Cormack) created a construct in which EPA1 tagged with 3HA (17) was under EPA1 promoter control. This construct was excised as an XbaI/EcoRI fragment and ligated into pGEM7 (Promega). The URA3 marker (from pHOBST-URA3 [35]) was ligated between the SmaI and EcoRI sites of the vector, and a 1-kb sequence immediately downstream of the EPA1 ORF was amplified from genomic DNA and inserted at the PacI site, creating pMS15. pMS15 contained an extended region of genomic sequence around EPA1(-3HA) which, together with the internal URA3 marker positioned immediately downstream of the EPA1 ORF, was excised as a ∼6.8-kb fragment by digestion with XbaI and PacI and transformed into C. glabrata using a modified lithium acetate method (8). (The URA3 marker added 1.2 kb distance between EPA1 and the telomere, but this was too small to affect silencing significantly [see Discussion]. Moreover, key results were confirmed with cells preserving a wild-type EPA1 region [see Fig. 4C, 5B, and 8]). Ura+ transformants were selected on YNB plates lacking uracil or uridine. Correct integration of the transforming fragment was verified with diagnostic PCR.
Fig 4.

Epigenetic regulation of Epa1 expression heterogeneity in C. glabrata BG2. (A) Time-lapse fluorescence microscopy tracking the retention of surface Epa1-HA by parent cells during division. Cells were stained once with anti-HA, Alexa Fluor 488 conjugate antibody and then incubated in YNB broth at 30°C. (B) Cells expressing Epa1-HA were probed with the anti-HA antibody and examined by fluorescence microscopy. The images show examples of cells showing a high (top) or low (bottom) correlation in Epa1-HA expression between parent cell and bud. Fluorescence due to Epa1-HA expression was quantified in parents and buds of 100 budding cells, and the data for each pair are presented. (C) Flow cytometric analysis of Epa1 expression in wild-type or sir3Δ cells of the BG2 background either expressing the EPA1-GFP transcriptional fusion construct (left) or probed with anti-Epa1 antibody (right).
Fig 5.

Nonepigenetic regulation of Epa1 expression heterogeneity in C. glabrata CBS138. (A) Flow cytometric histograms of Epa1-HA expression heterogeneity among cells of exponential-phase CBS138 or BG2 cultures. (B) Flow cytometric analysis of wild-type or sir3Δ cells of the CBS138 background expressing Epa1-HA and either probed with anti-HA, Alexa Fluor 488 conjugate antibody (left) or probed with anti-Epa1 antibody (right). (C) CBS138 cells expressing Epa1-HA were probed with anti-HA antibody and examined by fluorescence microscopy. Fluorescence due to Epa1-HA expression was quantified in parents and buds of 100 budding cells, and the data for each pair are presented.
Fig 8.

Variation in EPA1 silencing and heterogeneity between isolates of C. glabrata. (A) RNA was isolated from BG2 and CBS138 cells cultured in control SC medium (open bars) or nicotinic acid (NA)-limited SC medium (5% of the normal NA content; filled bars). EPA1 mRNA was quantified by qRT-PCR. Total RNA added to all RT-PCRs was normalized, and the data shown are means of the five median values from nine replicate determinations ± SEMs. (B) EPA1 mRNA levels were compared as described for panel A across 11 C. glabrata isolates; results are shown for 3 representatives. (C) Single-cell Epa1 expression across 11 C. glabrata isolates was probed with anti-Epa1 antibody and analyzed flow cytometrically; results are shown for 3 representatives. (D) Relationship between silencing and heterogeneity of Epa1 expression across all tested strains. BG2-like strains were BG2, NCYC388, and isolates 21, 32, 146, and 172. CBS138-like strains were CBS138 and isolates 31, 39, and 82. The other strains were isolates 22, 105, and 134.
To disrupt SIR3 in the different backgrounds, the SIR3 ORF, together with 1 kb each of upstream and downstream sequences, was amplified from CBS138 genomic DNA and ligated between the NsiI and SphI sites of pGEM7. The URA3 marker (from pMS15) was ligated into the MscI-cut vector, and an ∼3.8-kb sir3::URA3 fragment was released by digestion with BamHI and HpaI and integrated into genomic DNA via transformation. To disrupt SIR3 in Ura+ Epa1-HA expressing cells, a HIS3 marker excised by XhoI digestion of pCgACH-3 (a gift from Ken Haynes, University of Exeter) was ligated into the XhoI site of SIR3 within pGEM7. An MscI/PmlI fragment was used for SIR3 disruption via homologous recombination. To express SIR3 from a single-copy plasmid, the C. glabrata SIR3 ORF, together with ∼1 kb each of the upstream and downstream sequences, was amplified from CBS138 genomic DNA and ligated into SacI-digested pCgACT-14 (from Ken Haynes).
A yps7Δ deletion strain in the CBS138 background was kindly provided by Ken Haynes. A deletion cassette was amplified from this strain as an ∼3.2-kb fragment, encompassing the NATR marker together with flanking sequences corresponding to 1 kb upstream and 1 kb downstream of the YPS7 ORF. This fragment was transformed into the EPA1-HA-expressing strain derived from C. glabrata CBS138 (see above). To delete YPS1, the YPS1 ORF and two 1-kb flanking sequences were amplified as a 3.8-kb fragment, with each terminus tagged by a SacI site, and the fragment was inserted into the pJET2.1 vector (Fermentas Life Sciences). The HIS3 marker from pCgACH-3 was inserted into the plasmid as a BamHI/NheI fragment, and a 2.3-kb fragment released with SacI was used for transformation to produce yps1Δ cells. Escherichia coli XLI-Blue was used as the host for all DNA manipulations, with transformation being performed by electroporation (2). Restriction enzymes and DNA ligase were from New England BioLabs.
Growth conditions for experiments.
Unless stated otherwise, cells were grown overnight at 30°C to stationary phase in standard YEPD medium (2% [wt/vol] bacteriological peptone [Oxoid], 1% yeast extract [Oxoid], 2% d-glucose), before dilution in fresh medium and further growth for 3 h before experimental use (21). Synthetic complete (SC) medium was used for experiments involving NA limitation and comprised yeast nitrogen base (YNB) without amino acids and NA (Formedium) supplemented with SC medium amino acid mixture (Formedium). The medium was supplemented with 5% (0.167 μM) or 100% (3.25 μM) of the normal SC medium nicotinic acid concentration. Cultures grown overnight at 5% or 100% NA were diluted in the same medium and grown for a further 3 h.
Probing for Epa1.
Freshly harvested cells (1.5 × 106) were used for staining procedures, after 5 min incubation at 30°C with 50 mM EDTA to prevent cell aggregation. Cells were collected by centrifugation (8,000 × g, 2 min), and pellets were resuspended in 300 μl phosphate-buffered saline (PBS). HA-tagged Epa1 was probed with anti-HA, Alexa Fluor 488 conjugate (Molecular Probes, Invitrogen) by incubation with 3.5 μg ml−1 antibody for 30 min at 4°C. Samples were washed twice with 300 μl PBS before analysis.
An anti-Epa1 rabbit polyclonal antibody was a gift from Brendan Cormack. To avoid cross-reactivity, the antibody was preabsorbed against C. glabrata epa1Δ cells before use: cultures were grown in SC medium, 5% NA medium (see above) to induce EPA gene expression (14). Cells (1 × 108) were suspended in 500 μl PBS, and preabsorption was performed through 10 1-h incubations on ice with antibody (1/5 dilution), with each incubation being with a fresh batch of cells. The final supernatant was confirmed to have Epa1-specific reactivity by probing wild-type and epa1Δ cells, with analysis by flow cytometry (see below). This antibody was used to probe Epa1 by incubating 1.5 × 106 cells for 30 min at 4°C in the presence of antibody (1/5,000 dilution). Cells were washed twice with PBS before incubation with 3.5 μg ml−1 anti-rabbit, Alexa Fluor 488 conjugate secondary antibody (Molecular Probes, Invitrogen). After 20 min at 4°C, cells were washed twice with PBS before analysis.
Flow cytometry and cell sorting.
Analysis by flow cytometry was with a BD FACSCanto instrument equipped with a 488-nm argon ion laser (BD Biosciences). Cell samples were suspended in 300 μl PBS in flow cytometry tubes (BD Biosciences). Fluorescence was recorded via a fluorescein isothiocyanate filter for 10,000 cells per sample at ≤5,000 cells s−1. Data were stored and analyzed using FacsDIVA software (BD Biosciences), with additional analysis performed using Weasel software (Walter and Eliza Hall Institute of Medical Research, Australia). Fluorescence measurements with the FACSCanto instrument were normalized for cell size by forward scatter (FSC-A) correction. Unless indicated otherwise (34), these normalized data were used to determine coefficients of variation (CVs), excluding the outlying top and bottom 0.1% of data. Cell sorting was with a Coulter Altra fluorescence-activated cell sorter (FACS), after gating into the lowest- or highest-fluorescence subpopulations. Each subpopulation comprised 13% of all cells.
Fluorescence microscopy.
Cells were examined through a ×100 objective lens with a Nikon Eclipse Ti microscope equipped with a DS-Qil camera and temperature-controlled chamber. Image acquisition was with NIS elements Br software (Nikon). For time-lapse studies, cells were adhered for 20 min at 4°C to glass chambers (WAKI) precoated with 1 mg ml−1 concanavalin A and air dried. The supernatant above adhered cells was removed and the chamber was washed gently with 1 ml PBS to remove nonadherent cells, before adding 1 ml of prewarmed (30°C) YNB broth with appropriate amino acid supplements. Images were captured at intervals during incubation at 30°C. Cell fluorescence was quantified per unit cell area with Image J software, using the region-of-interest (ROI) tool. Cells at all stages of budding were examined.
Adherence assay.
HEp-2 epithelial cells were grown (5% CO2, 37°C) to confluence in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) fetal calf serum, 100 U ml−1 penicillin, 0.1 mg ml−1 streptomycin, and 2 mM l-glutamine, before splitting with 0.25% (wt/vol) trypsin, 0.53 mM EDTA. After incubation for 24 to 48 h in DMEM, 24-well tissue culture plates (Nunclon) were seeded with 1 × 104 cells ml−1 in DMEM and incubated at 37°C for 2 days. Confluent monolayers were washed twice with Hank's balanced salt solution (HBSS; Sigma) and fixed for 10 min in 4% (vol/vol) paraformaldehyde, before three 5-min washes with PBS. C. glabrata was diluted to 2,000 cells ml−1 in HBSS, before 1 ml of cells was added to individual wells containing the HEp-2 cell monolayers. Plates were centrifuged (500 × g, 1 min) and incubated at 37°C for 30 min to allow adherence. Supernatant was removed by pipetting, followed by three washes with 500 μl HBSS to remove unbound cells. Monolayers and bound C. glabrata cells were released by incubation at 37°C for 15 min with 500 μl 2% (wt/vol) trypsin in PBS. After addition of 10 μl of 10% (vol/vol) Triton X-100, 50 μl 100 mM EDTA, and 40 μl YEPD broth, released cells were plated in replicate on YEPD agar and incubated at 30°C for at least 2 days. Colonies were counted to determine adherent cell numbers.
RNA extraction and qRT-PCR.
Total RNA was isolated from either 1 × 106 (after FACS analysis) or 1 × 107 cells using an RNeasy minikit (Qiagen). Reverse transcription (RT) was performed using SuperScript III reverse transcriptase, and oligo(dT)20 primer (Invitrogen). RNase-free conditions were maintained with RNaseOUT recombinant RNase inhibitor (Invitrogen), and nuclease-free microcentrifuge tubes were used throughout. cDNA was purified (Geneflow PCR purification kit) and quantified with a NanoDrop spectrophotometer (NanoDrop Technologies). Samples were stored at −20°C. Quantitative RT-PCR (qRT-PCR) was performed in triplicate reactions, each comprising 30 ng cDNA, 100 nM each gene-specific primer (sequences available on request), and 1× Fast SYBR green master mix (Applied Biosystems), made up to 10 μl with RNase-free water. Reactions were performed in sealed 96-well plates and monitored with a 7500 Fast real-time PCR system (Applied Biosystems). The annealing temperature was 56°C. Amplification was quantified from a standard curve constructed from reactions with defined cDNA copy number. Results were analyzed with 7500 software, version 2.0.4 (Applied Biosystems).
RESULTS
Expression of Epa1 is heterogeneous and is related to adherence by individual cells.
During flow cytometric analyses of C. glabrata BG2 cells modified to express an EPA1-GFP transcriptional fusion construct, we noted that GFP reporter levels varied by >100-fold between individual cells (Fig. 1A). Two peaks of frequency could be distinguished, suggesting a bimodal distribution among cells from the asynchronous batch cultures (the heterogeneity was neither growth phase nor cell cycle dependent; see later). The degree of variation was more marked than that associated with autofluorescence or with other GFP transcriptional reporter constructs that we examine routinely; the result for ACT1-GFP in S. cerevisiae (30) is shown as a standard (Fig. 1A). Calculation of the CV according to previously published criteria (34) yielded a value of ∼106.3 for EPA1-GFP. This was higher than the CVs obtained for all 2,212 other yeast genes that have been tested in a similar way in asynchronous cultures, where values ranged from 8.4 to 77.3 (34). The CV for ACT1-GFP from the present data was 13.1.
Fig 1.

Single-cell Epa1 expression is heterogeneous and correlates with single-cell adherence phenotype. (A) Flow cytometric analysis of cellular fluorescence from either wild-type C. glabrata BG2 (autofluorescence), C. glabrata expressing an EPA1-GFP transcriptional fusion construct, or S. cerevisiae expressing an ACT1-GFP transcriptional fusion. (B) C. glabrata BG2 cells expressing an Epa1-HA construct were gated and sorted by FACS analysis according to Epa1-HA expression level, as indicated with anti-HA, Alexa Fluor 488 conjugate antibody (see Fig. 3A for example of gating). Sorted cells of the high- and low-Epa1-HA-expressing subpopulations were tested for adherence to HEp-2 epithelial cells, in comparison with unsorted C. glabrata BG2 and the isogenic epa1Δ mutant. Mean data from replicate determinations are shown ± SD.
To test the hypothesis that this marked cell-to-cell variation in EPA1 expression would predict adherence by individual C. glabrata cells, we expressed an Epa1 protein that was HA tagged at its N-terminal end (17). Probing with an anti-HA, Alexa Fluor 488 conjugate antibody enabled subsequent FACS analysis of cells according to cell surface Epa1 level. Sorted cell subpopulations comprising the lower and upper 13% of all Epa1-HA-expressing cells were tested for adherence to HEp-2 epithelial cells. Percent adherence was more than threefold greater by the higher- than the lower-Epa1-HA-expressing cells (Fig. 1B). (Adherence by the unsorted wild type was most similar to that of the high-expressing cells, as these comprised a majority of the population.) An epa1Δ null mutant was adherence defective, as reported previously (11, 17). To substantiate that the single-cell Epa1 expression level was transient, consistent with a nongenotypic rather than genotypic basis for the variation (3), low- and high-expressing cells were sorted and monitored for Epa1-HA at intervals following subculture to fresh medium. In both cases, initially skewed populations reverted to a mixed phenotype within 20 h of subculture (Fig. 2). Therefore, any inheritance of the Epa1-HA expression phenotype was not discernible beyond a few cell generations (the cell doubling time was ∼1.3 h).
Fig 2.

Dynamics of single-cell Epa1 expression. C. glabrata BG2 cells expressing high (left) or low (right) levels of Epa1-HA were gated and sorted by FACS analysis. The sorted subpopulations were incubated in fresh medium and reanalyzed by flow cytometry for Epa1-HA expression at the indicated intervals.
Epa1 expression heterogeneity is Sir3 dependent in C. glabrata BG2.
The data for the EPA1-GFP transcriptional fusion (Fig. 1A) suggested that cell-to-cell variation in Epa1 expression may be regulated at the mRNA level. We tested whether variation in the levels of Epa1-HA protein and EPA1 transcript was correlated in single cells, by measuring the latter in subpopulations sorted according to Epa1-HA content. The level of EPA1 mRNA was >7-fold greater in high- than in low-Epa1-HA-expressing cells, indicating that the Epa1-HA protein heterogeneity was at least partly described by EPA1 transcript heterogeneity (Fig. 3).
Fig 3.
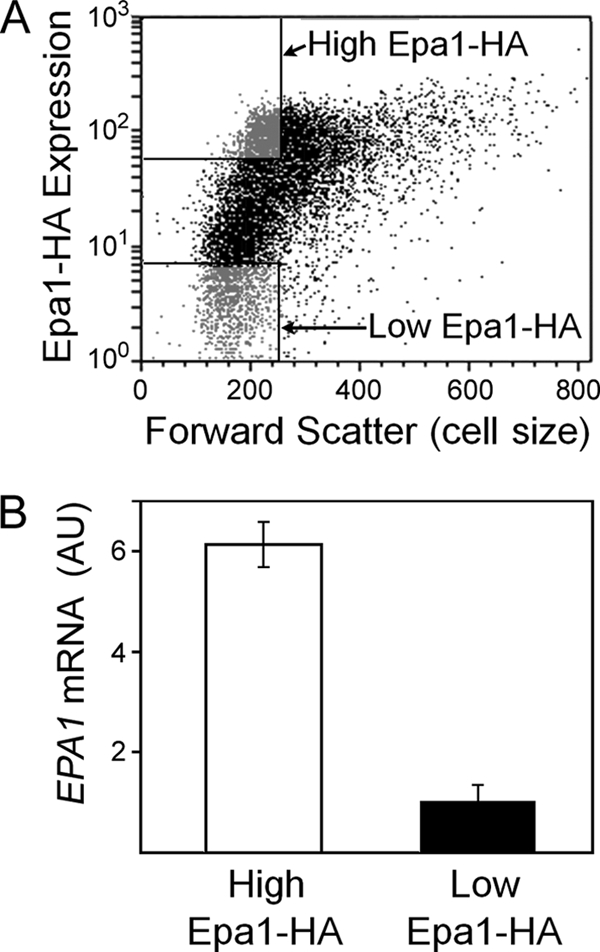
Correlation between Epa1 protein and EPA1 mRNA levels of individual C. glabrata BG2 cells. (A) Cells were gated and sorted by FACS analysis, according to Epa1-HA expression level (indicated with anti-HA, Alexa Fluor 488 conjugate antibody). The gates (rectangles at upper and lower left) were set to sort cells within the same forward scatter (cell size) range. (B) RNA was isolated from ∼1 × 106 cells of the high- and the low-Epa1-HA-expressing subpopulations (A), and EPA1 mRNA was quantified with qRT-PCR using standardized RNA additions in all reactions. The data shown are means from three independent experiments (each analyzed in triplicate) ± SEMs. AU, arbitrary units.
The phenotype reversion assay (Fig. 2) established that the single-cell EPA1 expression level was transient, reverting during several hours' growth. This timescale was relatively long compared to cell cycle- and rhythm-driven heterogeneity (47, 50) and suggested some inheritance of the expression state over a limited timescale. This was examined further by comparing Epa1-HA expression of parent cells and their offspring (before cell separation) during cell division. First, to show that the measured Epa1-HA in a budding cell reflected solely de novo synthesis in that cell, i.e., to discount a contribution from presynthesized protein derived from the parent, the fate of parental Epa1 protein was tracked during cell division (Fig. 4A). Antibody that was used to immunodetect Epa1-HA in individual cells was retained by the parental cell during subsequent budding and division. Therefore, Epa1-HA does not partition to offspring and the anti-HA antibody provided a snapshot of the cells' own Epa1-HA expression states. Subsequent comparison of bud and parent cell Epa1-HA levels revealed that these were partly correlated (Fig. 4B). This substantiated that the Epa1-HA expression state is partly inherited in C. glabrata BG2.
Given the bimodality and partial heritability of the Epa1 expression state (Fig. 1A, 2, and 4B), combined with the subtelomeric localization of EPA1 and its regulation by transcriptional silencing (9, 13), we hypothesized that differential gene silencing was a driver of heterogeneous EPA1 expression. To test this, heterogeneity was compared in wild-type C. glabrata BG2 and a sir3Δ deletion strain, defective for EPA1 silencing (9, 13). Mean EPA1-GFP expression was higher in the sir3Δ mutant, as expected (Fig. 4C). Moreover, the distribution of single-cell GFP expression levels was markedly narrower in the mutant (CV, ∼46) than the wild type (CV, ∼107), largely due to the absence of cells expressing small amounts of GFP from the EPA1 promoter. Such a contribution of gene silencing to heterogeneity has not previously been quantified in this way. We experienced difficulties deleting SIR3 in the Epa1-HA-expressing background. Therefore, we supported the above-mentioned results by probing Epa1 protein levels with an anti-Epa1 antibody (25). The results were similar qualitatively to those with the EPA1-GFP construct (Fig. 4C), indicating that Sir3-dependent silencing is a primary mechanism driving Epa1 expression heterogeneity in C. glabrata BG2. (Note that the relative proportions of cells occupying low- or high-Epa1-expressing subpopulations in the wild type [Fig. 4C] could vary between experiments and were not a function of the method used to determine expression.)
Silencing independent Epa1 heterogeneity in C. glabrata CBS138.
As part of this study, we also expressed the Epa1-HA construct in the C. glabrata reference strain CBS138. Single-cell Epa1-HA expression across the population was more uniform in the CBS138 strain (CV, ∼53) than in the BG2 strain (CV, ∼94) (Fig. 5A). As CBS138 cells expressed relatively large amounts of Epa1-HA, with no evidence of the silencing indicated in BG2 cells (Fig. 4C), it was reasoned that Epa1-HA silencing may be weak or absent in CBS138. This was borne out by the fact that SIR3 deletion did not further decrease heterogeneity or increase the mean Epa1 expression in CBS138 (Fig. 5B); indeed, the sir3Δ mutant of CBS138 exhibited a small decrease in mean Epa1 expression. Consistent with this evidence, the partial heritability of the Epa1-HA expression state apparent in single cells of the BG2 strain (Fig. 4B) was absent in CBS138 (Fig. 5C).
Despite the absence of Sir-dependent EPA1 silencing in CBS138, EPA1 expression was still sufficiently heterogeneous in this strain to place it within the top percentile of all similarly tested yeast genes (34). Accordingly, CBS138 provided a model to elucidate factors other than gene silencing that contribute to heterogeneity. GPI-anchored proteins commonly localize to lipid rafts, which themselves are heterogeneous among cells (31, 36). Treating cells with 100 μM ketoconazole and 2 μM myriocin, which inhibit biosynthesis of major lipid raft components (sterols and sphingolipids) (29), had no discernible effect on Epa1-HA expression heterogeneity, suggesting that the heterogeneity is not lipid raft related (data not shown). Yps1 and Yps7 are extracellular GPI-anchored aspartyl proteases which cleave Epa1 from the cell surface of C. glabrata (4, 25). The possibility that these protein activities determine heterogeneous cellular Epa1-HA levels was discounted with the observation that YPS1 and/or YPS7 deletion had no significant effect on heterogeneity (data not shown). As Epa1 is a mannoprotein, it could be subject to any general heterogeneity in mannoprotein synthesis or incorporation. Probing with 70 μg ml−1 of a fluorescent concanavalin A conjugate (Molecular Probes, Invitrogen) (6) revealed relatively uniform mannoprotein staining across cells of the population (CV, ∼29) (data not shown). This contrasted with the heterogeneity of Epa1-HA expression evident in the CBS138 strain (CV, ∼53). As the above-described posttranscriptional factors did not appear to be relevant to heterogeneous Epa1-HA expression, we considered whether silencing-independent heterogeneity in CBS138 may also be determined at the mRNA level. CBS138 cells sorted according to high Epa1-HA expression exhibited ∼3-fold higher levels of EPA1 mRNA than sorted low-Epa1-HA-expressing cells (Fig. 6). Therefore, noise at the mRNA level can account for Epa1-HA expression heterogeneity in both CBS138 and BG2 but is Sir regulated only in the latter strain, which is the more heterogeneous.
Fig 6.
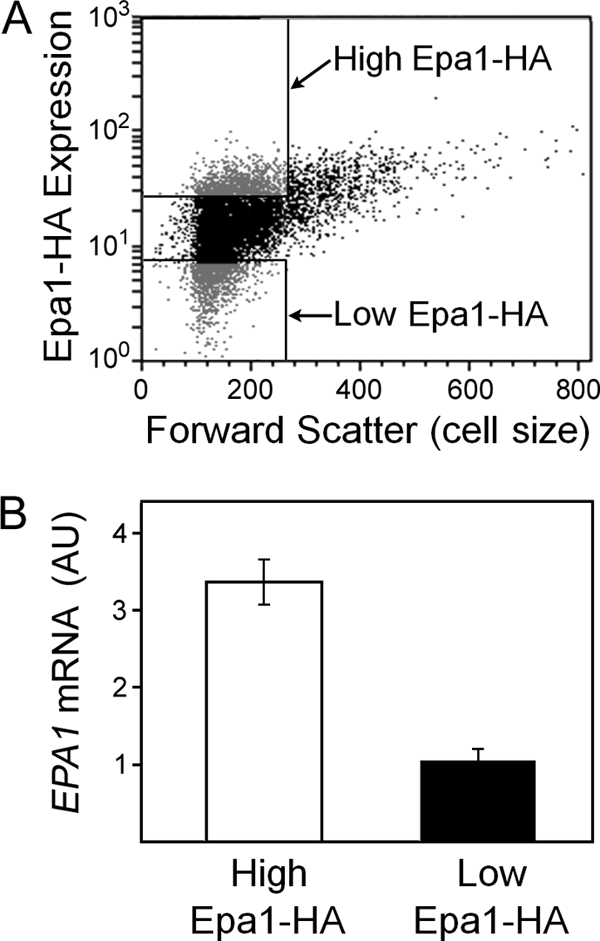
Correlation between Epa1 protein and EPA1 mRNA levels of individual C. glabrata CBS138 cells. (A) Cells were gated and sorted by FACS analysis, according to Epa1-HA expression level. The gates (rectangles at upper and lower left) were set to sort cells within the same forward-scatter (cell size) range. (B) RNA was isolated from ∼1 × 106 cells of the high- and the low-Epa1-HA-expressing subpopulations (A), and EPA1 mRNA was quantified with qRT-PCR using standardized RNA additions in all reactions. The data shown are means from three independent experiments (each analyzed in triplicate) ± SEMs.
Variation in EPA1 silencing between strains of C. glabrata.
It was striking that Sir-dependent silencing was a major source of Epa1-HA expression heterogeneity in one (BG2) but not another (CBS138) common C. glabrata strain. Most previous studies of EPA gene regulation have been with BG2, whereas CBS138 was the reference strain used for the genome sequence. The DNA sequence from the region 500 bp upstream to 500 bp downstream of the EPA1 ORF is 99% identical between the strains (http://cbi.labri.fr/Genolevures; www.ncbi.nlm.nih.gov). The sequence discrepancies are outside functionally important regions, like the promoter, the N-terminal ligand-binding domain, or the GPI-anchor sequences. Moreover, the heterogeneity difference was retained when the same Epa1-HA construct was expressed in both strains. Genomic polymorphism involving translocations and recombination between repeat sequences in the (sub)telomeric regions is widespread in C. glabrata (32, 37, 53). Any such events leading to an altered distance of the EPA1 locus from the chromosome end could explain the present strain difference: the strength of Sir-dependent silencing decreases with distance from telomeres (41, 51). We estimated that the scale of difference in silencing strength apparent in this study would require EPA1 to be at least 2.6 kb closer to the chromosome end in BG2 than in CBS138 (13, 42). Such a difference was not evident from probing the large, terminal EPA1-containing chromosome E fragment following PmeI or StuI digestion of C. glabrata genomic DNA and separation by field inversion gel electrophoresis (data not shown). Consistent with this, the total length of chromosome E is the same in BG2 and CBS138 (32).
We introduced an extra copy of the SIR3 gene to CBS138 with the centromeric plasmid pCgACT-14-SIR3. This established a silencing-like effect on EPA1, broadening the Epa1-HA expression profile and increasing the heterogeneity (Fig. 7). The fact that an extra SIR3 copy was required to establish this silencing in CBS138 (whereas the extant chromosomal SIR3 was insufficient, as its deletion did not increase EPA1 expression; Fig. 5B) indicated that natural EPA1 silencing is weak in this strain. SIR3 mRNA levels were no lower in CBS138 than in BG2 according to qRT-PCR (data not shown), suggesting that weak silencing was not due to defective SIR3 expression.
Fig 7.

Weak EPA1 silencing in C. glabrata CBS138. Flow cytometric histograms of Epa1-HA expression heterogeneity among cells of exponential-phase CBS138 cells either transformed (+SIR3) or not with pCgACT-14-SIR3.
There are many potential causes of inefficient gene silencing (see Discussion), and its further elucidation here was outside the scope of this study's focus on heterogeneity. Instead, considering the relevance to virulence-associated properties (Fig. 1B), the prevalence of Sir-dependent or -independent Epa1 expression heterogeneity was investigated across a range of clinical and nonclinical C. glabrata isolates. As genetic manipulation of these isolates was not feasible, nicotinic acid (NA) limitation was used to block Sir-dependent Epa1 silencing (14). We validated the approach with the BG2 and CBS138 strains, where NA limitation gave, respectively, increased and decreased culture-averaged EPA1 expression (measured with qRT-PCR) (Fig. 8A), in agreement with the results for sir3Δ mutants (Fig. 4C and 5B). Similar effects of NA were observed by measuring Epa1-HA (data not shown). It was verified previously that the effect of NA on EPA gene expression is Sir mediated, as the effect was abolished in a sir2Δ mutant (14). To gauge the Sir dependency of EPA1 expression across a range of isolates, we quantified EPA1 mRNA as described above during growth in NA-replete and NA-limited medium. Each strain exhibited one of three responses to NA, and a typical example of each is presented (Fig. 8B). Five of the 11 test strains (including isolate 172) exhibited EPA1 silencing that was relieved by NA limitation, similar to BG2. The effect of NA limitation mirrored that in CBS138 in three of the other test strains (isolates 31, 39, and 82), while NA limitation had no significant effect on EPA1 expression in the remaining isolates (P > 0.05, according to Student's t test). The clinical strains were isolates from different types of patient samples (blood, urine, etc.), but we could not discern any relationship between isolate source and the responses to NA limitation.
Based on preceding data for BG2 and CBS138, we predicted that the apparent marked variation in silencing-dependent EPA1 expression across the isolates should have implications for heterogeneity. This was tested by probing Epa1 expression with anti-Epa1 antibody and analysis by flow cytometry (for representative plots, see Fig. 8C). Like the BG2 strain, all isolates in which EPA1 silencing was suppressible with low NA (e.g., isolate 172) exhibited both low- and high-Epa1-expressing subpopulations under NA-replete conditions, hence a broad heterogeneity (Fig. 8C and D). In contrast, the other isolates exhibited a single dominant peak of high Epa1 expression and less heterogeneity. A positive relationship between silencing-dependent EPA1 expression (low NA/high NA expression ratio) and heterogeneity (CV) was elucidated across the range of isolates examined (Fig. 8D), indicating that strain-specific gene silencing is a key factor determining strain-to-strain variation in the heterogeneity of Epa1 expression in C. glabrata.
DISCUSSION
Research efforts in recent years have improved our understanding of the molecular mechanisms of fungal pathogenesis, including the gene products that contribute to virulence. The use of large sample sizes is a necessity for many such studies, generating culture-averaged data. Although valuable, these mask underlying variation in the phenotype. Here we showed that Epa1, a well-studied virulence-related adhesin of C. glabrata, is subject to marked variation in expression between cells. Furthermore, as the expression level of Epa1 in individual cells was correlated with adherence to epithelial cells, this establishes a link between EPA1 expression noise and virulence potential. The threat of serious infection will increase with C. glabrata inoculum size, one reason for which could be an increased likelihood that the inoculum will include more virulent cell variants, akin to the inoculum effect (49). In principle, it could take just a few such variant cells masked within a much larger, apparently avirulent, yeast population to initiate infection.
The level of cell-to-cell variation in EPA1 expression was higher than we or others have previously reported for a yeast gene (34). Expression noise is reported to be a trait selected in genes that respond to environmental change (16, 34). It is tempting to speculate that similar evolutionary pressures may also favor expression noise in virulence factors. The EPA1 expression variation appeared to be mediated primarily at the mRNA level, consistent with transcriptional regulation being a major contributor to gene expression noise generally (3, 48). The partial inheritability of cellular EPA1 expression state combined with its Sir dependency revealed transcriptional silencing to be a primary contributor to heterogeneity in some strains of C. glabrata. Epigenetic regulation is recognized to be a major source of cell-to-cell variation more widely (3), and gene silencing mediates differential expression of the S. cerevisiae FLO genes, functional homologues of the EPA gene family (20). Here, changes in Epa1 expression state occurred at a relatively high frequency, as evidenced by the fact that high- or low-expressing subpopulations reverted to a mixed phenotype within a few cell generations. In the context of cell-to-cell heterogeneity, more rapid phenotypic transitions are predicted to be increasingly favored with increasing frequency at which an organism's environment changes (1). Such changes in the environment of C. glabrata are expected during the course of an infection, as cells traverse barriers within the body, invading the bloodstream and/or colonizing different epithelia and organs.
Sir-dependent silencing accounted for only approximately half of the variation in EPA1 expression of the BG2 strain. Previous studies have not quantified the contribution of gene silencing to expression heterogeneity in this way. The degree of heterogeneity that remained in the sir3Δ mutant (and in the CBS138 strain) still placed EPA1 within the top percentile of 2,212 yeast genes ranked according to the degree of noise in their expression (34). This Sir-independent EPA1 expression heterogeneity also appeared to have a transcriptional basis. Transcriptional noise in yeast genes is commonly extrinsic in origin, e.g., arising from cell-to-cell regulatory variation, as opposed to being stochastic events at the smallest spatial scales (10, 40). Potential sources of regulatory variation are difficult to predict in the present case as, other than silencing, EPA regulation is not well characterized. The transcription factors Flo8 and Mss11 regulate FLO gene expression in S. cerevisiae (55), and the corresponding regulators in C. glabrata have been implicated in EPA gene activation (33). Intriguingly, Flo8 has been identified to be a mediator of gene expression diversity among yeasts (56). Furthermore, many yeast strains carry a nonsense mutation in FLO8 which, it has been proposed, may be suppressed by the unstable PSI+ prion (45). The reversibility of this prion function determines readthrough, or not, of in-frame nonsense codons and a resultant heterogeneity in the expression of affected genes (54). This potential source of Flo8 expression heterogeneity would be expected to be transmitted to any Flo8-regulated genes. The noninheritance of the Epa1 expression state in CBS138 is consistent with expression changes that occur over short (≤1 generation) timescales to determine heterogeneity (3). Our data indicate that neither epigenetic silencing nor the cell cycle or cell aging (S. C. Halliwell and S. V. Avery, unpublished data) is a driver of EPA1 expression noise in the CBS138 strain.
Previous insights into Sir-dependent EPA gene regulation were gained from studies using the BG2 strain background (9, 13), and our results for BG2 were consistent with these. However, EPA1 silencing is strain specific, as neither the CBS138 strain nor several clinical isolates exhibited Sir-dependent EPA1 repression. Therefore, a key aspect of our existing understanding of EPA1 regulation does not apply to all C. glabrata strains. As discussed further below, this could explain some of the marked variation in adherence abilities of different C. glabrata strains reported previously (12, 28). The establishment of silencing in the CBS138 strain required the introduction of only an additional copy of the SIR3 gene, suggesting that EPA1 silencing is merely weakened (not blocked) in the wild type and that this weakening might be SIR3 centered. Sir3 along with Sir4 in yeast is considered to help remodel the structure of chromatin regions (which are subject to silencing) to a repressive state (43).
SIR3 mRNA levels were actually approximately 1.5-fold higher in CBS138 than in BG2. This could suggest a response to compensate for weak silencing more generally in CBS138, consistent with the fact that no EPA1-specific features that might explain differential silencing between CBS138 and BG2 have been identified. A previous study assigned the differing responses to osmotic stress between these two C. glabrata strains to a sequence difference in a MAPKKK gene (19). Sequences in and around EPA1 are near identical in the two strains, including the number of repeats of the EPA1 120-bp minisatellite (13, 32, 46, 52). The heterogeneity difference was retained when the same Epa1-HA construct was expressed in both strains. Our data did not support the possibility that insertions or deletions between EPA1 and the chromosome ends may have produced differing mean strengths of the telomere position effect at EPA1. (That is not to say that cell-to-cell variation in telomere length [9, 23] could not contribute to the heterogeneity within a strain.) Thirty-five negative regulators of Sir activity have been identified in S. cerevisiae (38), and the potential mechanistic bases for a nonspecific weakening of gene silencing in one strain versus another are manifold; this study has highlighted a major phenotypic consequence of such strain-to-strain variation. It is uncertain why several of the test strains, including CBS138, actually exhibited decreased EPA1 expression when Sir-dependent silencing was suppressed. One possibility is loss of silencing of a repressor of EPA1 expression. We have noted a putative binding site for the C. glabrata α2 repressor protein in the EPA1 promoter (at −101 to 109 bp), and the α2-encoding gene is subject to transcriptional silencing (39).
The relevance of heterogeneity to the adherence (virulence) potential of individual C. glabrata cells, shown here, suggests that the strain specificity of this heterogeneity may be an important factor in a strain's virulence. Similarly, heterogeneity in bacterial antibiotic resistance is a factor that can exacerbate bacterial infectious disease (27). Adhesion capacity is known to vary between strains of C. glabrata (12). With increasing heterogeneity, there should be an increased risk of variant hypervirulent cells arising in a population. To that end, it might be predicted that isolates with the greatest heterogeneity in this study (i.e., those exhibiting Sir-mediated EPA silencing) could have the greatest virulence potential (26). This would not be the only factor, as both strain types were represented among the range of clinical isolates studied here. Furthermore, the impact of silencing on heterogeneity here results from a tendency toward increased numbers of cells expressing low rather than high levels of EPA1. Indeed, the low-heterogeneity, high-EPA1-expressing strain CBS138 exhibits particularly strong adherence to plastic surfaces (12), i.e., the type of substrate favored by Epa1 (11). Nevertheless, where Epa1 is not incorporated in the cell wall, other Epa proteins may take its place, so adding another layer of diversity. The different Epa proteins have different ligand-binding specificities (57) and, it is thought, may be differentially regulated to facilitate adherence to different host surfaces during the progress of infection (9, 12, 57). A future challenge will be to determine the heterogeneity of expression of all key Epa proteins across individual cells and how this impacts population-level virulence in vivo.
ACKNOWLEDGMENTS
This work was funded by research grants from the NERC (NE/E005969/1) and the BBSRC (BB/C506656/1). S.C.H. was supported by a BBSRC Ph.D. research studentship.
We thank Neil Gow for initially suggesting Epa1 as a model for our heterogeneity studies and Brendan Cormack for C. glabrata strains and the anti-Epa1 antibody. The clinical isolates were a gift from Michael Petrou, and some additional reagents were from Ken Haynes. We thank Ed Louis for valuable discussions and Lee Shunburne for expert technical assistance.
Footnotes
Published ahead of print 2 December 2011
REFERENCES
- 1. Acar M, Mettetal JT, van Oudenaarden A. 2008. Stochastic switching as a survival strategy in fluctuating environments. Nat. Genet. 40:471–475 [DOI] [PubMed] [Google Scholar]
- 2. Ausubel FM, et al. 2011. Current protocols in molecular biology. John Wiley & Sons, New York, NY [Google Scholar]
- 3. Avery SV. 2006. Microbial cell individuality and the underlying sources of heterogeneity. Nat. Rev. Microbiol. 4:577–587 [DOI] [PubMed] [Google Scholar]
- 4. Bairwa G, Kaur R. 2011. A novel role for a glycosylphosphatidylinositol-anchored aspartyl protease, CgYps1, in the regulation of pH homeostasis in Candida glabrata. Mol. Microbiol. 79:900–913 [DOI] [PubMed] [Google Scholar]
- 5. Bayliss CD. 2009. Determinants of phase variation rate and the fitness implications of differing rates for bacterial pathogens and commensals. FEMS Microbiol. Rev. 33:504–520 [DOI] [PubMed] [Google Scholar]
- 6. Biondo C, et al. 2005. Characterization of two novel cryptococcal mannoproteins recognized by immune sera. Infect. Immun. 73:7348–7355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blake WJ, et al. 2006. Phenotypic consequences of promoter-mediated transcriptional noise. Mol. Cell 24:853–865 [DOI] [PubMed] [Google Scholar]
- 8. Castano I, et al. 2003. Tn7-based genome-wide random insertional mutagenesis of Candida glabrata. Genome Res. 13:905–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castano I, et al. 2005. Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol. Microbiol. 55:1246–1258 [DOI] [PubMed] [Google Scholar]
- 10. Colman-Lerner A, et al. 2005. Regulated cell-to-cell variation in a cell-fate decision system. Nature 437:699–706 [DOI] [PubMed] [Google Scholar]
- 11. Cormack BP, Ghori N, Falkow S. 1999. An adhesin of the yeast pathogen Candida glabrata mediating adherence to human epithelial cells. Science 285:578–582 [DOI] [PubMed] [Google Scholar]
- 12. de Groot PW, et al. 2008. The cell wall of the human pathogen Candida glabrata: differential incorporation of novel adhesin-like wall proteins. Eukaryot. Cell 7:1951–1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. De Las Penas A, et al. 2003. Virulence-related surface glycoproteins in the yeast pathogen Candida glabrata are encoded in subtelomeric clusters and subject to RAP1- and SIR-dependent transcriptional silencing. Genes Dev. 17:2245–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Domergue R, et al. 2005. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Science 308:866–870 [DOI] [PubMed] [Google Scholar]
- 15. Fidel PL, Jr, Vazquez JA, Sobel JD. 1999. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 12:80–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fraser HB, Hirsh AE, Giaever G, Kumm J, Eisen MB. 2004. Noise minimization in eukaryotic gene expression. PLoS Biol. 2:834–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frieman MB, McCaffery JM, Cormack BP. 2002. Modular domain structure in the Candida glabrata adhesin Epa1p, a beta1,6 glucan-cross-linked cell wall protein. Mol. Microbiol. 46:479–492 [DOI] [PubMed] [Google Scholar]
- 18. Geisel N, Vilar JM, Rubi JM. 2011. Optimal resting-growth strategies of microbial populations in fluctuating environments. PLoS One 6:e18622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gregori C, Schueller C, Schwarzmuller T, Ammerer G, Kuchler K. 2007. The high-osmolarity glycerol response pathway in the human fungal pathogen Candida glabrata strain ATCC 2001 lacks a signaling branch that operates in baker's yeast. Eukaryot. Cell 6:1635–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Halme A, Bumgarner S, Styles C, Fink GR. 2004. Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast. Cell 116:405–415 [DOI] [PubMed] [Google Scholar]
- 21. Holland SL, Avery SV. 2009. Actin-mediated endocytosis limits intracellular Cr accumulation and Cr toxicity during chromate stress. Toxicol. Sci. 111:437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horn DL, et al. 2009. Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin. Infect. Dis. 48:1695–1703 [DOI] [PubMed] [Google Scholar]
- 23. Kachouri-Lafond R, et al. 2009. Large telomerase RNA, telomere length heterogeneity and escape from senescence in Candida glabrata. FEBS Lett. 583:3605–3610 [DOI] [PubMed] [Google Scholar]
- 24. Kaur R, Domergue R, Zupancic ML, Cormack BP. 2005. A yeast by any other name: Candida glabrata and its interaction with the host. Curr. Opin. Microbiol. 8:378–384 [DOI] [PubMed] [Google Scholar]
- 25. Kaur R, Ma B, Cormack BP. 2007. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. U. S. A. 104:7628–7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumamoto CA, Pierce JV. 2011. Immunosensing during colonization by Candida albicans: does it take a village to colonize the intestine? Trends Microbiol. 19:263–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lewis K. 2010. Persister cells. Annu. Rev. Microbiol. 64:357–372 [DOI] [PubMed] [Google Scholar]
- 28. Luo G, Samaranayake LP. 2002. Candida glabrata, an emerging fungal pathogen, exhibits superior relative cell surface hydrophobicity and adhesion to denture acrylic surfaces compared with Candida albicans. APMIS 110:601–610 [DOI] [PubMed] [Google Scholar]
- 29. Martin SW, Konopka JB. 2004. Lipid raft polarization contributes to hyphal growth in Candida albicans. Eukaryot. Cell 3:675–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mateus C, Avery SV. 2000. Destabilized green fluorescent protein for monitoring dynamic changes in yeast gene expression with flow cytometry. Yeast 16:1313–1323 [DOI] [PubMed] [Google Scholar]
- 31. Mishra S, Joshi PG. 2007. Lipid raft heterogeneity: an enigma. J. Neurochem 103(Suppl 1):135–142 [DOI] [PubMed] [Google Scholar]
- 32. Muller H, et al. 2009. Genomic polymorphism in the population of Candida glabrata: gene copy-number variation and chromosomal translocations. Fungal Genet. Biol. 46:264–276 [DOI] [PubMed] [Google Scholar]
- 33. Mundy RD, Cormack B. 2009. Expression of Candida glabrata adhesins after exposure to chemical preservatives. J. Infect. Dis. 199:1891–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Newman JR, et al. 2006. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441:840–846 [DOI] [PubMed] [Google Scholar]
- 35. Payne T. 2007. Protein secretion in Saccharomyces cerevisiae. Ph.D. thesis University of Nottingham, Nottingham, United Kingdom [Google Scholar]
- 36. Pike LJ. 2004. Lipid rafts: heterogeneity on the high seas. Biochem. J. 378:281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Polakova S, et al. 2009. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc. Natl. Acad. Sci. U. S. A. 106:2688–2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raisner RM, Madhani HD. 2008. Genomewide screen for negative regulators of sirtuin activity in Saccharomyces cerevisiae reveals 40 loci and links to metabolism. Genetics 179:1933–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramirez-Zavaleta CY, Salas-Delgado GE, De Las Penas A, Castano I. 2010. Subtelomeric silencing of the MTL3 locus of Candida glabrata requires yKu70, yKu80, and Rif1 proteins. Eukaryot. Cell 9:1602–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raser JM, O'Shea EK. 2004. Control of stochasticity in eukaryotic gene expression. Science 304:1811–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Renauld H, et al. 1993. Silent domains are assembled continuously from the telomere and are defined by promoter distance and strength, and by SIR3 dosage. Genes Dev. 7:1133–1145 [DOI] [PubMed] [Google Scholar]
- 42. Rosas-Hernandez LL, et al. 2008. yKu70/yKu80 and Rif1 regulate silencing differentially at telomeres in Candida glabrata. Eukaryot. Cell 7:2168–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rusche LN, Kirchmaier AL, Rine J. 2003. The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu. Rev. Biochem. 72:481–516 [DOI] [PubMed] [Google Scholar]
- 44. Scherf A, Lopez-Rubio JJ, Riviere L. 2008. Antigenic variation in Plasmodium falciparum. Annu. Rev. Microbiol. 62:445–470 [DOI] [PubMed] [Google Scholar]
- 45. Serio TR, Lindquist SL. 1999. PSI+: an epigenetic modulator of translation termination efficiency. Annu. Rev. Cell Dev. Biol. 15:661–703 [DOI] [PubMed] [Google Scholar]
- 46. Sherman D, et al. 2006. Genolevures complete genomes provide data and tools for comparative genomics of hemiascomycetous yeasts. Nucleic Acids Res. 34:D432–D435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Smith MC, Sumner ER, Avery SV. 2007. Glutathione and Gts1p drive beneficial variability in the cadmium resistances of individual yeast cells. Mol. Microbiol. 66:699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Snijder B, Pelkmans L. 2011. Origins of regulated cell-to-cell variability. Nat. Rev. Mol. Cell Biol. 12:119–125 [DOI] [PubMed] [Google Scholar]
- 49. Steels H, James SA, Roberts IN, Stratford M. 2000. Sorbic acid resistance: the inoculum effect. Yeast 16:1173–1183 [DOI] [PubMed] [Google Scholar]
- 50. Sumner ER, Avery AM, Houghton JE, Robins RA, Avery SV. 2003. Cell cycle- and age-dependent activation of Sod1p drives the formation of stress resistant cell subpopulations within clonal yeast cultures. Mol. Microbiol. 50:857–870 [DOI] [PubMed] [Google Scholar]
- 51. Talbert PB, Henikoff S. 2006. Spreading of silent chromatin: inaction at a distance. Nat. Rev. Genet. 7:793–803 [DOI] [PubMed] [Google Scholar]
- 52. Thierry A, Bouchier C, Dujon B, Richard GF. 2008. Megasatellites: a peculiar class of giant minisatellites in genes involved in cell adhesion and pathogenicity in Candida glabrata. Nucleic Acids Res. 36:5970–5982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thierry A, Dujon B, Richard GF. 2010. Megasatellites: a new class of large tandem repeats discovered in the pathogenic yeast Candida glabrata. Cell. Mol. Life Sci. 67:671–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. True HL, Berlin I, Lindquist SL. 2004. Epigenetic regulation of translation reveals hidden genetic variation to produce complex traits. Nature 431:184–187 [DOI] [PubMed] [Google Scholar]
- 55. van Dyk D, Pretorius IS, Bauer FF. 2005. Mss11p is a central element of the regulatory network that controls FLO11 expression and invasive growth in Saccharomyces cerevisiae. Genetics 169:91–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zheng W, Zhao HY, Mancera E, Steinmetz LM, Snyder M. 2010. Genetic analysis of variation in transcription factor binding in yeast. Nature 464:1187–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zupancic ML, et al. 2008. Glycan microarray analysis of Candida glabrata adhesin ligand specificity. Mol. Microbiol. 68:547–559 [DOI] [PubMed] [Google Scholar]