

Abstract
A hepatitis B virus (HBV) vaccine has been developed using a new adjuvant and HBV surface antigens produced from a CHO cell line. The purified HBV surface antigens are composed of L protein, M protein, and S protein in a mixture of 20- and 40-nm-diameter particles and filamentous forms. This HBV surface antigen, formulated with L-pampo, a proprietary adjuvant, induced 10 times more antibody than the same antigen with alum and was capable of inducing strong immune responses in three different HBV transgenic mice. In spite of the presence of a large amount of HBV antigen in the blood, no antibody against HBV surface antigen was normally detected in these transgenic mice. After immunization, the HBV antigen was also cleared from the blood.
INTRODUCTION
Hepatitis B virus (HBV) is a major cause of liver disease worldwide (3, 10, 24). Efficient vaccines against HBV infection are widely available, yet HBV infection is still a major problem in Asia, Latin America, Africa, and Eastern Europe. Every year, 50 million people are infected with HBV, and 5 to 10% become chronically infected. Vertical infection and infection from mother to neonate, however, lead to chronic infections in almost 100% of cases. Additionally, more than 90% of HBV infections in babies younger than 10 months result in chronic infection. Therefore, an improved HBV vaccine that can elicit protective immunity within 1 to 2 months would be beneficial, since currently available vaccines take 7 to 10 months to produce protective immunity.
Considerable efforts have been made to improve prophylactic HBV vaccines: mainly to achieve faster and better protection, to seroconvert those who do not respond to currently available vaccines, and to meet the demands of special groups of people, such as health care workers and immune-suppressed individuals (22, 30). In these efforts to develop advanced vaccines, the major strategy for improvement has been to supplement the small HBV surface antigen (8, 14, 25), the antigen used in most of the currently available vaccines, with the pre-S1 and pre-S2 portions of the HBV surface antigen (HBsAg).
HBsAg is composed of three kinds of envelope proteins: the S protein, consisting of 226 amino acids (aa); the 281-aa M protein, formed by the S protein linked to pre-S2 (55 aa); and the 389- or 400-aa L protein, formed by the M protein linked to pre-S1 (108 or 119 aa, depending on the HBV serotype). Glycosylation of these proteins yields six different molecules: two S proteins, a nonglycosylated 24-kDa protein (P24) and a glycosylated 27-kDa protein (GP27); two M proteins glycosylated on one (GP33) or two (GP36) glycosylation sites; and two L proteins, a nonglycosylated 39-kDa protein (P39) and a glycosylated 42-kDa protein (GP42) (7, 16, 23). In addition to these six proteins, one more 46-kDa protein band is routinely observed.
We have developed a CHO cell line that produces all three forms of HBV surface antigens, the L protein, the M protein, and the S protein, in three different particle forms. These particle forms of the HBV envelope antigen, when formulated in aluminum hydroxide (alum), are highly immunogenic in mice, inducing more HBV surface antigen-specific antibodies than any HBV vaccine we have tested. This new vaccine has been further improved by using an adjuvant that we have developed. When used with the new adjuvant, the new vaccine efficiently induced strong HBV-specific antibodies in three different HBV gene transgenic mice.
MATERIALS AND METHODS
Animals.
Female C57BL/6 mice or BALB/c mice (Charles River, Japan) aged 6 to 8 weeks were used for immunization. Three different HBV gene transgenic mouse models expressing HBV surface antigen (HBsAg) were also used for immunization. One of the transgenic mouse models was the HBsAg/HLA-A2 transgenic (Tg) mouse generated by Loirat et al. (9, 11) and given to Y. C. Sung at the Pohang University of Science and Technology (POSTECH), Pohang, South Korea. The Tg mice in this model continuously express HBsAg in their liver cells and human HLA-A2 major histocompatibility complex (MHC) class I molecules on the surfaces of all cells. The sera from these mice contain HBsAg in the form of 22-nm-diameter particles but have no detectable HBV-specific antibody. These mice were immunized, and their sera were collected, at the POSTECH animal facility according to animal care guidelines. The other two HBV transgenic mouse models used contain the whole HBV genome (1.3 copy); sera from these mice contain HBsAg and HBeAg (29, 31). The mice in one of these models were provided by the 458 Hospital of PLA in Guangzhou, China, and experiments were performed at the hospital's facility; serum samples were analyzed in our laboratory. The other Tg mice with the complete HBV gene (Tg[HBV 1.3 genome]Chi32), used in immune tolerance-breaking studies (5), were developed at Francis Chisari's laboratory, Scripps Research Institute, La Jolla, CA, and were donated to the Institute for Antiviral Research, Utah State University.
Preparation of recombinant L-HBsAg.
The entire coding region of the HBV envelope gene (pre-S1–pre-S2–S) was amplified by PCR and was ligated into the pMSG vector (South Korean patent application 10-2000-0043996), resulting in the recombinant vector pMSG-L-HBsAg. From the CHO cells transformed with the recombinant vector construct, the transformant that showed the highest expression level [CHO DG44/L-HBsAg(J2.1)-G101] was selected and deposited in the Korean Collection for Type Cultures.
L-HBsAg, which is composed of L protein, M protein, and S protein was initially purified from the culture medium of the recombinant CHO cell line by centrifugation at 3,600 × g for 20 min to remove the cell debris, followed by filtration through a 0.22-μm-pore-size filter to remove impurities. Then the L-HBsAg was purified by a combination of ion-exchange chromatography and gel filtration chromatography.
Test vaccines for animal experiments.
Depending on the experiments, 0.5 to 5 μg of antigens was formulated with alum (Alhydrogel; Brenntag Biosector, Denmark) or L-pampo (Korean patent 10-0900837; PCT and U.S. patent applications pending), containing a lipopeptide and synthetic RNA, in 200 μl of buffer containing 150 mM NaCl and 5 mM sodium phosphate (pH 6.9). The amount of alum or L-pampo in each dose (200 μl) was 125 μg or 45 μg, respectively. The mixed antigen and adjuvant in the buffer were kept overnight at 4°C with gentle rocking. As a commercial vaccine control, an HBV S protein vaccine produced in yeast (Berna Biotech, South Korea) was used.
Western blot analysis of the purified HBV surface antigen.
Western blot analysis of the purified recombinant HBV surface antigen was carried out as described by Heermann et al. (7), except that the monoclonal antibodies used were produced in our laboratory. The monoclonal anti-S protein antibody was against recombinant HBV (adr) S protein from yeast (Green Cross Corp., South Korea), and the epitope was not defined. The monoclonal antibody against pre-S1 was prepared using a synthetic peptide composed of amino acids 37 to 47 (NSNNPDWDFNP), and the anti-pre-S2 monoclonal antibody was against a synthetic peptide comprising aa 132 to 143 (LDPRVRGLYFPA). Hybridoma cell lines producing these monoclonal antibodies have been established by Adipogen (Incheon, South Korea).
Immunization of animals and analysis of samples.
Animals at the age of 6 to 8 weeks were injected intramuscularly with various test vaccines formulated in alum or L-pampo at 2-week or 1-month intervals. The negative-control group received phosphate-buffered saline (PBS). Blood samples were taken from mice before immunization and then every 2 weeks to 1 month during the immunization process.
Antibody titers were determined by using commercial enzyme-linked immunosorbent assay (ELISA) kits (ETI-AB-AUK-3; Diasorin, Italy) and were expressed in milli-international units (mIU) per milliliter, according to the manufacturer's instructions. For routine experiments where antibody titers did not have to be in international units, in-house-prepared ELISA kits were used as follows. Briefly, 96-well microplates were coated with HBsAg (S protein from yeast) at a concentration of 100 ng/well and were blocked with bovine serum albumin (1%) for 1 h. After the microplates were washed, diluted serum was added to each well and was incubated at 37°C for 2 h. Then horseradish peroxidase (HRP)-conjugated anti-mouse IgG (KPL) was added as a secondary antibody, and the plates were incubated at 37°C for 1 h. After washing, a color-developing agent (tetramethylbenzidine [TMB]-peroxidase solution; KPL) was added, and the mixture was incubated at room temperature for 20 min. Then the optical density (OD) of each well was measured at 450 nm using an automatic ELISA plate reader. The antibody titer (geometric mean titer [GMT]) was defined as the inverse of the fold dilution at which the OD value was 3-fold greater than that of the negative control. To analyze the production of the anti-pre-S antibody, recombinant pre-S rather than HBsAg was used to coat the ELISA plate, and the procedure described above was employed. To determine the titers of antibody isotypes IgG1, IgG2a, and IgG2b, mouse monoclonal antibody isotyping reagents (Sigma) were used. Analyses of HBsAg in preimmune sera and in sera after each immunization were carried out using the Genedia HBsAg ELISA kit, version 3.0 (Green Cross Corp., South Korea), according to the manufacturer's instructions.
Statistical analysis.
For comparison of HBsAg titers before and after immunization, we performed statistical analysis using the paired t test. P values less than 0.01 were considered to indicate significant decreases in HBsAg levels in the blood (99% confidence limit).
Nucleotide sequence accession number.
The CHO DG44/L-HBsAg(J2.1)-G101 transformant has been deposited in the Korea Research Institute of Bioscience and Biotechnology (Korean Collection for Type Cultures, Ueun-dong, Yusung-gu, Daejeon-si, South Korea) under accession number KCTC 11058BP.
RESULTS
Characterization of the HBV surface antigen produced from a CHO cell line.
HBV surface antigens produced from the recombinant CHO cell line transfected with an expression vector containing the entire HBV surface antigen gene were purified and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) as shown in Fig. 1. The recombinant HBV surface antigen spread on an SDS-PAGE gel as six major protein bands, representing a nonglycosylated 24-kDa S protein (P24), a glycosylated 27-kDa S protein (GP27), two glycosylated M proteins of 33 kDa (GP33) and 36 kDa (GP36), a nonglycosylated L protein of 39 kDa (P39), and a glycosylated L protein of 42 kDa (GP42). In addition to these, one minor band larger than any of the six proteins was repeatedly observed. N-terminal sequence analysis revealed that this protein is an authentic translation product of the HBV surface antigen gene, utilizing the AUG codon present on the upstream sequence of the regular initiation codon. It is not known at this point whether this happens in natural HBV infection. Western blot analysis using S protein- and pre-S-specific antibodies confirmed that all seven protein bands were HBV surface antigens (Fig. 1). This SDS-PAGE separation pattern is similar to that of the HBV surface protein purified from the blood of chronic human patients (7, 23). This HBV surface antigen from the CHO cell line is designated L-HBsAg.
Fig 1.
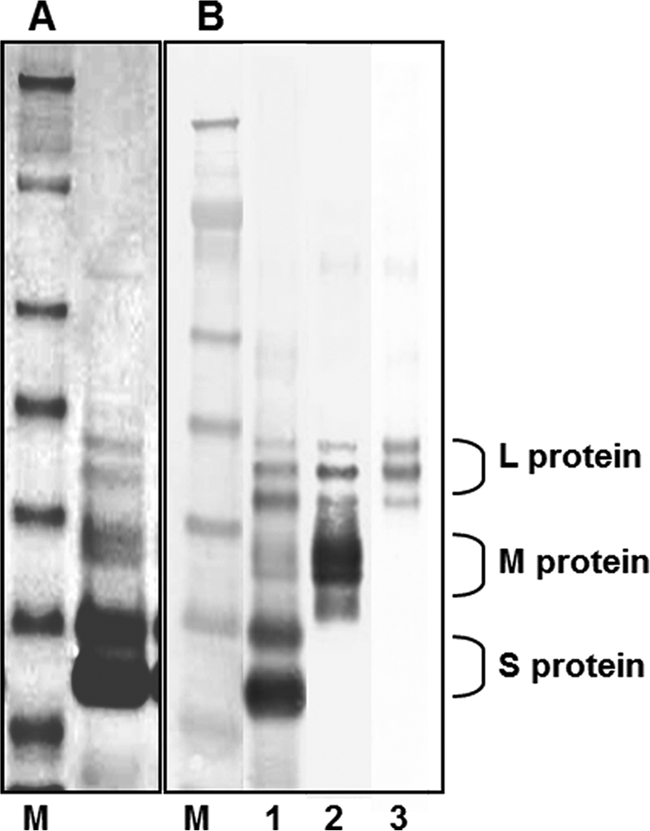
SDS-PAGE (A) and Western blot (B) analyses of purified recombinant L-HBsAg. M, protein size markers (198, 98, 62, 49, 38, 28, and 17 kDa from the top). (A) The six major bands represent the 24-kDa S protein, the glycosylated 27-kDa S protein, glycosylated M proteins of 33 and 36 kDa, the 39-kDa L protein, and the glycosylated 42-kDa L protein. (B) Anti-HBsAg (lane 1), anti-pre-S2 (lane 2), and anti-pre-S1 (lane 3) antibodies were used for Western blotting.
The average content of pre-S protein in each batch of the three different preparations was about 25%, and the distributions of L protein, M protein, and S protein were ∼8%, ∼17%, and ∼75%, respectively (data not presented). The electron micrograph of the purified surface antigen shows 20- and 40-nm-diameter particles and filamentous forms (Fig. 2). This is interesting because, according to previous findings (18, 20), the pre-S region should inhibit the secretion of HBV surface antigen when it is linked to the S protein.
Fig 2.

Electron micrograph of L-HBsAg. Purified recombinant L-HBsAg formed 20-nm- and 40-nm-diameter particles, as well as filamentous forms, as revealed by electron microscopy. A Tecnai G2 Spirit Bio transmission electron microscope (FEI Company) was used.
Glycosylation of L-HBsAg was confirmed by N-glycosidase F, O-glycosidase, and sialidase treatment, as performed previously by Schmitt et al. (23). N-glycosylation sites on the M protein were positively identified at amino acid positions 4 and 146 by introducing mutations (Asn→Gly) at one or both sites and analyzing the proteins from CHO cells transfected with mutated HBV surface antigen genes. Of note, the HBV surface antigens that were glycosylated at only one of the two possible glycosylation sites were unable to form regular particles. Further, the immunogenicity of the antigens was severely compromised, showing only about 10% of the level exhibited by the fully glycosylated antigen (data not shown).
Improvement of the L-HBsAg-containing vaccine by a new adjuvant.
The new adjuvant used in this study is called L-pampo. It is a coprecipitate consisting of a lipopeptide and a synthetic double-stranded RNA (dsRNA). L-pampo has a powerful immune-stimulatory effect when combined with an antigen.
As shown in Fig. 3, the overall adjuvant effect of L-pampo on HBV-specific antibody induction was 10-fold higher than that of alum, and the level of increase was consistent throughout the experimental period of 34 weeks. Another interesting fact is that boosting at the 16-week point, after three consecutive injections at 2-week intervals, dramatically increased antibody production to levels 2- to 5-fold higher than those obtained previously, while maintaining levels at least 10-fold-higher than those with the alum-formulated vaccine. The boosting effect appears to be more dramatic with a smaller amount of antigen (0.5 μg).
Fig 3.

Antibody responses induced by the novel adjuvant L-pampo. Anti-S antibody titers induced by alum-based and L-pampo-based L-HBsAg formulations were monitored for 34 weeks. Each vaccine formulation contained 0.5 μg or 5.0 μg L-HBsAg. Immunization was performed three times at 2-week intervals, and single boosting was performed at week 16. Arrows indicate intramuscular vaccine administrations.
L-pampo also dramatically changes the isotype of the antibody induced, as shown in Fig. 4. The alum-formulated vaccine induced mainly IgG1, whereas the L-pampo-formulated vaccine induced mainly IgG2b and some IgG2a and IgG1, in C57BL/6 mice, indicating that the immune response was skewed toward a Th1 response by L-pampo. This isotype switching to IgG2a or IgG2b seemed to be dependent on the mouse strain: L-pampo induced mainly IgG2b in a C57BL/6 mouse (Fig. 4) and predominantly IgG2a in a BALB/c mouse (Fig. 5B).
Fig 4.

Antibody isotype patterns. Patterns of induction of anti-S IgG isotypes were compared after three immunizations with alum-based or L-pampo-based L-HBsAg formulations. Each formulation contained 0.5 μg of L-HBsAg. L-HBsAg formulated with L-pampo induced predominantly anti-S IgG2b, whereas the alum-based L-HBsAg formulation induced predominantly anti-S IgG1. Six- to 8-week-old C57BL/6 mice were used.
Fig 5.

Immune responses and clearance of the antigen in HBsAg/HLA-A2 transgenic mice. Transgenic mice were injected with 5 μg of yeast-derived S-HBsAg or L-HBsAg formulated with alum or L-pampo. Immunizations were performed three times at 2-week intervals. Antibody titers, antibody isotypes, and antigen clearance were determined 2 weeks after the final injection. (A) Anti-S antibody titers (GMT); (B) anti-S antibody isotypes; (C) HBsAg titers in sera before and after immunization in each group; (D) HBsAg and anti-S antibody titers for individual transgenic mice before (open bars) and after (filled bars) immunization. Statistically significant decreases in HBsAg levels were observed in the groups immunized with L-HBsAg formulated with either alum (n = 7) (P < 0.01) or L-pampo (n = 7) (P < 0.01), whereas the conventional yeast-derived vaccine was not effective at clearing HBsAg from the blood (n = 7) (P > 0.01).
Induction of HBV-specific antibody in HBsAg/HLA-A2 double transgenic mice and clearance of HBV antigen from the blood.
Loirat et al. (9) established an HBsAg/HLA-A2 double transgenic mouse strain that produced HBV surface antigen in the blood as 22-nm-diameter particles; however, this Tg mouse produced no detectable levels of HBV antibody in the blood. In this regard, the HBsAg/HLA-A2 double transgenic mouse strain resembles a patient with chronic HBV infection, in whom HBV-specific immune cells become nonresponsive to the HBV antigen; that is, HBV-specific immune cells become immune tolerant or anergic to HBV antigens present in the patient's body. Although the state of HBV-specific immune cells in this Tg mouse is unknown, we wanted to find out whether our improved HBV vaccine could reactivate those non-HBV antigen-responsive immune cells and induce an immune response. Further, if an HBV-specific immune response could be induced, we wanted to see whether that HBV-specific antibody would clear HBV antigen from the blood. To our surprise, both of these tasks—inducing a large amount of HBV-specific antibody (Fig. 5A) and clearing HBV surface antigen from the blood of the mice (Fig. 5C and D)—could be accomplished quite efficiently.
Three injections of a conventional HBV vaccine containing the S protein produced from yeast into the Tg mice failed to induce detectable amounts of HBV surface antigen-specific antibody (Fig. 5A), whereas L-HBsAg formulated with alum or L-pampo induced significant amounts. The S protein-specific antibody titer, expressed as a reciprocal of the endpoint dilution (GMT), was 39,000 for L-HBsAg formulated with alum and 560,000 for L-HBsAg formulated with L-pampo (Fig. 5A). The distribution of IgG isotypes (Fig. 5B) clearly demonstrates that L-pampo-induced IgG is dominated by the IgG2a and IgG2b isotypes.
The pattern of clearance of antigen from the blood of immunized Tg mice is shown in Fig. 5C. The level of HBV antigen in serum ranged widely, from 100 ng/ml to 300 ng/ml, and tended to decrease as the age of the Tg mice increased. Loirat et al. (9) also noticed this tendency toward a spontaneous decrease in antigen levels with increasing age; however, in their study, antigen levels never fell below 50% of the starting level. Therefore, a <50% reduction in the antigen level, as seen in the mice that received the conventional vaccine, cannot be considered significant. The control group that received PBS without any antigen showed exactly the same pattern as the group that received the conventional yeast vaccine (data not shown). In the group of Tg mice that received L-HBsAg formulated with L-pampo, five mice out of seven cleared HBV antigen from the blood to below the detection limit, and one additional mouse cleared more than 80% of its circulating HBV antigen. The group of Tg mice receiving an alum-formulated L-HBsAg vaccine also efficiently cleared circulating HBV antigen from the blood: one mouse completely cleared HBV antigen, and five showed a >50% reduction in the level of HBV antigen in the blood. This clearance of the HBV antigen from the blood by vaccination with L-HBsAg either with alum (n = 7) (P < 0.01) or with L-pampo (n = 7) (P < 0.01) was statistically significantly greater than that for the group that received the conventional vaccine.
Induction of HBV-specific antibody in HBV whole-gene transgenic mouse.
HBV whole-gene transgenic mice (29, 31) displayed wide-ranging amounts of HBV antigen, from 50 to 650 ng/ml of blood. Of the HBV antigen particles, about 40% had HBV DNA. However, these mice, like the HBV.S2.S/HLA-A2 double transgenic mice, produced no antibody. In contrast to that for the double Tg mouse with the envelope antigen-coding DNA sequence only, the level of HBV antigen in serum in the HBV whole-gene transgenic mouse is robust throughout the aging process, maintaining less than 20% variation. Three injections of alum-formulated L-HBsAg vaccines induced HBV-specific antibody GMTs of <10,000, whereas L-HBsAg formulated with L-pampo induced very high levels of HBV-specific antibody (average GMT, 160,000) (Fig. 6A). Despite this high-titer antibody induction, the amount of HBV antigen in the blood changed little, if any, in the HBV whole-gene transgenic mice. Therefore, in the next experiment, Tg mice were injected five times with vaccines at 2-week intervals; the antibody titers increased to GMTs of 130,000 and 4.3 million for alum and L-pampo formulations, respectively (Fig. 6A). A significant amount of pre-S-specific antibody was also induced by L-pampo-formulated L-HBsAg (data not shown). Further, the HBV antigen level in the blood started to decrease (Fig. 6B and C). In this experiment, a decrease of >30% in the serum HBsAg level was considered significant, since the general variation was about 20%. As shown in Fig. 6C, more than 30% clearance of HBV antigen from the blood was obvious in four of the nine mice (mice 5, 6, 7, 9) in the group that received the vaccine formulated with L-HBsAg and alum. However, this antigen reduction was not statistically significant (n = 9) (P > 0.01). The clearing efficiency was higher in the group that received L-HBsAg and L-pampo: 8 out of 10 mice (mice 1, 2, 4, 5, 6, 8, 9, and 10) cleared more than 30% of the HBV antigen from the blood, and the remaining 2 had decreases of >20% in HBV circulating antigen levels. Unlike the alum-formulated vaccine, the L-pampo-formulated vaccine could result in statistically significant (n = 10) (P < 0.01) reductions of HBV antigen levels in the blood.
Fig 6.

Immune responses and antigen clearance in HBV whole-gene transgenic mice. Five micrograms of L-HBsAg formulated with alum or L-pampo was injected five times at 2-week intervals. (A and B) The titers of anti-S antibody (A) and HBsAg (B) in sera were determined. (C) HBsAg and anti-S antibody titers before (open bars) and after (filled bars) immunization are shown for individual mice. HBsAg titers were statistically significantly decreased only in the group vaccinated with L-HBsAg and L-pampo (n = 10) (P < 0.01). Although >30% decreases in serum HBsAg levels were detected in 4 out of 9 test animals (mice 5, 6, 7, and 9) immunized with alum-formulated L-HBsAg, the overall decrease for this group was not statistically significant (n = 9) (P > 0.01).
DISCUSSION
It has been well established that the HBV surface antigen has three components: the S protein (major protein), the M protein, and the L protein (7, 16, 23). Glycosylation of these components makes them separate on an SDS-PAGE gel as six distinct bands of nonglycosylated 24-kDa and glycosylated 27-kDa S proteins, 33-kDa and 36-kDa glycosylated M proteins, and nonglycosylated 39-kDa and glycosylated 42-kDa L proteins. As shown in Fig. 1, our HBV surface antigen produced from the recombinant CHO cell line containing the entire HBV envelope gene gave an SDS-PAGE profile remarkably similar to those found previously, where the HBV surface antigen purified from chronic HBV carriers showed six major protein bands (7). However, we observed one additional protein of 46 kDa, synthesized in a small but significant quantity and represented as a minor band on the SDS-PAGE gel (Fig. 1). A study to determine whether this is an artifact or is also produced by natural infection of liver cells with HBV is under way. Shouval et al. (26) also reported the production of pre-S1- and pre-S2-containing HBV surface antigen from a CHO cell line (Bio-Hep-B/Sci-B-Vac; SciGen, Hong Kong), but their preparation yielded 20-nm-diameter particles only. In contrast, our HBV antigen with the entire HBV surface antigen gene, purified from a CHO cell line, had 20-nm-diameter particles, 40-nm-diameter particles, and filamentous forms. Initially, it was suspected that this filamentous form might be from broken cells, since previous studies showed that pre-S inhibits the secretion of HBV surface antigen, and the filamentous form contains more pre-S than any other particles (7, 18). However, this appears not to be true, since more pre-S was obtained from attached cell cultures in flasks than from suspension cultures. The suspension cultures had a greater tendency to produce more broken cells, and three different particles were consistently obtained from all our preparations regardless of culture methods. A possible reason why Shouval et al. obtained only 20-nm-diameter particles could be the very small amount of L protein present in their preparation, since 20-nm-diameter particles should contain only about 1% pre-S, whereas 40-nm-diameter infectious virus particles and filamentous forms contain 20 times more pre-S (7).
The immunogenicity of L-HBsAg prepared with alum is very high compared to that of the S protein from the CHO cell line or from yeast. L-HBsAg prepared with alum induced >10,000 times more HBV S protein-specific antibody than the S protein vaccine from yeast and 30 times more than the S protein from the CHO cell line. According to Milich et al. (15), S protein was not immunogenic in B10.S and B10.M mice, but L-HBsAg containing the L protein, M protein, and S protein induced antibodies not only against pre-S1 and pre-S2 but also against S protein. These data suggest that the pre-S portion somehow rendered the S protein immunogenic in those congenic mice. Therefore, we believe that the exceptionally high immunogenicity of our L-HBsAg from CHO cells may be attributable to the presence of pre-S, which possibly helps form all three particle types. Appropriate glycosylation of the antigen appears to play some role, too, since the S protein from the CHO cell line was much more immunogenic than the same S protein produced in yeast (data not shown). Our further studies also support these notions. Abolishing the glycosylation site by substitution of amino acids (Asn→Gly) located on one or both glycosylation sites of the M protein (aa 4 and 145) via mutagenesis had a profound effect both on particle formation by the HBV surface antigen and on its immunogenicity. Mutagenesis reduced immunogenicity 10-fold, and the antigen formed an amorphous slush instead of the organized particles seen in glycosylated forms of the HBV surface antigen (data not shown).
Our L-HBsAg vaccine has been improved further by the use of a new adjuvant named L-pampo. Based on the amount of HBV-specific antibody induced, the L-pampo-based vaccine was about 10-fold improved over the alum-formulated vaccine (Fig. 3). Further, the isotype of the antibody was dramatically changed: the L-pampo-formulated vaccine induced mainly IgG2a and IgG2b, while the alum-formulated vaccine induced predominantly IgG1 (Fig. 4 and 5B). L-pampo contains a lipopeptide and a dsRNA, which are toll-like receptor 2 (TLR2) and TLR3 ligands, respectively. TLRs are a family of pathogen recognition receptors (PRR) that recognize pathogen-associated molecular patterns (PAMP) and generate signals for the activation of immune response genes upon appropriate ligand engagement, inducing cytokines and costimulatory molecules. In general, a group of TLRs, such as TLR2, that mediate signals through myeloid differentiation primary-response gene 88 (MyD88) and other groups, such as TLR3, that generate MyD88-independent signals should be synergistic in inducing cytokines (28). However, further studies on the mechanisms of L-pampo functions in stimulating adaptive immune responses and switching IgG isotypes are required.
The efficacies of the different isotypes in viral and cancer immunity seem to differ on a case-by-case basis, favoring IgG2a and IgG2b over IgG1 in mice for certain infections (12, 13, 17, 27) but not others (1, 2, 4, 19). In our study, the clearance of HBV antigen from the HBV Tg mice appeared to depend more on the total amount of HBV-specific antibody induced than on the IgG isotypes, as previously observed by Bachmann et al. (1) for viral infections. Thus, whether the vaccine was formulated with alum or with L-pampo, when the antibody titer was above a certain level, higher than a GMT of 40,000 with the HBsAg/HLA-A2 Tg mouse, HBV antigen was cleared from the blood (Fig. 5A and D). It is not known whether this is due to the fact that expression of the HBsAg gene from the transgene is different from actual virus infection.
The two different HBV Tg mice used in this study are not perfect models of the chronic HBV patient. Nevertheless, they are useful in the development of an immune therapy enabling determination of whether any new vaccine is able to overcome immune tolerance or not. Reactivation of silenced or anergic immune cells and induction of strong immune responses against HBV are the ultimate goals of immune therapy. Furthermore, the immune response should lead to the clearance of infectious virus from the patient's system and should control virus growth in the liver. Information regarding the size of the total HBV-specific T cell and B cell clonal repertoire, which epitope-specific T and/or B cells become anergic in patients with chronic HBV infections, and which epitope-specific immune cells can be reactivated is not available yet. It is likely that some of the low-affinity antigen binding receptor-bearing T and/or B cell clones that have survived the continuous presence of HBV antigen in the patient's system, and some of the T and/or B cell clones that have become anergic but have not been deleted from the repertoire, might be revived by a strong vaccine formulated with a good antigen and adjuvant. Therefore, we think that a good antigen should encompass a whole set of HBV surface antigen-specific epitopes in a highly immunogenic form and that a strong adjuvant should be able to reactivate some HBV surface antigen-specific immune cells, probably by upregulating the expression of costimulatory molecules as well as proper cytokines.
It was also noted that the whole-HBV Tg mice required a higher titer of antibody for the clearance of serum HBV antigen than did the partial HBV surface antigen gene Tg mice (Fig. 5 and 6). With the HBsAg/HLA-A2 gene Tg mice, a GMT of <50,000 was sufficient to clear the antigen from the blood, but the antigen was never completely cleared from the sera of whole-HBV-gene Tg mice, even with 100 times more antibody induced. This lack of complete clearance could be due to differences in the molecular structure of the HBV surface antigen in these two different Tg mice (6), since the HBsAg/HLA-A2 Tg mouse produces only 20-nm-diameter particles in the blood, whereas the whole-HBV-gene Tg mouse produces both 20- and 40-nm-diameter particles, as well as filamentous forms. The status of the cleared antigen–whether it is completely destroyed by phagocytes or whether it remains in the blood as an immune complex undetectable by our assay method—is also not known at this point.
In the experiment with whole-HBV-gene Tg mice (5), the alanine aminotransferase (ALT) level in the blood was assessed after vaccination, and no significant difference from that in normal unvaccinated mice was found, suggesting that the vaccination inflicted little, if any, liver damage. Therefore, the clearance of the HBV antigen from the blood may not be entirely due to T-cell-mediated and/or antibody-dependent cellular cytotoxicity (ADCC)-mediated lysis of liver cells harboring the HBV gene. According to Guidotti et al. (5), HBV-specific effector cells (CD8 T cells) control virus replication predominantly through cytokine production instead of infected-cell lysis. While further studies are required, the clearance of HBV antigen after vaccination might be due to a combined effect of induced antibody (2, 21) and cell-mediated immunity.
ACKNOWLEDGMENTS
This work was supported by the Next Generation New Technology Development Program of the Korean Ministry of Knowledge, Economy (grant 10024062), and the Health and Medical Research and Development Program of the Korean Ministry of Health and Welfare (grant A111058). The experiment carried out at the Institute for Antiviral Research at Utah State University was supported by NIAID, U.S. NIH (HNS272201000039I and HNS27200001).
We thank Bo Cheng for kindly arranging for the use of Chinese Tg mice and for help in handling samples. We also thank Diana Berard at the U.S. NIH for reading the manuscript and giving us editorial help.
Footnotes
Published ahead of print 7 December 2011
REFERENCES
- 1. Bachmann MF, et al. 1997. The role of antibody concentration and avidity in antiviral protection. Science 276:2024–2027 [DOI] [PubMed] [Google Scholar]
- 2. Burton DR. 2002. Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2:706–713 [DOI] [PubMed] [Google Scholar]
- 3. Chisari FV, Ferrari C. 1995. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 13:29–60 [DOI] [PubMed] [Google Scholar]
- 4. Fleming JO, Shubin RA, Sussman MA, Casteel N, Stohlman SA. 1989. Monoclonal antibodies to the matrix (E1) glycoprotein of mouse hepatitis virus protect mice from encephalitis. Virology 168:162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Guidotti LG, et al. 1996. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity 4:25–36 [DOI] [PubMed] [Google Scholar]
- 6. Hangartner L, Zinkernagel RM, Hengartner H. 2006. Antiviral antibody responses: the two extremes of a wide spectrum. Nat. Rev. Immunol. 6:231–243 [DOI] [PubMed] [Google Scholar]
- 7. Heermann KH, et al. 1984. Large surface proteins of hepatitis B virus containing the pre-S sequence. J. Virol. 52:396–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones CD, et al. 1999. T-cell and antibody response characterisation of a new recombinant pre-S1, pre-S2 and SHBs antigen-containing hepatitis B vaccine; demonstration of superior anti-SHBs antibody induction in responder mice. Vaccine 17:2528–2537 [DOI] [PubMed] [Google Scholar]
- 9. Loirat D, Mancini-Bourgine M, Abastado J-P, Michel M-L. 2003. HBsAg/HLA-A2 transgenic mice: a model for T cell tolerance to hepatitis B surface antigen in chronic hepatitis B virus infection. Int. Immunol. 15:1125–1136 [DOI] [PubMed] [Google Scholar]
- 10. Lok ASF. 2002. Chronic hepatitis B. N. Engl. J. Med. 346:1682–1683 [DOI] [PubMed] [Google Scholar]
- 11. Mancini M, et al. 1996. DNA-mediated immunization in a transgenic mouse model of the hepatitis B surface antigen chronic carrier state. Proc. Natl. Acad. Sci. U. S. A. 93:12496–12501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Markine-Goriaynoff D, Coutelier J-P. 2002. Increased efficacy of the immunoglobulin G2a subclass in antibody-mediated protection against lactate dehydrogenase-elevating virus-induced polioencephalomyelitis revealed with switch mutants. J. Virol. 76:432–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKendall RR, Woo W. 1988. Murine IgG subclass response to herpes simplex virus type 1 and polypeptides. J. Gen. Virol. 69:847–857 [DOI] [PubMed] [Google Scholar]
- 14. Milich DR, et al. 1985. Enhanced immunogenicity of the pre-S region of hepatitis B surface antigen. Science 228:1195–1199 [DOI] [PubMed] [Google Scholar]
- 15. Milich DR, McLachlan A, McNamara MK, Chisari FV, Thornton GB. 1986. New approaches to immunization, p 377–382 In Brown F, Chanock RM, Lerner RA. (ed), Vaccine 86. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 16. Neurath AR, Kent SB, Strick N, Taylor P, Stevens CE. 1985. Hepatitis B virus contains pre-S gene-encoded domains. Nature 315:154–156 [DOI] [PubMed] [Google Scholar]
- 17. Nimmerjahn F, Ravetch RV. 2005. Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science 310:1510–1512 [DOI] [PubMed] [Google Scholar]
- 18. Ou J-H, Rutter WJ. 1987. Regulation of secretion of the hepatitis B virus surface antigen by the preS-1 protein. J. Virol. 61:782–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Palladino G, Mozdzanowska K, Washko G, Gerhard W. 1995. Virus-neutralizing antibodies of immunoglobulin G (IgG) but not of IgM or IgA isotypes can cure influenza virus pneumonia in SCID mice. J. Virol. 69:2075–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Persing DH, Varmus HE, Ganem D. 1986. Inhibition of secretion of hepatitis B surface antigen by a related presurface polypeptide. Science 234:1388–1391 [DOI] [PubMed] [Google Scholar]
- 21. Ravetch JV, Bolland S. 2001. IgG Fc receptors. Annu. Rev. Immunol. 19:275–290 [DOI] [PubMed] [Google Scholar]
- 22. Rendi-Wagner P, et al. 2006. Comparative immunogenicity of a preS/S hepatitis B vaccine in non- and low responders to conventional vaccine. Vaccine 24:2781–2789 [DOI] [PubMed] [Google Scholar]
- 23. Schmitt S, et al. 1999. Analysis of the pre-S2 N- and O-linked glycans of the M surface protein from human hepatitis B virus. J. Biol. Chem. 274:11945–11957 [DOI] [PubMed] [Google Scholar]
- 24. Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shapira MY, Zeira E, Adler R, Shouval D. 2001. Rapid seroprotection against hepatitis B following the first dose of a pre-S1/pre-S2/S vaccine. J. Hepatol. 34:123–127 [DOI] [PubMed] [Google Scholar]
- 26. Shouval D, et al. 1994. Improved immunogenicity in mice of a mammalian cell-derived recombinant hepatitis B vaccine containing pre-S1 and pre-S2 antigens as compared with conventional yeast-derived vaccines. Vaccine 12:1453–1459 [DOI] [PubMed] [Google Scholar]
- 27. Smucny JJ, Kelly EP, Macarthy PO, King AD. 1995. Murine immunoglobulin G subclass responses following immunization with live dengue virus or a recombinant dengue envelope protein. Am. J. Trop. Med. Hyg. 53:432–437 [DOI] [PubMed] [Google Scholar]
- 28. Trinchieri G, Sher A. 2007. Cooperation of toll-like receptor signals in innate immune defence. Nat. Rev. Immunol. 7:179–190 [DOI] [PubMed] [Google Scholar]
- 29. Xiong Y, et al. 2001. Hepatitis B virus transgenic mice for the model of anti-hepatitis B virus drug study. Zhonghua Gan Zang Bing Za Zhi (Chin. J. Hepatol.) 9:19–21 [PubMed] [Google Scholar]
- 30. Young MD, et al. 2001. Adult hepatitis B vaccination using a novel triple antigen recombinant vaccine. Hepatology 34:372–376 [DOI] [PubMed] [Google Scholar]
- 31. Zheng S-J, et al. 2003. Distribution and anti-HBV effects of antisense oligodeoxy nucleotides conjugated to galactosylated poly-l-lysine. World J. Gastroenterol. 9:1251–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]