

Abstract
Unlike apoptosis, necrotic cell death is characterized by marked loss of plasma membrane integrity. Leakage of cytoplasmic material to the extracellular space contributes to cell demise, and is the cause of acute inflammatory responses, which typically accompany necrosis. The mechanisms underlying plasma membrane damage during necrotic cell death are not well understood. We report that endocytosis is critically required for the execution of necrosis. Depletion of the key endocytic machinery components dynamin, synaptotagmin and endophilin suppresses necrotic neurodegeneration induced by diverse genetic and environmental insults in C. elegans. We used genetically encoded fluorescent markers to monitor the formation and fate of specific types of endosomes during cell death in vivo. Strikingly, we find that the number of early and recycling endosomes increases sharply and transiently upon initiation of necrosis. Endosomes subsequently coalesce around the nucleus and disintegrate during the final stage of necrosis. Interfering with kinesin-mediated endosome trafficking impedes cell death. Endocytosis synergizes with autophagy and lysosomal proteolytic mechanisms to facilitate necrotic neurodegeneration. These findings demonstrate a prominent role for endocytosis in cellular destruction during neurodegeneration, which is likely conserved in metazoans.
Keywords: autophagy, C. elegans , intracellular trafficking, necrosis, neurodegeneration
Introduction
Necrotic cell death contributes extensively to the pathogenesis of human neurodegenerative diseases in stroke, trauma and other pathological conditions. In sharp contrast to apoptosis, one of the defining features of necrosis is the loss of plasma membrane integrity and the consequent leakage of cellular contents, which triggers inflammatory responses. The molecular mechanisms that transpire during necrotic cell death to cause catastrophic perforation of the plasma membrane and cellular destruction are poorly understood. In the nematode Caenorhabditis elegans, neurons expressing hyperactive ion channels undergo necrotic cell death with morphological and ultrastructural characteristics reminiscent of neuronal excitotoxicity following ischaemia in mammals. Electron microscopy studies of dying neurons have shown that the first detectable abnormality is an infolding of the plasma membrane and the production of small electron-dense whorls (Hall et al, 1997). This feature suggests that initiation of neurodegeneration may involve abnormal endocytosis at the plasma membrane.
In eukaryotic cells, endocytosis serves important functions, including the uptake of extracellular molecules and ligands, the internalization of plasma membrane proteins and lipids, the regulation of cell signalling pathways, the recycling of proteins to the Golgi and plasma membrane, and the degradation of proteins from the secretory and endocytic pathways. In neurons, clathrin-mediated endocytosis also mediates the retrieval of synaptic vesicle components after release of neurotransmitters in response to action potentials (Cremona and De Camilli, 1997; Harris et al, 2001). The process involves five main steps, initiating with the formation of a putative nucleation module that defines the plasma membrane site for clathrin recruitment and vesicle budding (the nucleation step). Subsequently, components of the nucleation module recruit cargo-specific adaptor proteins, which mediate selection of the cargo to be endocytosed (cargo selection step). After cargo is selected, the clathrin coat is assembled by clathrin triskelia recruited to adaptor localization sites (clathrin coat assembly step). The vesicle is then separated from the membrane (vesicle scission step) and the clathrin coat is disassembled into clathrin triskelia (uncoating and clathrin component recycling step). The clathrin lattice is removed and the vesicle fuses with an early endosome. The released clathrin machinery is then reused for another round of clathrin-coated pit formation (reviewed by Takei and Haucke, 2001; McMahon and Boucrot, 2011).
Defects in endocytosis and trafficking of the endocytic vesicles are apparent and have been implicated in many human neurodegenerative conditions. For example, abnormally enlarged endosomes and aggregates containing vesicles, organelles and kinesin have been found in neurons from Alzheimer's disease patients (Cataldo et al, 1997; Stokin et al, 2005). In addition, mutations in a subunit (p150) of the transporter protein dynactin have been found in a family with autosomal dominant lower motor neuron disease (Forman et al, 2004) and in patients suffering from Amyotrophic Lateral Sclerosis (ALS; Munch et al, 2004). Endocytic processes defects are also observed in animal models of neurodegenerative diseases, such as the WOBBLER mouse (a mouse model for ALS; Schmitt-John et al, 2005), the Motor Neuron Degeneration (MND) mouse (a model for neuronal ceroid lipofuscinosis; reviewed by Syntichaki and Tavernarakis, 2003) and mouse models of motor neuron disease expressing mutant forms of SOD1 (Chevalier-Larsen and Holzbaur, 2006; De Vos et al, 2008). Moreover, recent studies have revealed that in the brains of mice with Alzheimer's disease, the levels of the endocytotic protein endophilin I are increased (Ren et al, 2008). However, the molecular mechanisms that engage endocytosis and trafficking during neurodegeneration are still unknown.
We utilized the well-characterized C. elegans model of necrotic cell death to investigate the involvement of clathrin-mediated endocytosis and intracellular trafficking in necrosis. We systematically examined the contribution of clathrin-coated pit formation and maturation, fission and uncoating of endocytic vesicles, and kinesin-mediated movement of endosomes and other cargoes in necrosis. In addition, we examined the interaction of endocytosis and intracellular trafficking, with lysosomal proteolytic degradation and autophagy, two processes implicated in the execution of necrotic cell death.
Results
Inhibition of endocytosis suppresses necrosis induced by diverse genetic and environmental insults in C. elegans
The snt-1 gene encodes the C. elegans orthologue of synaptotagmin (SNT-1), a protein that plays a role both in exocytosis of neurotransmitters (Fernandez-Chacon et al, 2001; Koh and Bellen, 2003) and in the retrieval of synaptic vesicle proteins through clathrin-mediated endocytosis (Poskanzer et al, 2003). It comprises an N-terminal transmembrane segment followed by the cytoplasmic domain, incorporating two Ca2+- and phospholipid-binding C2 domains. SNT-1 interacts with the adaptor protein 2 (AP2) complex, a protein complex that initiates recruitment of the endocytic machinery via the second C2 domain (C2B; Jarousse and Kelly, 2001). We find that SNT-1 deficiency strongly suppresses necrosis induced by diverse genetically encoded insults in the nematode. Deletion of snt-1 ameliorates necrosis induced by the hyperactive ion channel subunit MEC-4(d) in touch receptor neurons (Figure 1A). Time-course analysis of cell death revealed that suppression is not a consequence of delaying neurodegeneration to later developmental stages in SNT-1-deficient mutants (Supplementary Figure S1). We considered whether suppression of neurodegeneration by lack of SNT-1 is an indirect consequence of eliminating the toxic MEC-4(d) ion channel. We assayed the relative expression, stability and localization of full-length MEC-4 fused to GFP. Touch receptor neurons of snt-1 mutants express the fusion protein at typical, wild-type levels (Figure 1B). Moreover, snt-1 mutants respond normally to light mechanical stimuli, indicating that the touch receptor neurons are functional and thus the expression and transfer of the MEC-4 at their membrane occurs normally (Supplementary Figure S2). Therefore, SNT-1 does not interfere with the biogenesis or proper localization of the necrosis-triggering, hyperactive MEC-4(d) ion channel subunit.
Figure 1.

Clathrin-mediated endocytosis is required for necrotic cell death in C. elegans. (A) Number of cell corpses at the L1 stage of development per 100 animals carrying the neurotoxic mec-4(d) or deg-3(d) alleles in SNT-1-deficient mutants. For mec-4(d) mutant animals, bars denote touch receptor neuron corpses. For deg-3(d) mutants, bars denote inner labial neuron 1 (L1) sensory neuron and PVC interneuron corpses. (B) Expression of a full-length MEC-4::GFP reporter fusion under the control of the mec-4 promoter in touch receptor neurons of wild-type, snt-1 and unc-57 mutant animals. Representative photos of touch receptor neuron cell bodies are shown. Bar denotes 6 μm. The quantification (%) of GFP signal intensity from the animals examined is graphed below (n=100 neurons per assay; P<0.05 compared with wild type; t-test). (C) Percentage of SNT-1-deficient animals that survive near-lethal treatment with sodium azide. This chemical inhibits the activity of the respiratory electron transport complex IV (cytochrome C oxidase) and simulates hypoxia. (D) Number of touch receptor neuron corpses, at the L1 stage of development, per 100 animals carrying the neurotoxic mec-4(d) or deg-3(d) alleles in UNC-57-deficient animals. (E) Percentage of UNC-57-deficient animals that survive after the hypoxia-inducing treatment with sodium azide. (F) Number of touch receptor neuron corpses, at the L1 stage of development, per 100 animals carrying the neurotoxic mec-4(d) or deg-3(d) alleles together with lesions in genes encoding key proteins that participate in all four steps of clathrin-mediated endocytosis. UNC-11 and DPY-23 are adaptor proteins required for the formation of clathrin-coated pits, DYN-1 is a GTPase necessary for fission of clathrin-coated vesicles and UNC-26 participates in the uncoating of vesicles. Error bars denote s.e.m. values (n>250 for all populations examined; P<0.001, compared with wild-type animals, unpaired t-test).
We next determined whether removing synaptotagmin is generally protective against necrotic cell death. To this end, we induced necrosis by using other genetic and environmental insults. Gain-of-function mutations in the α-7 nicotinic acetylcholine receptor Ca2+ channel subunit gene deg-3 induce degeneration of specific sets of neurons expressing the toxic gene (Treinin and Chalfie, 1995). We find that SNT-1 deficiency ameliorates deg-3(d)-induced neurodegeneration (Figure 1A). Hypoxia, a condition of low oxygen availability that transpires in ischaemic episodes and stroke, also induces necrotic cell death in the nematode (Scott et al, 2002). SNT-1 deficiency protects animals from hypoxia-induced death triggered either by the chemical sodium azide (Figure 1C) or by low oxygen concentration (Supplementary Figure S3A). Moreover, the dopaminergic neurons of snt-1 mutants are more resistant to cell death induced by the neurotoxin 6-hydroxydopamine (6-OHDA; Nass et al, 2002), compared with wild-type control animals (Supplementary Figure S3B). In summary, our observations reveal a broad requirement for a functional SNT-1 in necrosis.
In addition to synaptotagmin, endophilin plays a crucial role in clathrin-mediated endocytosis. Endophilin is a cytoplasmic protein containing an N-terminal BAR domain and a C-terminal SH3 domain involved in membrane dynamics. The N-terminal BAR domain is important for the generation of the membrane curvature (Gallop et al, 2006; Masuda et al, 2006). The SH3 domain mediates interaction with dynamin, a GTPase important for neck constriction and scission of nascent clathrin-coated vesicles. It is also involved in the recruitment of synaptojanin, a phosphatase that contributes to the uncoating of the vesicles at the final stage of the endocytic process (Ringstad et al, 1999; Schuske et al, 2003). We find that loss of endophilin (UNC-57) suppresses mec-4(d)-induced degeneration (Figure 1D), without delaying cell death (Supplementary Figure S1). Moreover, endophilin depletion does not interfere with the expression or the localization of the toxic channel at the membrane; unc-57 mutants are sensitive to light mechanical stimuli (Supplementary Figure S2) and express a full-length MEC-4::GFP protein at typical wild-type levels (Figure 1B). Furthermore, UNC-57 deficiency suppresses deg-3(d)-induced degeneration (Figure 1D), protects from hypoxia (Figure 1E; Supplementary Figure S3A) and increases resistance of dopaminergic neurons to the neurotoxin 6-OHDA (Supplementary Figure S3B), indicating that endophilin is generally required for necrotic cell death in the nematode.
Clathrin-mediated endocytosis is a process that involves many steps, orchestrated by the combined action of several proteins (Takei and Haucke, 2001). We investigated whether necrosis requires the activity of additional proteins that mediate these steps, including formation of clathrin-coated pits, fission of clathrin-coated vesicles from the plasma membrane and vesicle uncoating. The adaptor protein 2 (AP2) is a protein complex consisting of four subunits linking the clathrin shell to the plasma membrane through interactions with membrane proteins and lipids (Takei and Haucke, 2001; Mousavi et al, 2004). AP2 is essential for the nucleation and assembly of clathrin-coated vesicles, and interacts with numerous other proteins that participate in the process, such as synaptotagmin (Jarousse and Kelly, 2001). We find that dysfunction of the AP2 protein complex (by inactivation of the μ2 subunit-dpy-23) strongly suppresses mec-4(d)-induced degeneration (Figure 1F). Clathrin adaptor protein AP180 is additional monomeric adaptor protein that binds clathrin, phosphoinositides and interacts with AP-2 complex during the clathrin-coat nucleation and assembly (Morgan et al, 1999, 2000; Nonet et al, 1999; Takei and Haucke, 2001). Elimination of AP180, encoded by unc-11, protects neurons from mec-4(d)-induced necrosis (Figure 1F). In addition, UNC-11 depletion promotes animal survival under hypoxia (Supplementary Figure S3A) and increases resistance of dopaminergic neurons to the neurotoxin 6-OHDA (Supplementary Figure S3B).
Dynamin (DYN-1) is a GTPase that polymerizes into oligomeric rings at the neck of invaginating buds and cooperates with endophilin to catalyse scission of vesicles from the plasma membrane (Ringstad et al, 1999; Takei and Haucke, 2001). Inactivation of dynamin ameliorates necrotic cell death (Figure 1E). Similarly, depletion of synaptojanin (UNC-26), a phosphatidylinositol phosphatase that regulates PIP2 metabolism and the stability of clathrin-AP-2 coats (Verstreken et al, 2003), protects from necrosis (Figure 1E). Our observations, in their totality, demonstrate that disruption of clathrin-mediated endocytosis interferes with the execution of necrotic cell death.
Endosome formation increases transiently early during necrosis
Our genetic analysis reveals a contribution of clathrin-mediated endocytosis in necrotic cell death. To gain insight into the role of endocytic processes in necrosis, we first monitored clathrin-mediated endocytosis during neurodegeneration in vivo. To visualize clathrin-coated pits and clathrin-coated vesicles, we fused GFP to the C-terminus of APS-2, the σ subunit of the AP2 adaptor complex that recruits clathrin at the plasma membrane (Ehrlich et al, 2004). The fluorescent marker was expressed specifically in the six touch receptor neurons of wild-type and mec-4(d) mutant animals. We observed similar numbers of fluorescent puncta in neurons of wild-type animals and mec-4(d) mutants during the early stage of degeneration, when cells still maintain their shape (1–3 h following expression of the necrotic stimulus; Figure 2A). By contrast, the number of fluorescent puncta declines considerably during late necrosis, when cells have lost their normal morphology and have swollen into a vacuolated form (5–7 h post initiation of cell death). The number of fluorescent puncta is also significantly reduced in neurons of unc-57(e1190) endocytic mutants, indicating that they correspond to clathrin-coated pits and vesicles (Figure 2A). Thus, clathrin-mediated endocytosis is not obstructed early upon induction of neurodegeneration.
Figure 2.
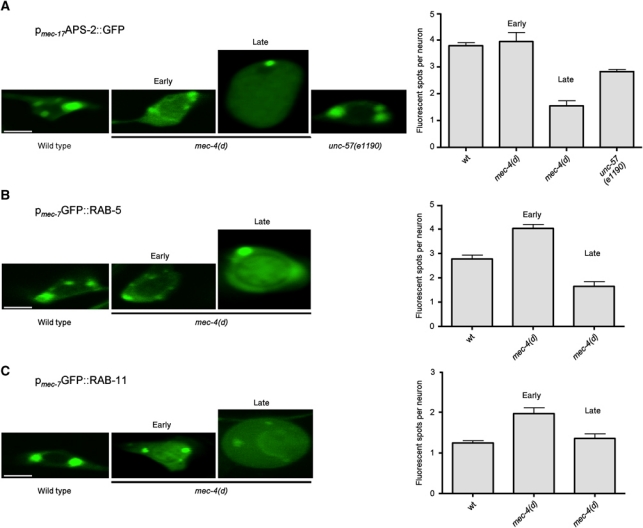
Endosome formation is induced early during necrosis. (A) Confocal images of touch receptor neurons expressing a pmec-17APS-2::GFP reporter transgene. During early necrosis, the number of APS-2::GFP puncta, corresponding to clathrin-coated pits and vesicles, in touch receptor neurons of mec-4(d) animals does not change significantly, whereas at later stages it decreases. A similar decrease is also observed in unc-57(e1190) mutants, defective for clathrin-mediated endocytosis. (B) Confocal images of touch receptor neurons expressing a pmec-7GFP::RAB-5 reporter transgene. The number of fluorescent puncta that correspond to early endosomes in touch receptor neurons of mec-4(d) animals significantly increases during early necrosis, whereas it declines later. (C) Confocal images of touch receptor neurons expressing a pmec-7GFP::RAB-11 reporter transgene. The number of GFP::RAB-11 puncta that correspond to recycling endosomes in touch receptor neurons of mec-4(d) animals increases early during cell death, while it declines as degeneration proceeds. Both early and recycling endosomes tend to accumulate around a swollen structure that is probably the nucleus. Bars denote 4 μm. Error bars denote s.e.m. values (n>250 for all populations examined; P<0.001, compared with control animals, unpaired t-test).
To detect early endosomes, we expressed a GFP::RAB-5 reporter fusion in touch receptor neurons of wild-type and mec-4(d) mutant animals. RAB-5 is a protein that localizes to the membrane of early endosomes (Zerial and McBride, 2001; Rink et al, 2005). We observed fluorescent puncta corresponding to early endosomes, close to the plasma membrane of wild-type neurons. In neurons of mec-4(d) mutants undergoing degeneration, the number of fluorescent puncta increased during early stages of death. Late during necrosis, the number of endosomes decreases below that observed in wild-type neurons (Figure 2B). We also monitored endocytic recycling during necrosis by fusing GFP to the N-terminus of RAB-11, which is localized specifically to the membrane of recycling endosomes (Sato et al, 2008). We found that similarly to early endosomes, the number of fluorescent spots corresponding to recycling endosomes increases transiently early in neurodegeneration and drops during late cell death (Figure 2C). In both cases, vesicles appear to coalesce around a distended nucleus before cell destruction and corpse removal (7–9 h after necrosis has been triggered). Taken together, these findings indicate that initiation of necrotic cell death is accompanied by enhanced formation of early and recycling endosomes.
Endocytosis and lysosomal proteolysis contribute in the same pathway to mediate necrotic cell death
Early endosomes are the major sorting stations in the endocytic pathway. Depending on their cargo, received at the cell surface, they may follow two different routes. One involves their maturation to late endosomes, while the other leads to the formation of recycling endosomes (Cavalli et al, 2001). In the first case, late endosomes are ultimately delivered to lysosomes, where their contents are degraded. We examined the interaction of clathrin-mediated endocytosis with lysosomal proteolytic mechanisms that contribute to cellular destruction during necrosis (Syntichaki et al, 2002). cad-1(j1) mutants exhibit 90% reduced activity of the lysosomal aspartyl proteases (Jacobson et al, 1988). Necrotic cell death is suppressed in these protease-deficient mutants (Syntichaki et al, 2002). In addition to aspartyl protease enzymes, acidification of lysosomes by the V-ATPase is required for necrosis (Syntichaki et al, 2005; Artal-Sanz et al, 2006). To moderate the activity of this pump, we targeted genes encoding key V-ATPase subunits by RNAi. Knockdown of vha-2, which encodes the essential c subunit of the membrane-integral, V0 domain of V-ATPase suppresses necrotic cell death (Syntichaki et al, 2005). We asked whether endocytic proteins are required in the lysosomal degradation pathway to mediate necrosis. Elimination of SNT-1 or endophilin (UNC-57) does not further enhance suppression of necrotic cell death by aspartyl protease depletion or perturbation of lysosomal acidification (Figure 3A–C).
Figure 3.

Endocytosis functions together with lysosomal proteolysis to facilitate necrotic cell death. (A) Depletion of synaptotagmin (SNT-1) or endophilin (UNC-57) does not further suppress MEC-4(d)-induced necrosis in cad-1 mutants with reduced cathepsin activity or with V-ATPase dysfunction (B, C). Similarly, no synthetic reduction of necrotic cell death is observed in calpain-deficient animals that also carry lesions in the synaptotagmin (D) or endophilin (E) genes. Error bars denote s.e.m. values (n>250 for all populations examined; P>0.5, compared with single mutant control animals, unpaired t-test).
Necrosis is characterized by rupture of lysosomes and release of their acidic contents and catabolic cathepsin proteases into the cytoplasm (Artal-Sanz et al, 2006). Lysosomal rupture is mediated by specific, Ca2+-dependent caplain proteases, which function in the same pathway together with lysosomal aspartyl proteases to mediate cell destruction during necrosis (Syntichaki and Tavernarakis, 2003; Yamashima, 2004). Loss of caplain (CLP-1) activity significantly suppresses necrotic cell death (Syntichaki et al, 2002). We find that the combined inactivation of CLP-1 and key endocytic proteins (synaptotagmin and endophilin) required for necrosis does not result in synthetic enhancement of neurodegeneration (Figure 3D and E). Our observations indicate that both endocytosis and lysosomal proteolytic mechanisms act in the same pathway to facilitate necrotic cell death.
Clathrin-mediated endocytosis synergizes with autophagy to facilitate necrosis
Autophagy is a cellular catabolic process contributing to protein and organelle recycling under extracellular or intracellular stress (Kourtis and Tavernarakis, 2009). Previous studies have revealed that autophagy is required for the execution of necrotic cell death and becomes hyperactivated early during necrosis in C. elegans (Toth et al, 2007; Samara and Tavernarakis, 2008). We examined the interaction of autophagy and clathrin-mediated endocytosis during necrosis. We assayed neurodegeneration induced by the toxic MEC-4(d) degenerin ion channel subunit in animals, deficient for both autophagy and endocytosis. To interfere with autophagy, we targeted lgg-1 by RNAi. lgg-1 encodes the orthologue of Atg8/LC3, a ubiquitin-like, microtubule-associated protein that facilitates autophagic vesicle growth and is essential for autophagy (Xie and Klionsky, 2007). We observed significant synergistic protection against neurodegeneration, in autophagy-deficient animals that also carry lesions in synaptotagmin or endophilin genes (Figure 4A and B). Furthermore, we knocked down two key autophagy regulators: bec-1, which encodes the C. elegans orthologue of the yeast autophagy gene Atg6/Vps30 (Melendez et al, 2003), and unc-51, which encodes the Atg1 serine threonine kinase (Ogura et al, 1994). We observed a similar synergistic protection against necrosis (Supplementary Figure S4A). These findings suggest that autophagy and endocytosis function in parallel to contribute to necrotic cell death.
Figure 4.

Endocytosis synergizes with autophagy to mediate necrotic cell death. (A) Synaptotagmin (SNT-1) deficiency further suppresses MEC-4(d)-induced necrosis in animals with impaired autophagy. (B) Endophilin dysfunction enhances suppression of necrosis in mec-4(d);lgg-1(RNAi) animals, where autophagy is impaired. Error bars denote s.e.m. values (n>250 for all populations examined; P<0.01, compared with single mutant control animals, unpaired t-test).
Kinesin-mediated intracellular vesicle trafficking contributes together with the endocytic pathway to promote necrotic cell death in C. elegans
We have noted that late endosomes and lysosomes accumulate around the misshapen nucleus during the course of neurodegeneration, indicating intracellular trafficking defects (Figure 2; Artal-Sanz et al, 2006). Interestingly, failure in axonal transport of vesicles has been implicated in a variety of neurodegenerative diseases (Chevalier-Larsen and Holzbaur, 2006; De Vos et al, 2008). Kinesins are molecular motors that mediate vesicular anterograde transport, moving organelles and proteins towards the plus ends of microtubules (Hirokawa and Takemura, 2005). The conventional kinesin 1 is a heterotetramer composed of two kinesin heavy chain (KHC) and two kinesin light chain (KLC) subunits. Kinesin 1 transports mitochondria, lysosomes, tubulin oligomers, synaptic vesicle precursors or vesicles and vesicles that contain the amyloid precursor protein (APP) or the apolipoprotein E receptor 2 across axons (Hirokawa and Takemura, 2005). In addition, it transports receptor-containing vesicles and RNA-containing granules to dendrites (Hirokawa and Takemura, 2005). In C. elegans, KHC is encoded by the unc-116 gene (Patel et al, 1993). We found that UNC-116 dysfunction suppresses neurodegeneration triggered by the toxic MEC-4(d) and DEG-3(d) ion channel subunits (Figure 5A). unc-116 knockdown does not delay the progression of mec-4(d)-induced degeneration (time-course analysis shown in Supplementary Figure S1). We also checked whether the dysfunction of kinesin 1 (UNC-116 deficiency) affects the expression or localization of the toxic MEC-4(d) channel to the plasma membrane using a full-length MEC-4::GFP reporter fusion. Touch receptor neurons of unc-116 mutants express the reporter at typical, wild-type levels, indicating that dysfunction of kinesin 1 does not affect the stability or transfer of MEC-4 (Figure 5B). Furthermore, unc-116 mutants respond normally to gentle mechanical stimuli, indicating that their touch receptor neurons are functional. Thus, MEC-4 is localized properly at the plasma membrane (Supplementary Figure S2). In addition to necrosis induced by hyperactive ion channels, UNC-116 deficiency protects worms from hypoxia-induced death (Figure 5C).
Figure 5.

Intracellular trafficking by kinesins UNC-116 and UNC-104 is required for necrotic cell death. (A) Depletion of the kinesin 1 heavy chain UNC-116 suppresses necrosis induced by both mec-4(d) and deg-3(d) toxic alleles. (B) Expression of a full-length MEC-4::GFP reporter fusion under the control of the mec-4 promoter in touch receptor neurons of wild-type, unc-116 and unc-104 mutant animals. Representative photos of touch receptor neuron cell bodies are shown. Bar denotes 6 μm. The quantification (%) of GFP signal intensity from the animals examined is graphed below (n=100 neurons per assay; P<0.05 compared with wild type; t-test). (C) Percentage of unc-116 animals that survive after treatment with sodium azide. (D) Depletion of the monomeric kinesin UNC-104 protects neurons against MEC-4(d)-induced degeneration. (E) unc-104 mutant animals are more resistant to hypoxic death induced by sodium azide. (F) Combined inactivation of endocytosis and kinesin-mediated intracellular trafficking does not enhance protection of touch receptor neurons against MEC-4(d)-induced degeneration. Error bars denote s.e.m. values (n>250 for all populations examined; P<0.001, compared with control animals, unpaired t-test).
We also tested the contribution of the kinesin-related protein UNC-104, which plays a role in the axonal transport of synaptic vesicles in C. elegans (Hall and Hedgecock, 1991). We found that, similarly to UNC-116, depletion of UNC-104 ameliorates MEC-4(d)-induced degeneration (Figure 5D), without delaying the progress of death (Supplementary Figure S1). UNC-104 deficiency does not interfere with the expression or proper localization of the toxic trigger, as the unc-104 mutants express the full-length MEC-4::GFP reporter at normal levels (Figure 5B) and are touch sensitive (Supplementary Figure S2). In addition, loss of UNC-104 is protective against hypoxic death (Figure 5E).
Finally, we examined the genetic interaction between kinesin-mediated intracellular trafficking and the endocytic pathway. To this end, we assayed neurodegeneration in double mutant animals carrying the toxic mec-4(d) allele and lesions in either of two endocytic genes (snt-1 or unc-57) after knock down of the unc-116 or unc-104 kinesin genes. We found that the combined inactivation of endocytosis and kinesin-mediated trafficking does not enhance suppression of necrotic cell death, indicating that these two processes act in the same pathway to contribute to cellular destruction during necrosis (Figure 5F).
Kinesin-mediated intracellular trafficking couples with lysosomal proteolysis and autophagy to mediate necrotic cell death
We examined the involvement of kinesin-mediated intracellular trafficking in cellular degradation mechanisms that contribute to cell destruction during necrosis. We found that dysfunction of kinesin 1 (UNC-116) marginally enhances suppression of cell death in animals with compromised lysosomal cathepsin proteolysis (Figure 6A). We observed no synthetic enhancement in double UNC-104 and aspartyl protease-deficient mutants (Figure 6B). Similarly, dysfunction of either kinesin did not significantly increase suppression of necrosis in animals with defective lysosomal V-ATPase, or in calpain protease mutants (Figure 6C and F).
Figure 6.

Genetic interaction between kinesin-mediated intracellular trafficking and cellular proteolytic pathways mediating necrotic cell death. Dysfunction of either kinesin 1 heavy chain (UNC-116) or the monomeric kinesin UNC-104 does not significantly enhance suppression of neurodegeneration in aspartyl protease-deficient mutant animals (A, B), in animals with compromised lysosomal acidification (C, D), or in calpain protease-deficient mutants animals (E, F). Error bars denote s.e.m. values (n>250 for all populations examined; P>0.5, compared with single mutant control animals, unpaired t-test).
We also tested whether kinesins synergize with autophagy during necrotic cell death. RNAi knockdown of the lgg-1 autophagy gene in either unc-116(e2310) or unc-104(e1265) mutants resulted in a slight but not significant protection against necrotic insults (Figure 7A and B). We also impaired autophagy by downregulating bec-1 or unc-51 in kinesin double mutants and obtained similar results (Supplementary Figure S4B). Combined, our observations indicate that kinesin-mediated intracellular trafficking contributes both in autophagic degradation and in lysosomal proteolysis pathways to facilitate necrotic cell death.
Figure 7.

Kinesin-mediated intracellular trafficking functions together with autophagy to facilitate necrotic cell death. Number of neuron corpses, at the L1 larval stage of development, per 100 animals carrying the neurotoxic mec-4(d) allele in genetic backgrounds deficient for both intracellular trafficking and autophagy. (A) Kinesin 1 heavy chain (UNC-116) deficiency. (B) Monomeric kinesin UNC-104 deficiency. In both cases, no significant synthetic protection of neurons is observed in LGG-1/LC3-depleted animals with impaired autophagy. Error bars denote s.e.m. values (n>250 for all populations examined; P>0.5, compared with single mutant control animals, unpaired t-test).
Discussion
Endocytic processes and intracellular trafficking are of particular importance in neurons, both because of their specialized signal transduction functions, and their intricate and highly differentiated morphology. Synaptic transmission is contingent on endocytosis to maintain an active pool of synaptic vesicles. In addition, the distinctively elongated shape and arborization of neuronal cells places high demand on intracellular trafficking processes to distribute cargo to specialized sites, both in anterograde and in retrograde directions. Therefore, neurons are particularly sensitive to perturbations of endocytosis and intracellular trafficking (Nixon, 2005). Indeed, accumulating evidence indicates that endocytosis and trafficking are disturbed in many neurodegenerative diseases. However, the role of endocytic mechanisms in necrotic cell death remained unclear.
In this study, we utilized well-characterized necrosis models in C. elegans to dissect the involvement of endocytosis and intracellular trafficking by kinesin motor proteins in cellular destruction during necrotic death. Our findings reveal for the first time that both endocytosis and trafficking are required for neuron necrosis in the nematode. Downregulation of endocytosis or kinesin-mediated trafficking by interfering with key proteins regulating these processes, including SNT-1, endophilin (UNC-57), AP180 (UNC-11), synaptojanin (UNC-26), heavy chain of kinesin 1 (UNC-116) and the monomeric kinesin UNC-104 significantly suppresses neurodegeneration induced by hyperactive ion channels without affecting the expression, the localization or the function of the toxic insults. Suppression is not a result of a mere delay of the cell death. We note that responsiveness to touch is not restored in mec-4(d) mutants upon suppression of neurodegeneration because, although touch receptor neurons survive, they still express the mutant MEC-4(d) subunit and thus, lack a functional touch transduction ion channel (data shown in Supplementary Figure S2). This is the case for all genetic suppressors of necrosis induced by mec-4(d) reported in the literature (Xu et al, 2001; Syntichaki et al, 2002, 2005; Artal-Sanz et al, 2006; Samara et al, 2008). In addition to mec-4(d)-induced degeneration, downregulation of endocytosis or trafficking suppresses necrosis induced by diverse genetic and environmental insults such as the hyperactive DEG-3(d) Ca2+ channel, hypoxia and the neurotoxin 6-OHDA. Given that 6-OHDA is taken up by dopaminergic neurons through the dopamine transporter (DAT-1 in C. elegans), it is possible that suppression of necrosis by endocytosis or trafficking defects is due to impaired plasma membrane surface expression of DAT-1 and consequent lower uptake of the neurotoxin. Although this scenario cannot be excluded, we consider it unlikely because these mutants do not display phenotypes associated with defective dopaminergic signalling, and the localization of other membrane proteins such as MEC-4 is not affected (Figure 1B). Therefore, both endocytosis and trafficking are not specific to one particular necrotic insult but rather contribute together, downstream of multiple insults to facilitate cell destruction.
Interestingly, the endosomal sorting complexes required for transport (ESCRT) are not involved in neurodegeneration. Knock down of the vps-32.2 and vps-34 genes, related to the yeast Vacuolar Protein Sorting factors, does not suppress necrotic cell death (Supplementary Figure S5). In addition to its involvement in ESCRT function, VPS-34 also plays a role in autophagy (Kihara et al, 2001; Tassa et al, 2003), a process required for necrotic cell death (Toth et al, 2007; Samara et al, 2008). The marginal reduction of necrosis in vps-34(RNAi) animals may result from autophagy impairment. We also considered whether suppression of neurodegeneration by endocytosis and trafficking mutants is merely a consequence of interfering with synaptic activity. We tested the involvement of two proteins essential for synaptic function and neurotransmitter exocytosis, synaptobrevin (SNB-1) and syntaxin (UNC-64). None of these proteins is required for necrosis, since their knock down did not suppress cell death (Supplementary Figure S6). Thus, suppression of neurodegeneration is specific to impairment of endocytosis and not a collateral effect of defective synaptic function.
A potential caveat of interfering with endocytic and trafficking processes relates to the pleiotropic effects of such manipulations that cumulatively result in reduced animal fitness. In turn, suppression of necrosis might be a secondary consequence of endocytosis and trafficking downregulation, originating from non-specific metabolic alterations in the corresponding mutants. However, we consider this scenario unlikely because several genetic lesions affecting energy metabolism either directly or indirectly (insulin/IGF-1 pathway and mitochondrial mutants) do not suppress necrosis (Supplementary Figure S7).
In-vivo monitoring of necrotic cell death revealed that the number of both early and recycling endosomes increases upon initiation of neurodegeneration, indicating that the endosomal pathway is upregulated early during necrosis. Interestingly, the number and volume of early endosomes increases in human pyramidal neurons of Alzheimer's disease patients at early stages of disease (Cataldo et al, 1997). As necrosis progresses, endosome number decreases sharply, probably because cells cannot sustain energy demanding processes such as endocytosis for long. Our data indicate that endocytosis functions together with lysosomal proteolytic mechanisms and synergizes with autophagy to promote cell demise. Indeed, when clathrin-coated vesicles lose their coat they fuse with early endosomes. In turn, early endosomes may either be channelled for cargo degradation and fuse with lysosomes, or they may follow the recycling route to recycle membrane components. Monitoring of endosome formation during necrosis reveals that the number of recycling endosomes (labelled by GFP::RAB-11) increases early in cell death, indicating upregulation of endosomes recycling. To investigate the involvement of endosome recycling in neurodegeneration, we knocked down RME-1, a protein essential for receptor recycling from the endosome to the plasma membrane (Shi et al, 2007). We found that RME-1 deficiency significantly suppresses mec-4(d)-induced neurodegeneration (Supplementary Figure S8). Thus, the totality of our findings suggests that both endocytosis and autophagy converge on the lysosomal degradation pathway to mediate necrotic cell death.
Neuronal hyperexcitation observed in neurodegenerative disorders such as ischaemia, epilepsy, stroke, trauma and Alzheimer's disease results in massive Ca2+ influx. Elevated cytoplasmic Ca2+ levels cause neurotransmitter exocytosis, followed by clathrin-mediated endocytosis of synaptic vesicle proteins. Excessive Ca2+ entry may also increase endocytosis by promoting the oligomerization of clathrin and the synthesis of PI(4,5)P2, which mediates formation of the clathrin lattice (Nathke et al, 1990; Wenk et al, 2001). Ca2+ also activates calpain cytoplasmic proteases that function to compromise lysosomal membrane integrity. In addition, calpain blocks vesicle endocytosis by cleaving the endocytotic protein amphiphysin I (Wu et al, 2007). Amphiphysin mediates invagination and fission of synaptic vesicles, senses and facilitates membrane curvature and also stimulates the activity of dynamin GTPase (Zhang and Zelhof, 2002; Peter et al, 2004; Yoshida et al, 2004; Yoshida and Takei, 2005). Consistent with the role of calpains in moderating endocytosis, we observed a reduction of clathrin-coated vesicles and endosomes during late stages of necrotic cell death. Hence, endocytic processes contribute to cellular destruction in necrosis. The implication of endocytosis and intracellular trafficking suggests that interfering with induction of endocytosis may reduce cell damage during acute neurodegenerative episodes such as ischaemia, epilepsy and stroke.
Materials and methods
Strains and genetics
We followed standard procedures for C. elegans strain maintenance, crosses and other genetic manipulations. Nematode rearing temperature was kept at 20°C, unless noted otherwise. The following strains were used in this study: N2 (wild type, Bristol isolate), TU1747: deg-3(u662)V, referred to in the text as deg-3(d), mec-4(u231)X, referred to in the text as mec-4(d), CX51: dyn-1(ky51)X, CB840: dpy-23(e840)X, DH1201: rme-1(b1045)V, NM467: snb-1(md247)V, NM204: snt-1(md290)II, MT6977: snt-1(n2665)II, CB1265: unc-104(e1265)II, NJ479: unc-104(rh43)II, FF41: unc-116(e2310)III, CB1190: unc-57(e1190)I, CB406: unc-57(e406)I. CB47: unc-11(e47)I, CB1196: unc-26(e1196)IV, DR97: unc-26(e345)IV, DR2: unc-26(m2)IV. MC339: unc-64(md130)III, The following double and multiple mutant strains were examined for neurodegeneration: age-1(hx546)II;mec-4(u231)X, clk-1(qm30)III; mec-4(u231)X, daf-16(m26)I;mec-4(u231)X, daf-2(e1370)III;mec-4(u231)X, deg-3(u662)V;dyn-1(ky51)X, dpy-23(e840)X;mec-4(u231)X, cad-1(j1)II;mec-4(u231)X, isp-1(qm150)IV;mec-4(u231)X, mev-1(kn1)III; mec-4(u231)X, rme-1(b1045)V;mec-4(u231)X, snb-1(md247)V;mec-4(u231)X, snt-1(md290)II;mec-4(u231)X, snt-1(n2665)II;mec-4(u231)X, snt-1(md290)II;deg-3(u662)V, unc-104(e1265)II;mec-4(u231)X, unc-104(rh43)II;mec-4(u231)X, unc-11(e47)I;mec-4(u231)X, unc-116(e2310)III; mec-4(u231)X, unc-116(e2310)III;deg-3(u662)V, unc-26(e1196)IV;mec-4(u231)X, unc-26(e345)IV;mec-4(u231)X, unc-26(m2)IV;mec-4(u231)X, unc-57(e1190)I;mec-4(u231)X, unc-57(e406)I;mec-4(u231)X, unc-57(e1190);deg-3(u662)V, unc-64(md130)III;mec-4(u231)X, cad-1(j1)II;snt-1(md290)II;mec-4(u231)X, cad-1(j1)II;unc-116(e2310)III;mec-4(u231)X, unc-104(e1265)II;cad-1(j1)II;mec-4(u231)V. To verify that mutations in snt-1, unc-57, unc-116 or unc-104 do not interfere with touch receptor neuron formation or expression of the toxic mec-4 allele, we examined the following strains, carrying extrachromosomal arrays: N2;Ex[pmec-4MEC-4::GFP], snt-1(n2665);Ex[pmec-4MEC-4::GFP], unc-57(e1190);Ex[pmec-4MEC-4::GFP], unc-116(e2310));Ex[pmec-4MEC-4::GFP] and unc-104(e1265);Ex[pmec-4MEC-4::GFP]. For the quantification of GFP signal intensity, ∼100 neurons were imaged per strain. To monitor and quantify the formation of clathrin-coated pits or vesicles in neurons, we examined the following strains, carrying extrachromosomal arrays: N2;Ex[pmec-17APS-2::GFP], mec-4(u231);Ex[pmec-17APS-2::GFP]. For the monitoring and quantification of early and recycling endosomes in neurons, we used the following strains, carrying extrachromosomal arrays: N2;Ex[pmec-7GFP::RAB-5], mec-4(u231);Ex[pmec-7GFP::RAB-5], N2;Ex[pmec-17GFP::RAB-11], mec-4(u231);Ex[pmec-17GFP::RAB-11].
Plasmid constructs and RNA interference
To generate the aps-2 reporter construct, we fused GFP at the carboxy-terminus of the C. elegans APS-2 (∼970 bp). The aps-2 gene was amplified from wild-type (N2) genomic DNA with the primers 5′-CCCCCCGGGAATGATCCGTTTTATTCTGATC-3′ and 5′-GGAACCGGTTCCAGGGAAGTAAGCATGAG-3′, and cloned into the gfp vector pPD95.77 harbouring the mec-17, promoter, which drives expression in the six touch receptor neurons. The reporter construct was injected into the gonads of N2 animals together with pRF4, a plasmid that carries the rol-6(su1006), dominant co-injection marker. Transgenic roller hermaphrodites were crossed with mec-4(d) males. To generate the rab-5 reporter construct, we fused GFP at the amino-terminus of C. elegans RAB-5. A 1776-bp fragment containing the rab-5 gene was PCR amplified from N2 genomic DNA, using the primers 5′-CGGAATTCATGGCCGCCCGAAACGCAG-3′ and 5′-CGGAATTCCGAACTTCAATTTATTCGGAT-3′, and inserted at the C-terminus of GFP. The reporter was placed under the control of the mec-7 promoter. As in the case of APS-2::GFP, the construct was injected into the gonads of N2 animals together with pRF4 as a co-transformation marker. Transgenic roller hermaphrodites were crossed with mec-4(d) males. In order to generate an endosomal marker for recycling endosomes, we fused GFP at the amino-terminus of C. elegans RAB-11 (779 bp). rab-11 was amplified from N2 genomic DNA using the primers 5′-CGGAATTCAATGGGCTCTCGTGACGAT-3′ and 5′-CGGAATTCTTATGGGATGCAACACTGC-3′. The fusion was placed under the control of the mec-7 promoter. For RNAi experiments, we used HT115(DH3) Escherichia coli bacteria, deficient for RNase-E, which were transformed with RNAi plasmids for clp-1, lgg-1, bec-1, unc-51 and vha-2 that direct the synthesis of double-stranded RNAs corresponding to these genes. Transformed bacteria were fed to animals according to a previously described methodology (Kamath et al, 2001). The construction of these plasmids has been previously described (Syntichaki and Tavernarakis, 2002; Syntichaki et al, 2005; Samara and Tavernarakis, 2008). Generation of HT115(DH3) bacteria carrying RNAi plasmids for the vps-32.2, vps-34, unc-104 and unc-116 genes has been described previously (Fraser et al, 2000; Kamath and Ahringer, 2003), Plasmids were obtained from Source BioScience-LifeSciences (Nottingham, UK).
Cell death assays
Neurodegeneration in animals bearing mec-4(d) or deg-3(d) alleles was generally scored during the L1 larval stage, as previously described (Treinin and Chalfie, 1995; Hall et al, 1997; Syntichaki et al, 2002; Samara et al, 2008). We prepared L1 larvae by washing gravid adults off plates and allowing the remaining eggs to hatch for 4–5 h at 20°C. We scored necrosis during early to mid L1 stage, by the characteristic vacuolated appearance of the six touch receptor neuron corpses in the case of mec-4(d) plus IL1 sensory neuron corpses in the head and PVC in the tail in deg-3(d), by using differential interference contrast (Nomarski) microscopy. To infer statistical significance, we scored neurodegeneration in 100 L1 animals and repeated the experiment for four times. For RNAi experiments, L4 animals grown at 20°C are transferred to empty NGM plates for 10 min and then to NGM plates containing RNAi bacteria and let to develop and become gravid adults at 15°C. Necrotic cell death is assayed as described above. To induce hypoxic conditions chemically, nematodes at the L4 stage of development were selected, washed 2–3 times with 1 ml M9 and incubated for 30 min at 20°C with freshly made NaN3 (Sigma, Munich, Germany) in M9 at final concentration of 0.5 M. Worms were washed with M9 and placed into NGM plates to recover. After 24 h at 20°C, the percentage of living worms was calculated. Alternatively, hypoxia was induced in a hypoxic chamber using low oxygen concentration following the previously described methodology (Scott et al, 2002). Specifically, adults 2 days post L4, were exposed to low oxygen conditions (<0.5%) at 25°C for 24 h. Animals were allowed to recover for 24 h and then scored for survival. Assaying dopaminergic neuron survival after treatment with the neurotoxin 6-OHDA was performed as described previously (Nass et al, 2002). To test the phenotypic consequence of the suppression of the mec-4(d)-induced degeneration, animals were assayed for responsiveness to light mechanical stimuli at the young adult stage. Animals were stimulated three repeated times with an eyelash in the middle part of the body and in the tail. A change of movement to a direction away from the stimulus was recorded as positive response (sensitivity): these animals were considered sensitive to touch. For time-course analysis of necrotic cell death, gravid adults were bleached and the synchronized populations were divided into three sub-populations that were assayed for necrosis at the L1, L2 and L3–L4 developmental stages. The experiment was repeated three times for each mutant, with >150 animals examined in each trial.
Microscopy and quantification of clathrin-coated pits/vesicles, early endosomes and late endosomes
Animals were mounted in 2% agarose pads, anaesthetized with 20 mM sodium azide and observed at room temperature. Worms carrying the APS-2 reporter construct were harvested at the L1–L2 developmental stages and observed using a high magnification oil-objective lens ( × 63, Zeiss Plan-NEOFLUAR; numerical aperture 1.4; Carl Zeiss, Jena, Germany), on a confocal microscope (Zeiss Axioskop with a Bio-Rad Radiance 2100 scanhead, Bio-Rad, Hercules, USA), using a Bio-Rad LaserSharp 2000 software package. To acquire images from individual neurons, animals were scanned with a 488-nm laser beam. Emitted light was gathered using a 515±15 nm band-pass filter. The number of APS-2::GFP spots in each touch receptor neuron was counted. In mec-4(d) mutants, neurons were divided into two groups: vacuolated and non-vacuolated. Dots were scored separately in each group. The number of animals that were examined was ∼100 and the total number of neurons was ∼500. The mean number of dots per neuron was then calculated. The same strategy was also followed for the RAB-5 reporter construct for early endosomes and for the RAB-11 reporter construct for recycling endosomes. For the quantification of GFP signal intensity in the transgenic animals expressing the full-length MEC-4::GFP, we processed at least 100 cell images (8-bit colour depth, 256 shades of grey), from three independent trials and calculated the pixel intensity for each cell using the Image J software (http://rsb.info.nih.gov/ij/).
Statistical analysis
Statistical analyses were carried out using the Prism software package (GraphPad Software Inc., San Diego, USA) and the Microsoft Office 2003 Excel software package (Microsoft Corporation, Redmond, WA, USA). Mean values were compared using unpaired t-tests. For multiple comparisons, we used the one-factor (ANOVA) variance analysis corrected by the post hoc Bonferroni test.
Supplementary Material
Acknowledgments
We are grateful to Dr Daniele Bano for generously providing RNAi plasmids and reagents. We thank Angela Pasparaki for expert technical assistance. Some nematode strains used in this work were provided by the C. elegans Gene Knockout Project at OMRF (http://www.mutantfactory.ouhsc.edu/), which is part of the International C. elegans Gene Knockout Consortium, the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR), and Dr Shohei Mitani (National Bioresource Project) in Japan. We thank Dr Andrew Fire for plasmid vectors. This work was funded by grants from the European Research Council (ERC) and the European Commission 7th Framework Programme.
Author contributions: KT and NT designed and performed the experiments, analysed data and wrote the manuscript. NT supervised the study.
Footnotes
The authors declare that they have no conflict of interest.
References
- Artal-Sanz M, Samara C, Syntichaki P, Tavernarakis N (2006) Lysosomal biogenesis and function is critical for necrotic cell death in Caenorhabditis elegans. J Cell Biol 173: 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Pieroni C, Nixon RA (1997) Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased beta-amyloidogenesis. J Neurosci 17: 6142–6151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli V, Corti M, Gruenberg J (2001) Endocytosis and signaling cascades: a close encounter. FEBS Lett 498: 190–196 [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen E, Holzbaur EL (2006) Axonal transport and neurodegenerative disease. Biochim Biophys Acta 1762: 1094–1108 [DOI] [PubMed] [Google Scholar]
- Cremona O, De Camilli P (1997) Synaptic vesicle endocytosis. Curr Opin Neurobiol 7: 323–330 [DOI] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC (2008) Role of axonal transport in neurodegenerative diseases. Annu Rev Neurosci 31: 151–173 [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Boll W, Van Oijen A, Hariharan R, Chandran K, Nibert ML, Kirchhausen T (2004) Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell 118: 591–605 [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, Sudhof TC (2001) Synaptotagmin I functions as a calcium regulator of release probability. Nature 410: 41–49 [DOI] [PubMed] [Google Scholar]
- Forman MS, Trojanowski JQ, Lee VM (2004) Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat Med 10: 1055–1063 [DOI] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, Ahringer J (2000) Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature 408: 325–330 [DOI] [PubMed] [Google Scholar]
- Gallop JL, Jao CC, Kent HM, Butler PJ, Evans PR, Langen R, McMahon HT (2006) Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J 25: 2898–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH, Gu G, Garcia-Anoveros J, Gong L, Chalfie M, Driscoll M (1997) Neuropathology of degenerative cell death in Caenorhabditis elegans. J Neurosci 17: 1033–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM (1991) Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell 65: 837–847 [DOI] [PubMed] [Google Scholar]
- Harris TW, Schuske K, Jorgensen EM (2001) Studies of synaptic vesicle endocytosis in the nematode C. elegans. Traffic 2: 597–605 [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R (2005) Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci 6: 201–214 [DOI] [PubMed] [Google Scholar]
- Jacobson LA, Jen-Jacobson L, Hawdon JM, Owens GP, Bolanowski MA, Emmons SW, Shah MV, Pollock RA, Conklin DS (1988) Identification of a putative structural gene for cathepsin D in Caenorhabditis elegans. Genetics 119: 355–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarousse N, Kelly RB (2001) The AP2 binding site of synaptotagmin 1 is not an internalization signal but a regulator of endocytosis. J Cell Biol 154: 857–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J (2003) Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30: 313–321 [DOI] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J (2001) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2: RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara A, Noda T, Ishihara N, Ohsumi Y (2001) Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol 152: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh TW, Bellen HJ (2003) Synaptotagmin I, a Ca2+ sensor for neurotransmitter release. Trends Neurosci 26: 413–422 [DOI] [PubMed] [Google Scholar]
- Kourtis N, Tavernarakis N (2009) Autophagy and cell death in model organisms. Cell Death Differ 16: 21–30 [DOI] [PubMed] [Google Scholar]
- Masuda M, Takeda S, Sone M, Ohki T, Mori H, Kamioka Y, Mochizuki N (2006) Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J 25: 2889–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Boucrot E (2011) Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat Rev Mol Cell Biol 12: 517–533 [DOI] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301: 1387–1391 [DOI] [PubMed] [Google Scholar]
- Morgan JR, Prasad K, Hao W, Augustine GJ, Lafer EM (2000) A conserved clathrin assembly motif essential for synaptic vesicle endocytosis. J Neurosci 20: 8667–8676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JR, Zhao X, Womack M, Prasad K, Augustine GJ, Lafer EM (1999) A role for the clathrin assembly domain of AP180 in synaptic vesicle endocytosis. J Neurosci 19: 10201–10212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mousavi SA, Malerod L, Berg T, Kjeken R (2004) Clathrin-dependent endocytosis. Biochem J 377: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63: 724–726 [DOI] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM 3rd, Blakely RD (2002) Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci USA 99: 3264–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathke I, Hill BL, Parham P, Brodsky FM (1990) The calcium-binding site of clathrin light chains. J Biol Chem 265: 18621–18627 [PubMed] [Google Scholar]
- Nixon RA (2005) Endosome function and dysfunction in Alzheimer's disease and other neurodegenerative diseases. Neurobiol Aging 26: 373–382 [DOI] [PubMed] [Google Scholar]
- Nonet ML, Holgado AM, Brewer F, Serpe CJ, Norbeck BA, Holleran J, Wei L, Hartwieg E, Jorgensen EM, Alfonso A (1999) UNC-11, a Caenorhabditis elegans AP180 homologue, regulates the size and protein composition of synaptic vesicles. Mol Biol Cell 10: 2343–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura K, Wicky C, Magnenat L, Tobler H, Mori I, Muller F, Ohshima Y (1994) Caenorhabditis elegans unc-51 gene required for axonal elongation encodes a novel serine/threonine kinase. Genes Dev 8: 2389–2400 [DOI] [PubMed] [Google Scholar]
- Patel N, Thierry-Mieg D, Mancillas JR (1993) Cloning by insertional mutagenesis of a cDNA encoding Caenorhabditis elegans kinesin heavy chain. Proc Natl Acad Sci USA 90: 9181–9185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT (2004) BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303: 495–499 [DOI] [PubMed] [Google Scholar]
- Poskanzer KE, Marek KW, Sweeney ST, Davis GW (2003) Synaptotagmin I is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature 426: 559–563 [DOI] [PubMed] [Google Scholar]
- Ren Y, Xu HW, Davey F, Taylor M, Aiton J, Coote P, Fang F, Yao J, Chen D, Chen JX, Yan SD, Gunn-Moore FJ (2008) Endophilin I expression is increased in the brains of Alzheimer disease patients. J Biol Chem 283: 5685–5691 [DOI] [PubMed] [Google Scholar]
- Ringstad N, Gad H, Low P, Di Paolo G, Brodin L, Shupliakov O, De Camilli P (1999) Endophilin/SH3p4 is required for the transition from early to late stages in clathrin-mediated synaptic vesicle endocytosis. Neuron 24: 143–154 [DOI] [PubMed] [Google Scholar]
- Rink J, Ghigo E, Kalaidzidis Y, Zerial M (2005) Rab conversion as a mechanism of progression from early to late endosomes. Cell 122: 735–749 [DOI] [PubMed] [Google Scholar]
- Samara C, Syntichaki P, Tavernarakis N (2008) Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ 15: 105–112 [DOI] [PubMed] [Google Scholar]
- Samara C, Tavernarakis N (2008) Autophagy and cell death in Caenorhabditis elegans. Curr Pharm Des 14: 97–115 [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K, Liou W, Pant S, Harada A, Grant BD (2008) Regulation of endocytic recycling by C. elegans Rab35 and its regulator RME-4, a coated-pit protein. EMBO J 27: 1183–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt-John T, Drepper C, Mussmann A, Hahn P, Kuhlmann M, Thiel C, Hafner M, Lengeling A, Heimann P, Jones JM, Meisler MH, Jockusch H (2005) Mutation of Vps54 causes motor neuron disease and defective spermiogenesis in the wobbler mouse. Nat Genet 37: 1213–1215 [DOI] [PubMed] [Google Scholar]
- Schuske KR, Richmond JE, Matthies DS, Davis WS, Runz S, Rube DA, van der Bliek AM, Jorgensen EM (2003) Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron 40: 749–762 [DOI] [PubMed] [Google Scholar]
- Scott BA, Avidan MS, Crowder CM (2002) Regulation of hypoxic death in C. elegans by the insulin/IGF receptor homolog DAF-2. Science 296: 2388–2391 [DOI] [PubMed] [Google Scholar]
- Shi A, Pant S, Balklava Z, Chen CC, Figueroa V, Grant BD (2007) A novel requirement for C. elegans Alix/ALX-1 in RME-1-mediated membrane transport. Curr Biol 17: 1913–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307: 1282–1288 [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Samara C, Tavernarakis N (2005) The vacuolar H+ -ATPase mediates intracellular acidification required for neurodegeneration in C. elegans. Curr Biol 15: 1249–1254 [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Tavernarakis N (2002) Death by necrosis. Uncontrollable catastrophe, or is there order behind the chaos? EMBO Rep 3: 604–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntichaki P, Tavernarakis N (2003) The biochemistry of neuronal necrosis: rogue biology? Nat Rev Neurosci 4: 672–684 [DOI] [PubMed] [Google Scholar]
- Syntichaki P, Xu K, Driscoll M, Tavernarakis N (2002) Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature 419: 939–944 [DOI] [PubMed] [Google Scholar]
- Takei K, Haucke V (2001) Clathrin-mediated endocytosis: membrane factors pull the trigger. Trends Cell Biol 11: 385–391 [DOI] [PubMed] [Google Scholar]
- Tassa A, Roux MP, Attaix D, Bechet DM (2003) Class III phosphoinositide 3-kinase – Beclin1 complex mediates the amino acid-dependent regulation of autophagy in C2C12 myotubes. Biochem J 376: 577–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth ML, Simon P, Kovacs AL, Vellai T (2007) Influence of autophagy genes on ion-channel-dependent neuronal degeneration in Caenorhabditis elegans. J Cell Sci 120: 1134–1141 [DOI] [PubMed] [Google Scholar]
- Treinin M, Chalfie M (1995) A mutated acetylcholine receptor subunit causes neuronal degeneration in C. elegans. Neuron 14: 871–877 [DOI] [PubMed] [Google Scholar]
- Verstreken P, Koh TW, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, Bellen HJ (2003) Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron 40: 733–748 [DOI] [PubMed] [Google Scholar]
- Wenk MR, Pellegrini L, Klenchin VA, Di Paolo G, Chang S, Daniell L, Arioka M, Martin TF, De Camilli P (2001) PIP kinase Igamma is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron 32: 79–88 [DOI] [PubMed] [Google Scholar]
- Wu Y, Liang S, Oda Y, Ohmori I, Nishiki T, Takei K, Matsui H, Tomizawa K (2007) Truncations of amphiphysin I by calpain inhibit vesicle endocytosis during neural hyperexcitation. EMBO J 26: 2981–2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ (2007) Autophagosome formation: core machinery and adaptations. Nat Cell Biol 9: 1102–1109 [DOI] [PubMed] [Google Scholar]
- Xu K, Tavernarakis N, Driscoll M (2001) Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron 31: 957–971 [DOI] [PubMed] [Google Scholar]
- Yamashima T (2004) Ca2+-dependent proteases in ischemic neuronal death: a conserved ‘calpain-cathepsin cascade’ from nematodes to primates. Cell Calcium 36: 285–293 [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Kinuta M, Abe T, Liang S, Araki K, Cremona O, Di Paolo G, Moriyama Y, Yasuda T, De Camilli P, Takei K (2004) The stimulatory action of amphiphysin on dynamin function is dependent on lipid bilayer curvature. EMBO J 23: 3483–3491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Takei K (2005) Stimulation of dynamin GTPase activity by amphiphysin. Methods Enzymol 404: 528–537 [DOI] [PubMed] [Google Scholar]
- Zerial M, McBride H (2001) Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2: 107–117 [DOI] [PubMed] [Google Scholar]
- Zhang B, Zelhof AC (2002) Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp. Traffic 3: 452–460 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.