

Abstract
Acquisition of death-resistance is critical in the evolution of neoplasia. Our aim was to model the early stages of carcinogenesis by examining intracellular alterations in cells that have acquired apoptosis-resistance after exposure to a complex genotoxin. We previously generated sub-populations of BJ-hTERT human diploid fibroblasts, which have acquired death-resistance following exposure to hexavalent chromium [Cr(VI)], a broad-spectrum genotoxicant. Long-term exposure to certain forms of Cr(VI) is associated with respiratory carcinogenesis. Here, we report on the death-sensitivity of subclonal populations derived from clonogenic survivors of BJ-hTERT cells treated with 5 μM Cr(VI) (DR1, DR2), or selected by dilution-based cloning without treatment (CC1). Following Cr(VI) treatment, CC1 cells downregulated expression of the anti-apoptotic protein Bcl-2 and exhibited extensive expression of cleaved caspase 3. In contrast, the DR cells exhibited no cleaved caspase 3 expression and maintained expression of Bcl-2 following recovery from 24 h Cr(VI) exposure. The DR cells also exhibited attenuated mitochondrial-membrane depolarization and mitochondrial retention of cytochrome c and SMAC/DIABLO following Cr(VI) exposure. The DR cells exhibited less basal mtDNA damage, as compared to CC1 cells, which correlates with intrinsic (non-induced) death-resistance. Notably, there was no difference in p53 protein expression before or after treatment among all cell lines. Taken together, our data suggest the presence of more resilient mitochondria in death-resistant cells, and that death-resistance can be acquired in normal human cells early after genotoxin exposure. We postulate that resistance to mitochondrial-mediated cell death and mitochondrial dysregulation may be an initial phenotypic alteration observed in early stage carcinogenesis.
Keywords: death-resistance, genotoxin, chromium, mitochondria, early-stage carcinogenesis
1. Introduction
Despite making huge strides in understanding the molecular basis for pre-malignant progression and conversion to neoplasia little is known about the earliest stages of carcinogenesis. It has been suggested that genotoxic exposure is sufficient to trigger a mutagenic “initiation step” [1]. However, the causal and temporal relationships between genotypic and phenotypic alterations, as well as the potential role of the cellular stress response in mediating or modulating early stage carcinogenesis, are not clear. Following extensive genetic damage by a genotoxicant, most normal cells will be eliminated from the proliferating population by either apoptosis or terminal growth arrest (TGA). This might constitute a selective pressure for expansion of cells with the ability to survive after exposure to apoptogenic levels of a DNA damaging agent; and may yield a precursor pool of cells from which death-resistant variants, that are more predisposed towards neoplastic evolution, may emerge. Escape from or resistance to apoptosis or TGA seems to be a requirement for cells to achieve limitless replicative potential, making acquisition of death-resistance one of the most critical characteristics attained by cancer cells [1].
Certain forms of hexavalent chromium [Cr(VI)] compounds are well known occupational human respiratory carcinogens [2]. These compounds are classified by the International Agency for Research on Cancer as group 1 human carcinogens [3, 4], as they were encountered by workers in the chromium mining and smelting industry. The intracellular metabolic reduction of Cr(VI) yields an array of structural genetic lesions, including direct DNA damage in the form of Cr(VI)-DNA adducts, DNA strand breaks and DNA crosslinks, as well as potential indirect damage through generation of reactive oxygen species (ROS) [5]. The damage disrupts cellular homeostasis and triggers both apoptosis and TGA, which have been previously established as the primary mode of Cr(VI)-induced cell death [6-8]. These characteristics of Cr(VI)-induced damage, make it a useful model genotoxin for investigating the balance between cell death and survival.
Apoptosis is the best characterized mechanism of controlled cell death. It occurs via two main pathways, the extrinsic/death receptor-mediated pathway and the intrinsic/mitochondrial-mediated pathway, the latter of which is found to be compromised in most cancer cells [9-11]. Once triggered, signals from both pathways can lead to the activation of a group of cysteine proteases known as caspases, and the initiation of apoptotic cell death via cleavage of effector caspases (i.e. caspase 3) [9]. The mitochondria are critical regulators of apoptosis, as they harbor apoptogenic factors such as cytochrome c and the second mitochondria-derived activator of caspases/direct IAP binding protein with low pI (SMAC/DIABLO) that initiate cell death upon release [9]. Moreover, the mitochondria are also the prime functional location of the apoptotic regulatory proteins of the Bcl-2 family, some of which have been shown to prevent apoptosis by inhibiting mitochondrial-membrane permeabilization [12], cytochrome c release and caspase activation [13], as well as oxidative stress-induced damage [14, 15].
The mitochondria contain and maintain their own genetic material, mitochondrial DNA (mtDNA). Dysregulation of mitochondria-controlled functions, such as apoptosis and energy metabolism, may be related to mtDNA defects that affect the synthesis and function of encoded electron transport chain protein subunits [16]. mtDNA damage can occur at high frequencies, due to the elevated resident levels of ROS produced by mitochondrial respiration, absence of histones, and limited DNA repair mechanisms functioning within the mitochondria [16, 17]. Notably, excessive oxidative damage to the mtDNA may be a trigger for apoptosis [16, 18].
The goal of this study was to examine the phenotypic alterations in cells that have acquired apoptosis-resistance as a result of an “initiating” genotoxin exposure. Previously, our lab has reported on a population of Cr(VI)-resistant human fibroblasts, B-5Cr cells, derived from survivors of BJ-hTERT cells (stably transfected with human telomerase (hTERT)) exposed to 5 μM Cr(VI) [19]. To ensure that the observed resistance to Cr(VI)-induced cell death was due to acquired apoptosis-resistance and not a result of clonogenic selection pressure, the B-5Cr cells, as well as untreated B-0Cr cells, were further subcloned. The subclonal populations include the clonogenic control cell line, CC1 (derived from B-0Cr), and the DR1 and DR2 cell lines derived from the B-5Cr cells.
We tested the hypothesis that survival after an initiating genotoxic insult may involve the selection of cells with mitochondrial-associated dysregulated apoptotic control, leading to death-resistance. In this study, we investigated cleaved caspase 3 expression following exposure to both Cr(VI) and cisplatin, Bcl-2 protein expression, mitochondrial-membrane depolarization, and the release of cytochrome c and SMAC/DIABLO following exposure of normal and death-resistant cells to Cr(VI). In addition, we evaluated mtDNA damage and copy number before and after Cr(VI) exposure. Our results show that resistance to apoptosis can be acquired in normal diploid human cells following an apoptogenic exposure to a chemical genotoxicant. These observations suggest that the acquisition of resistance to mitochondrial-mediated cell death, and/or selection for mitochondrial dysregulation, may contribute to the earliest stages of carcinogenesis.
2. Materials and Methods
2.1. Cell lines and culture
Cr(VI) resistant fibroblasts (B-5Cr) were derived from human foreskin fibroblasts transfected with the hTERT gene (BJ-hTERT; Geron Corp.), and further subcloned into both clonogenic control (death-sensitive) cell lines (CC1 from B-0Cr cells) and death-resistant cell lines (DR1 and DR2 from B-5Cr cells), as previously described [19]. All cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen Corporation), containing Medium 199 (Invitrogen Corporation), 10% fetal bovine serum (Hyclone Laboratories, Inc.), 5 μg/ml gentamicin (Life Technologies), and 0.75 μg/ml fungizone antimycotic (Invitrogen Corporation). The hTERT transgene was selected for by the addition of 10 μg/ml hygromycin B (Life Science Technologies) to the medium. All cell lines were incubated in a 95% air and 5% CO2 humidified atmosphere at 37°C, and the medium was replaced every 48 h.
2.2. Experimental treatment of cells
Sodium chromate [Cr(VI)] (Na2CrO4.4H2O, J.T. Baker Chemical Co.) was dissolved and diluted in DMEM and sterilized by passage through a 0.2 μm filter. Where indicated, some experiments included a recovery period, in which Cr(VI) treated medium was removed from cells, cells were rinsed with phosphate buffered saline (PBS), and were incubated for 24 h in untreated DMEM. For all experiments, cells were incubated at 37°C for 24 h prior to treatment.
2.3. Immunoblotting
Cells were seeded at a density of 4 × 105 cells/60 mm dish. Following treatment, cells were scraped from the dish in CHAPS cell lysis buffer (Cell Signaling Technology) supplemented with 50 mM NaF, 1 mM Na3V04, and 1 mM PMSF. The cells were lysed by freezing and thawing. The lysates were centrifuged and immunoblotting was performed as previously described [20]. Antibodies used were as follows: cleaved caspase 3, Bcl-2 (Cell Signaling Technology), hTERT (Rockland), p53 (EMD), and Bax (Calbiochem). An antibody to β-actin (Sigma-Aldrich) was used to confirm equal protein loading.
2.4. Cr(VI)-DNA adducts
Cells were seeded at 8 × 105 cells/100 mm dish and treated with Cr(VI) containing 5 μCi Na 512CrO4/dish for 2 h, 24 h, or 24 h followed by a 24 h recovery period in fresh DMEM. The cells were suspended in lysis buffer (10 mM Tris-HCl, 0.5% SDS, 0.5% Triton X-100) containing proteinase K at 37°C overnight. DNA was phenol:chloroform extracted, precipitated, and quantified using a multi-purpose scintillation counter LS 6500 (Beckman Coulter) and expressed as pmole 51Cr(VI)/μg DNA.
2.5. Immunofluorescence
Cells were seeded at a density of 1.5 × 104/well in 8-well chamber slides (Nunc International). Following treatment the cells were stained with 500 nM MitoTracker Red CMXRos (Mitotracker; Molecular Probes) for 10 min at 37°C, and fixed with 3.7% paraformaldehyde in DMEM for 20 min at room temperature (RT). The cells were permeabilized with 0.1% Triton-X, and blocked with 1% BSA/1% heat-inactivated FBS. The cells were incubated with cytochrome c (1.5 μg/ml) (Calbiochem), or SMAC/DIABLO (1:250) (Cell Signaling Technology) for 1 h at RT. The slides were rinsed, blocked, and incubated with goat anti-mouse IgG secondary antibody conjugated to Alexa 488 (1:500) (Molecular Probes). Hoechst dye was used to indicate the nucleus (1:1000) (Molecular Probes). The cells were visualized at 63X using a Zeiss laser scanning microscope, at excitation wavelengths of 543, 488, and 405 nm for MitoTracker, Alexa 488, and Hoechst, respectively. Cells were individually analyzed using Image-Pro Plus software (Media Cybernetics, Inc.). For each experiment twenty cells per condition were analyzed. Segmentation analysis was done to threshold the images with dynamic ranges of approximately 30-255 (Red channel) and 40-255 (Green channel). Pixel area was counted and measured for the selected cell in the specific dynamic ranges to analyze Red only, Green only, Green within red, and Green outside of Red. Values for Green outside of Red were normalized to Red only.
2.6. Mitochondrial DNA damage analysis
Cells were seeded at a density of 1 × 106/100 mm dish and incubated at 37°C for 24 h prior to treatment. Cells were treated with 0 or 6 μM Cr(VI) for 24 h, washed with PBS, trypsinized, and centrifuged at 1000 rpm for 5 min. DNA was extracted using a Qiagen QIAamp DNA Mini Kit, according to the manufacturer’s instructions (Qiagen Incorporated). The DNA concentration and integrity were determined, and mtDNA and nuclear DNA (nuDNA) copy number were assessed by real-time polymerase chain reaction (RT-PCR) using SYBR Green I Dye, AmpliTaq gold DNA polymerase (Applied Biosystems Incorporated), and primers for the amplification of mitochondrial NADH dehydrogenase 1 (ND1) and the β2microglobulin (β2m) gene to assess mtDNA and nuDNA, respectively. Cell copy number was measured assuming diploid cells and calculated by dividing nuDNA copy number by two. mtDNA damage was measured by the amplification of an 8.9-kb mtDNA target by quantitative PCR (QPCR) according to a modification of the method of Santos et al [21]. mtDNA damage was expressed as amount of 8.9-kb DNA/106 ND1 copies amplified, with amplification being inversely proportional to mtDNA damage.
2.7. Statistical analysis
To determine significant differences among experimental groups, statistical analyses were performed using GraphPad Prism version 4.00 as previously described [22].
3. Results
3.1. Resistance to genotoxin-induced caspase 3-mediated cell death acquired after Cr(VI) exposure
To investigate relative death-sensitivity in our model system, we assessed susceptibility to apoptosis. Cleavage of caspase 3 was measured after 24 h exposure to the apoptogenic concentration of 6 μM Cr(VI) followed by a 24 h recovery period (Figure 1). In the absence of Cr(VI) treatment, cleaved caspase 3 protein expression was undetectable in all of the cell lines. The CC1 cell line exhibited a 73-fold increase in cleaved caspase 3 expression after Cr(VI) treatment, as compared to the untreated control. In sharp contrast, the DR1 and DR2 cell lines showed significant resistance to Cr(VI)-induced caspase 3 cleavage, which was only 9.5 and 11-fold of the untreated control, respectively. The apoptosis-resistance observed in the DR cells was not Cr(VI)-specific: the BJ-hTERT parental cells exhibited a marked increase in cleaved caspase 3 expression, which was 99 and 857-fold of control, respectively, after 3 h treatment with apoptogenic doses of cisplatin (20 μM and 40 μM) followed by a 48 h cisplatin-free recovery period. In contrast, the respective cisplatin-induced incremental caspase 3 cleavage was sharply attenuated in the DR1 cells, at just 56 and 169-fold of control (Supplemental Figure 1). Notably, our data show similar sensitivity to Cr(VI) in the BJ-hTERT and CC1 cell lines (data not shown).
Fig. 1.
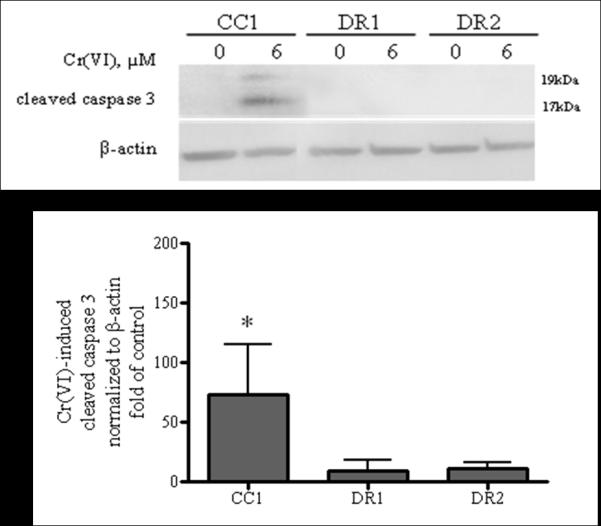
DR cells are resistant to Cr(VI)-induced caspase 3-mediated apoptosis. CC1, DR1 and DR2 cells (a) were treated with 0 or 6 μM Cr(VI) for 24 h, followed by a 24 h recovery period in fresh DMEM. Total cell protein was extracted after the recovery period. Proteins were separated by SDS-PAGE and cleaved caspase 3 expression was detected by immunoblotting at 17 and 19 kDa. The same amount of protein loading was confirmed by immunoblotting for β-actin. The data are expressed as percent of respective control, in the absence of Cr(VI) and are the mean +/− SE of four experiments. * indicates statistically significant difference from respective untreated control (p < 0.05).
To determine whether the observed death-resistance was only a consequence of decreased Cr(VI) uptake during the treatment, we measured Cr(VI)-DNA adduct formation, which serves as a rigorous, dose-sensitive biomarker for Cr(VI) exposure and uptake. There was no significant difference among the cell lines in Cr(VI)-DNA adduct formation after exposure to 6 μM Cr(VI), which was approximately 0.6 and 2.9 pmol/μg DNA after 2 h (Figure 2a) and 24 h (Figure 2b) exposure, respectively. The slight decrease in Cr(VI)-DNA adduct formation observed in both DR2 and CC1 cells following a 24 h recovery period is not statistically significant, and does not correlate with death sensitivity (Fig. 2b, striped bars).
Fig. 2.

DR cell resistance to Cr(VI)-induced cleaved caspase 3-mediated apoptosis is not related to decreased Cr(VI)-DNA adduct formation. CC1, DR1 and DR2 cells were treated with 0 or 6 μM Cr(VI) radiolabeled with Na251CrO4 for (a) 2 h, or (b) 24 h (solid bars) followed by a 24 h recovery period in fresh DMEM (hatched bars). DNA was isolated by phenol:chloroform extraction. The samples were lysed and analyzed for intracellular 51Cr using a multi-purpose scintillation counter. Data are mean +/− SE of 2 experiments done in triplicate and are expressed as picomole of 51Cr normalized to μg of DNA.
Apoptosis resistance in cancer cells has also been directly associated with the increased expression of Bcl-2 and/or the loss of Bax expression [23]. Therefore, we determined the protein expression levels of Bcl-2 after 24 h treatment with 0, 6, or 9 μM Cr(VI) followed by a 24 h recovery period, which temporally correlates with the observed cleaved caspase 3-mediated apoptosis resistance to Cr(VI). There was no significant difference in the basal expression levels among all cell lines. However, we observed a significant, 2-fold decrease in Bcl-2 protein following recovery from Cr(VI) treatment in the CC1 cells, which was not observed in the DR cells (Figure 3). We also determined the protein expression of Bax basally and after genotoxin exposure, but found no difference among DR and CC cell lines (data not shown).
Fig. 3.
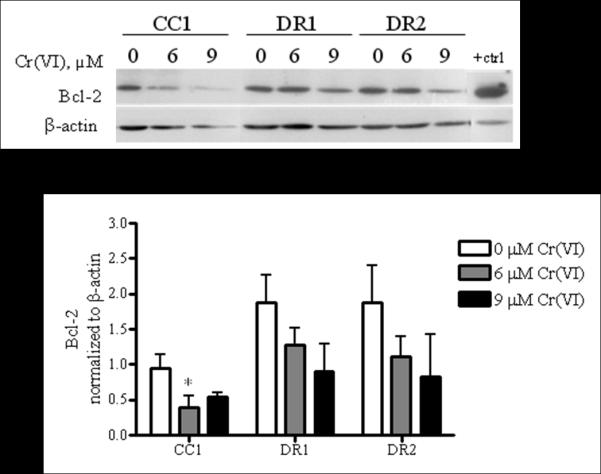
DR cells maintain Bcl-2 protein expression levels following post-Cr(VI) treatment recovery. CC1, DR1 and DR2 cells were treated with 0, 6, or 9 μM Cr(VI) for 24 h, followed by a 24 h recovery period in fresh DMEM. Total cell protein was extracted after recovery period. Proteins were separated by SDS-PAGE and Bcl-2 expression was detected by immunoblotting. The same amount of protein loading was confirmed by immunoblotting for β-actin. Data for 0 and 6 μM Cr(VI) exposure are mean +/− SE of three experiments, while 9 μM Cr(VI) exposure data are mean +/− SE of two experiments. All data are normalized to β-actin. * indicates a statistically significant difference from respective untreated control (p < 0.05)
3.2. DR cells are uniquely resistant to mitochondrial-mediated apoptosis after Cr(VI) exposure
To investigate the potential involvement of the death-receptor pathway of apoptosis in our death-resistant cell model system, each cell line was treated with or without 100 nM [24] of anti-Fas for 24 h, followed by a 24 h recovery period and analysis of cleaved caspase 3 protein expression by immunobloting (Supplemental Figure 2a). Our data show that the Fas ligand-mediated death-receptor pathway of apoptosis is not activated, as cleaved caspase 3 expression was not observed in the cells studied. Additionally, we investigated the expression levels of caspase 8 in our model system. Caspase 8 is critical for the activation of the death-receptor pathway of apoptosis and is activated upon cleavage [25]. We show that there is no decrease in caspase 8 expression after 24 h Cr(VI) treatment followed by a 24 h recovery period among all cell lines (Supplemental Figure 2b). These data are in keeping with previous reports from other laboratories, showing that fibroblasts are resistant to death-receptor-mediated apoptosis [26, 27].
The involvement of the mitochondrial-mediated pathway of apoptosis was assessed in the death-resistant cell model system, by investigating the dissipation of the mitochondrial-membrane potential (ΔΨm), as well as the release of the apoptogenic factors, cytochrome c and SMAC/DIABLO, from the mitochondrial inner membrane space (IMS) into the cytoplasm. Previous data from our lab using flow cytometric analysis of the JC-9 ΔΨm dependent dye show that BJ-hTERT parental cells exhibit dissipation of ΔΨm, resulting in a significant 1.7 and 4.9-fold increase in mitochondrial membrane depolarization following 24 h treatment with 9 μM Cr(VI) and 0.5 h treatment of 45 μM hydrogen peroxide (H2O2), respectively. In sharp contrast, the DR1 cells were unaffected by either treatment (Supplemental Figure 3). In light of these data, we determined the effect of Cr(VI) on the release of cytochrome c and SMAC/DIABLO from the mitochondrial IMS by immunofluorescent confocal microscopy.
As shown in Figure 4, cytochrome c in untreated CC1, DR1 and DR2 cells was localized to the mitochondria, as evidenced by the exclusive co-localization of the cytochrome c antibody with Mitotracker (far right panel, merge). Exposure of the CC1 cells to 6 μM Cr(VI) for 24 h induced a respective 3.3-fold increase in the mitochondrial release of cytochrome c into the cytosol, as compared to control. In contrast, cytochrome c remained predominantly localized to the mitochondria in the DR1 and DR2 cells following 24 h treatment with 6 μM Cr(VI), with just a 1.8 and 1.4-fold increase in cytosolic release as compared to untreated control, respectively. Similar results were observed when we measured the localization of SMAC/DIABLO following genotoxin exposure (Figure 5). When untreated, the CC1, DR1 and DR2 cells exhibited distinct mitochondrial localization of SMAC/DIABLO, as indicated by its exclusive co-localization with Mitotracker (far right panel, merge). However, following 6 μM Cr(VI) treatment, the CC1 cells showed a significant 11-fold increase in the release of SMAC/DIABLO into the cytosol, as compared to respective control and treated DR1 cells. Taken together, our data show that after Cr(VI) treatment, the release of both cytochrome c and SMAC/DIABLO was attenuated in the DR cells when compared to the CC1 cells.
Fig. 4.

DR cells show no release of cytochrome c from the inner mitochondrial membrane space following Cr(VI) treatment. CC1, DR1and DR2 cells were treated with 6 μM Cr(VI) for 24 h. After treatment, the cells were incubated with Mitotracker CMXRos, fixed and incubated with mouse anti-cytochrome c antibody and Alexa 488-conjugated goat anti-mouse secondary antibody, and DNA was stained with Hoechst. Cells were seeded on 8-well chamber slides prior to treatment. Mitochondria – red; cytochrome c – green; nucleus– blue. All images were taken on a Zeiss 510 laser scanning microscope at a magnification level of 63 X. Scale bar represents 10 microns. The release of cytochrome c as assessed by segmentation threshold analysis of mitochondrial versus cytosolic localization was quantified using Image Pro-Plus software. Data are mean +/− SE of three experiments and are normalized to Mitotracker CMXRos.
Fig. 5.

DR cells show no release of SMAC/DIABLO following Cr(VI) treatment. CC1, DR1and DR2 cells were treated with 6 μM Cr(VI) for 24 h. After treatment, the cells were incubated with Mitotracker CMXRos, fixed and incubated with mouse anti-SMAC/DIABLO antibody and Alexa 488-conjugated goat anti-mouse secondary antibody, and DNA was stained with Hoechst. Cells were seeded on 8-well chamber slides prior to treatment. Mitochondria – red; SMAC/DIABLO – green; nucleus – blue. All images were taken on a Zeiss 510 laser scanning microscope at a magnification level of 63 X. Scale bar represents 10 microns. The release of SMAC/DIABLO as assessed by segmentation threshold analysis of mitochondrial versus cytosolic localization was quantified using Image Pro-Plus software. Data are mean +/− SE of three experiments and are normalized to Mitotracker CMXRos. * indicates statistically significant difference from respective untreated control, ^ indicates statistically significant difference from DR1 cells treated with 6 μM Cr(VI) (p < 0.05)
3.3. DR cells display less basal mtDNA damage
Several studies have shown that in response to certain DNA damaging agents, mitochondrial biogenesis can be induced as a survival mechanism in an effort to repopulate damaged cells with functional mitochondria [28-32]. mtDNA content correlates with mitochondrial quantity, therefore, we measured mtDNA content before and after 24 h exposure to 6 μM Cr(VI), and found no significant difference among the cell lines (Figure 6a).
Fig. 6.

DR cells display no change in mtDNA copy number, but intrinsically less mtDNA damage. CC1, DR1, and DR2 cells were treated with 0 or 6 μM Cr(VI) for 24 h. Total DNA was extracted and quantified, (a) mtDNA and nuclear (nu) DNA copy number were determined by RT-PCR. Primers were generated for the 108-bp mitochondrial-encoded ND1 gene and an 89-bp β2 microglobulin (β2m) gene to assess mtDNA and nuDNA, respectively. Values are averages of three determinations obtained in one RT-PCR run. The copy number/cell was calculated by dividing nuDNA copy number by two (assuming the cells were diploid). mtDNA copy number was expressed as mtDNA copies/cell. (b) mtDNA damage was measured by the amplification of an 8.9-kb mtDNA target by QPCR. mtDNA damage was inversely proportional to amount of amplification , and was expressed as amount of 8.9-kb DNA/106 ND1 copies amplified. * indicates a statistically significant difference from CC1 cells, regardless of Cr(VI) treatment. Data are mean +/− SE of three experiments (0 μM Cr(VI), p = 0.0516; 6 μM Cr(VI) p=0.0430).
Loss of mitochondrial function, growth arrest, and apoptosis have been correlated with mtDNA damage [33]. Therefore, to further investigate the stability and potential involvement of mtDNA in death-resistance in our model system, we measured mtDNA damage before and after Cr(VI) treatment (Figure 6b). QPCR amplification of a primer designed to detect an 8.9-kb product along the circular mtDNA molecule is blocked by the presence of damage; therefore amplification is inversely proportional to mtDNA damage. Our data suggest that there is no direct effect of Cr(VI) exposure on mtDNA damage, as treatment caused no difference in mtDNA damage as compared to the respective untreated control among all cell lines. Intriguingly, however, we found that the CC1 cells display 20.5% and 10.8% less basal amplification of the 8.9-kb mtDNA product per 1 million copies of the ND1 gene, as compared to the DR1 and DR2 cells, respectively. These data indicate that the DR1 cells have significantly less intrinsic/steady-state mtDNA damage as compared to the CC1 cells, while the DR2 cells exhibit a moderate difference in damage as compared to the CC1 cells (Figure 6a).
4. Discussion
The selection of cells with the ability to survive after exposure to apoptogenic levels of a DNA damaging agent may yield a precursor pool of cells from which neoplastic variants can emerge [19]. The dysregulation of mitochondrial-mediated cellular mechanisms such as apoptosis, are frequently observed in malignant tumors, thus highlighting a potential connection between mitochondria, death-resistance and carcinogenesis [28, 30]. We postulate that survival after genotoxic insult may involve the selection of cells with mitochondrial dysregulation, leading to death-resistance. Our data demonstrate that the DR cells, as compared to the death-susceptible cells, are resistant to mitochondrial-mediated apoptosis and display differences in intrinsic mtDNA damage that correlate with death-resistance. The results of the present study provide insight into the possible role of mitochondrial dysregulation and apoptosis-resistance in the mechanisms of early stage carcinogenesis.
The use of human diploid fibroblasts in this study enables us to investigate the earliest genotoxin-induced events that may occur in normal cells, yielding a pro-survival phenotype. As part of the intricate cellular microenvironmental milieu, fibroblasts are associated with pathological conditions such as fibrosis and carcinogenesis, as they affect the behavior of neighboring cells, and contribute to the maintenance of the extracellular matrix [34, 35].
Our model system was derived from survivors of Cr(VI)-treated, immortalized human diploid fibroblasts (BJ-hTERT). BJ-hTERT cells were exposed to a single dose of Cr(VI), yielding 1% cell survival, from which a population of surviving (B-5Cr) cells was expanded [19]. The B-5Cr cells exhibited enhanced long term survival and growth potential (also observed in the subclonal populations examined in the current study; Supplemental Figure 4), resistance to apoptosis induction from a variety of stimuli (indicated by phosphtidlyserine translocation), and a stable p53 response [19]. Of note, the resistant phenotype of the B-5Cr cells diminished with time in cell culture. This phenomenon was attributed to the effect of cumulative cell divisions in culture and extensive expansion. Similar progressive phenotypic changes have been observed in human lung fibroblasts [36]. Therefore, to ensure that the observed resistance to Cr(VI)-induced cell death in the B-5Cr cells was due to acquired apoptosis-resistance and not a result of clonogenic selection pressure, early passages of the B-5Cr cells, that still exhibited the pro-survival phenotype, were subcloned to establish the DR1 and DR2 cell lines used in the present study. Untreated BJ-hTERT (B-0Cr) cells were also used to derive the clonogenic control cell line, CC1.
It is critical to emphasize that the observed death-resistant phenotype in our model system is a consequence of a single toxic and genotoxic exposure. In vitro models of cellular death-resistance generated via long-term, chronic, and slowly escalating exposure doses are advantageous when studying the role of altered transport and intracellular pharmacodynamics in response to cellular genotoxic stress [37-40]. However, our system is unique in that it models initial molecular events that occur in a normal cell that survived a single, acute, initiating genotoxic challenge. While the selection model in this study was generated by Cr(VI) treatment, it also exhibits a cross-resistance to the well-known chemotherapeutic agent, cisplatin, as well as to H2O2 (Supplemental Figures 1 and 3). This is in contrast to published reports where Cr(VI)-resistant cell models were generated and exhibit resistance only to Cr(VI) (reviewed in [41]). This underlines the utility of our cell model to assess loss of therapeutic efficacy and toxicity of chemotherapeutic agents in cells exhibiting the earliest hallmarks of cancer.
Disruption of mitochondrial-mediated apoptosis signaling in malignant tumors has been directly associated with the enhanced expression of Bcl-2 [23]. The requirement of Bcl-2 for resistance to Cr(VI)-induced death has been highlighted in studies in SV40-transformed lung epithelial cells (BEAS-2B) by Azad et al [42]. Our data are in keeping with these studies in that a maintenance of Bcl-2 protein expression correlated with Cr(VI)-induced cleaved caspase 3-mediated death-resistance as compared to a significant decrease in protein expression in the death-sensitive cells (Figure 3). Studies have also shown a relationship between Bcl-2 degradation and Cr(VI)-induced ROS production [14, 43], however; this correlation was not observed in our system. Moreover, it is generally accepted that ROS are mediators of cellular damage, including mtDNA damage and consequently mitochondrial function [44, 45]. Along these lines, a recent study by Indran et al. has demonstrated that the basal and endogenous production of ROS in cancer cells can be reduced as a consequence of hTERT overexpression and lead to the improvement of mitochondrial function [45]. As the cells in our system are derived from hTERT-immortalized fibroblasts, and exhibit phenotypic alterations suggestive of dysregulated mitochondrial-mediated cell death signaling, we measured the levels of hTERT protein expression. However, we found no difference basally or after genotoxin exposure among all cell lines, suggesting lack of TERT involvement in the observed DR phenotype (Supplemental Figure 5a).
In an effort to further investigate the possible link between mitochondrial-mediated apoptosis resistance, the observed phenotypic alterations, and a potential redox effect in our model system, we assessed mtDNA damage in the cell lines before and after Cr(VI) treatment. Our data were obtained by long-range QPCR amplification and analysis of an 8.9-kb mtDNA product that is inversely proportional to mtDNA damage (Figure 6b). Interestingly, we found no direct effect of Cr(VI) on mtDNA damage among all cell lines studied. However, in the absence of any Cr(VI) treatment, both DR1 and DR2 cells exhibited less inherent mtDNA damage as compared to the CC1 cells. The reduced levels of mtDNA damage observed in the DR cells uncover a potentially striking intrinsic mechanism for resistance. These data imply that the ability of a population of cells to maintain a more stable mitochondrial genome (or potentially enhanced ROS buffering capacity), may increase their ability to survive in the face of cellular oxidative stress. Therefore, we investigated the possibility that the DR cells may exhibit restricted generation of ROS, which would result in lower reduced intrinsic mtDNA damage. However, we found no difference in the basal levels of total cellular ROS, total cellular O2−, or mitochondria-specific O2− among all cell lines (data not shown). Similar results were reported in a study by Kulawiec et al., where breast cancer cell lines containing mutated mtDNA, as compared to wild-type cells containing undamaged mtDNA exhibited resistance to etopside-induced apoptosis, but demonstrated no association with increased ROS production [46]. Furthermore, the authors found that mutated mtDNA leads to the constitutive activation of the P13K/Akt pathway [46]. Further data from our laboratory show a significant induction of phosphorylated-Akt serine-473 protein expression following Cr(VI) genotoxic exposure in the DR1 cells, which was downregulated in the BJ-hTERT cells (Supplemental Figure 6). Of note, constitutive activation of Akt1, by myristolated Akt transfection, did not induce death-resistance in the BJ-hTERT cells (data not shown), potentially pointing to a further link between mtDNA dysregulation and death resistance in the DR1 cells, which exhibit decremental basal levels of mtDNA damage, as compared to the control cells.
5. Conclusion
Here we focused on mitochondrial involvement in the “black box” of early stage carcinogenesis and cellular death-resistance. Mitochondria are heterogeneous in nature, varying in content from cell to cell, and varying in genetic material from mitochondria to mitochondria. It is known that changes in critical cellular functions (ie. oxidative phosphorylation) can result from damaged or mutated mtDNA, and it has been hypothesized that these alterations correlate with tumorigenesis [47, 48]. Therefore, our hypothesis is supported by the idea that a selected pool of cells with an innately enhanced survival capacity (ie. lower ROS production or enhanced ROS buffering capacity), may support or lead to the generation of more robust mitochondria; and in the face of subsequent oxidative stress, these cells and their dysregulated mitochondria, are able to survive.
The present study provides evidence that resistance to mitochondrial-mediated apoptosis, dysregulation of mitochondrial associated proteins, and the stability of mtDNA, may be among the first phenotypic alterations observed in early stage carcinogenesis. We demonstrate that heritable resistance to apoptosis, one of the hallmarks of cancer, can be acquired in normal diploid human cells, following a single toxic and genotoxic exposure [1]. If death-resistant cells also harbor mutations, the inappropriate survival and replication of such genomically unstable cells may predispose them to neoplastic progression. Investigations into the potential role of mitochondria in early stage carcinogenesis are critical to our understanding of origins of tumor emergence and may implicate mitochondrial dysregulation as a carcinogenic biomarker.
Supplementary Material
Highlights.
> Normal cells made death-resistant after a single exposure to Cr(VI)
> Resistant cells retain Bcl-2 expression and show no caspase 3 cleavage after Cr(VI)
> Resistant cells do not undergo mitochondrial-mediated apoptosis after Cr(VI) exposure
> Resistant cells exhibit less basal mtDNA damage
> Mitochondrial dysregulation may be indicative of early stage carcinogenesis
Acknowledgments
The authors acknowledge Dr. Popratiloff for his help with the confocal microscopy studies and analysis, Dr. O’Brien for helpful discussions, and Dr. Lee for the generous use of his cell culture facility. We also thank Dr. Pritchard, Dr. Beaver and Dr. Lizardo-Escano for their earlier contributions to this study, as well as Dr. Lal-Nag and Dr. Camilli for their helpful insight.
Role of Funding Source This work was supported by the National Institutes of Health [RO1CA107972 to S.C., RO1CA107972 supplement to K.N., R01ES05304 and R01ES09961 to S.P.]. The funding source had no involvement in the conduct of this research or the preparation of this article.
Abbreviations
- Cr(VI)
hexavalent chromium
- TGA
terminal growth arrest
- ROS
reactive oxygen species
- SMAC/DIABLO
second mitochondria-derived activator of caspases/direct inhibitor of apoptosis binding protein
- hTERT
human telomerase
- mtDNA
mitochondrial DNA
- ΔΨm
mitochondrial membrane potential
- ND1
NADH dehydrogenase 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- [2].Gavin IM, Gillis B, Arbieva Z, Prabhakar BS. Identification of human cell responses to hexavalent chromium. Environ Mol Mutagen. 2007;48:650–657. doi: 10.1002/em.20331. [DOI] [PubMed] [Google Scholar]
- [3].Chromium, nickel and welding. IARC Monogr Eval Carcinog Risks Hum. 1990;49:1–648. [PMC free article] [PubMed] [Google Scholar]
- [4].NTP 11th Report on Carcinogens. Rep Carcinog. 2005:1–A32. [PubMed] [Google Scholar]
- [5].Jennette KW. Microsomal reduction of the carcinogen chromate produces chromium(V) Journal of the American Chemical Society. 1982;104:874–875. [Google Scholar]
- [6].Blankenship LJ, Carlisle DL, Wise JP, Orenstein JM, Dye LE, 3rd, Patierno SR. Induction of apoptotic cell death by particulate lead chromate: differential effects of vitamins C and E on genotoxicity and survival. Toxicol Appl Pharmacol. 1997;146:270–280. doi: 10.1006/taap.1997.8237. [DOI] [PubMed] [Google Scholar]
- [7].Blankenship LJ, Manning FC, Orenstein JM, Patierno SR. Apoptosis is the mode of cell death caused by carcinogenic chromium. Toxicol Appl Pharmacol. 1994;126:75–83. doi: 10.1006/taap.1994.1092. [DOI] [PubMed] [Google Scholar]
- [8].Ha L, Ceryak S, Patierno SR. Chromium (VI) activates ataxia telangiectasia mutated (ATM) protein. Requirement of ATM for both apoptosis and recovery from terminal growth arrest. J Biol Chem. 2003;278:17885–17894. doi: 10.1074/jbc.M210560200. [DOI] [PubMed] [Google Scholar]
- [9].Shi Y. A structural view of mitochondria-mediated apoptosis. Nat Struct Biol. 2001;8:394–401. doi: 10.1038/87548. [DOI] [PubMed] [Google Scholar]
- [10].Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- [11].Gimenez-Bonafe P, Tortosa A, Perez-Tomas R. Overcoming drug resistance by enhancing apoptosis of tumor cells. Curr Cancer Drug Targets. 2009;9:320–340. doi: 10.2174/156800909788166600. [DOI] [PubMed] [Google Scholar]
- [12].Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- [13].Srinivasan A, Foster LM, Testa MP, Ord T, Keane RW, Bredesen DE, Kayalar C. Bcl-2 expression in neural cells blocks activation of ICE/CED-3 family proteases during apoptosis. J Neurosci. 1996;16:5654–5660. doi: 10.1523/JNEUROSCI.16-18-05654.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Azad N, Iyer AK, Manosroi A, Wang L, Rojanasakul Y. Superoxide-mediated proteasomal degradation of Bcl-2 determines cell susceptibility to Cr(VI)-induced apoptosis. Carcinogenesis. 2008;29:1538–1545. doi: 10.1093/carcin/bgn137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- [16].Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 2002;286:127–134. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- [17].LeDoux SP, Wilson GL, Beecham EJ, Stevnsner T, Wassermann K, Bohr VA. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis. 1992;13:1967–1973. doi: 10.1093/carcin/13.11.1967. [DOI] [PubMed] [Google Scholar]
- [18].Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- [19].Pritchard DE, Ceryak S, Ramsey KE, O’Brien TJ, Ha L, Fornsaglio JL, Stephan DA, Patierno SR. Resistance to apoptosis, increased growth potential, and altered gene expression in cells that survived genotoxic hexavalent chromium [Cr(VI)] exposure. Mol Cell Biochem. 2005;279:169–181. doi: 10.1007/s11010-005-8292-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lal MA, Bae D, Camilli TC, Patierno SR, Ceryak S. AKT1 mediates bypass of the G1/S checkpoint after genotoxic stress in normal human cells. Cell Cycle. 2009;8:1589–1602. doi: 10.4161/cc.8.10.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- [22].Chun G, Bae D, Nickens K, O’Brien TJ, Patierno SR, Ceryak S. Polo-like kinase 1 enhances survival and mutagenesis after genotoxic stress in normal cells through cell cycle checkpoint bypass. Carcinogenesis. 2010;31:785–793. doi: 10.1093/carcin/bgq014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hasenjager A, Gillissen B, Muller A, Normand G, Hemmati PG, Schuler M, Dorken B, Daniel PT. Smac induces cytochrome c release and apoptosis independently from Bax/Bcl-x(L) in a strictly caspase-3-dependent manner in human carcinoma cells. Oncogene. 2004;23:4523–4535. doi: 10.1038/sj.onc.1207594. [DOI] [PubMed] [Google Scholar]
- [24].Mahalingam D, Natoni A, Keane M, Samali A, Szegezdi E. Early growth response-1 is a regulator of DR5-induced apoptosis in colon cancer cells. Br J Cancer. 2010;102:754–764. doi: 10.1038/sj.bjc.6605545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta. 2011;1813:558–563. doi: 10.1016/j.bbamcr.2011.01.026. [DOI] [PubMed] [Google Scholar]
- [26].Nesterov A, Nikrad M, Johnson T, Kraft AS. Oncogenic Ras sensitizes normal human cells to tumor necrosis factor-alpha-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2004;64:3922–3927. doi: 10.1158/0008-5472.CAN-03-2219. [DOI] [PubMed] [Google Scholar]
- [27].Tanaka T, Yoshimi M, Maeyama T, Hagimoto N, Kuwano K, Hara N. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur Respir J. 2002;20:359–368. doi: 10.1183/09031936.02.00252602. [DOI] [PubMed] [Google Scholar]
- [28].Fu X, Wan S, Lyu YL, Liu LF, Qi H. Etoposide induces ATM-dependent mitochondrial biogenesis through AMPK activation. PLoS ONE. 2008;3:e2009. doi: 10.1371/journal.pone.0002009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nisoli E, Clementi E, Moncada S, Carruba MO. Mitochondrial biogenesis as a cellular signaling framework. Biochem Pharmacol. 2004;67:1–15. doi: 10.1016/j.bcp.2003.10.015. [DOI] [PubMed] [Google Scholar]
- [30].Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- [31].Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, Cantoni O, Carruba MO, Moncada S, Clementi E. Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci U S A. 2004;101:16507–16512. doi: 10.1073/pnas.0405432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xu J, Shi C, Li Q, Wu J, Forster EL, Yew DT. Mitochondrial dysfunction in platelets and hippocampi of senescence-accelerated mice. J Bioenerg Biomembr. 2007;39:195–202. doi: 10.1007/s10863-007-9077-y. [DOI] [PubMed] [Google Scholar]
- [33].Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McAnulty RJ. Fibroblasts and myofibroblasts: their source, function and role in disease. Int J Biochem Cell Biol. 2007;39:666–671. doi: 10.1016/j.biocel.2006.11.005. [DOI] [PubMed] [Google Scholar]
- [35].Vaheri A, Enzerink A, Rasanen K, Salmenpera P. Nemosis, a novel way of fibroblast activation, in inflammation and cancer. Exp Cell Res. 2009;315:1633–1638. doi: 10.1016/j.yexcr.2009.03.005. [DOI] [PubMed] [Google Scholar]
- [36].Pritchard DE, Ceryak S, Ha L, Fornsaglio JL, Hartman SK, O’Brien TJ, Patierno SR. Mechanism of apoptosis and determination of cellular fate in chromium(VI)-exposed populations of telomerase-immortalized human fibroblasts. Cell Growth Differ. 2001;12:487–496. [PubMed] [Google Scholar]
- [37].Grishko VI, Rachek LI, Spitz DR, Wilson GL, LeDoux SP. Contribution of mitochondrial DNA repair to cell resistance from oxidative stress. J Biol Chem. 2005;280:8901–8905. doi: 10.1074/jbc.M413022200. [DOI] [PubMed] [Google Scholar]
- [38].Lu YY, Yang JL. Long-term exposure to chromium(VI) oxide leads to defects in sulfate transport system in Chinese hamster ovary cells. J Cell Biochem. 1995;57:655–665. doi: 10.1002/jcb.240570410. [DOI] [PubMed] [Google Scholar]
- [39].Rodrigues CF, Urbano AM, Matoso E, Carreira I, Almeida A, Santos P, Botelho F, Carvalho L, Alves M, Monteiro C, Costa AN, Moreno V, Alpoim MC. Human bronchial epithelial cells malignantly transformed by hexavalent chromium exhibit an aneuploid phenotype but no microsatellite instability. Mutat Res. 2009;670:42–52. doi: 10.1016/j.mrfmmm.2009.07.004. [DOI] [PubMed] [Google Scholar]
- [40].Son KH, Zhang M, Rucobo E, Nwaigwe D, Montgomery F, Leffert H. Derivation and study of human epithelial cell lines resistant to killing by chromium trioxide. J Toxicol Environ Health A. 2004;67:1027–1049. doi: 10.1080/15287390490447304. [DOI] [PubMed] [Google Scholar]
- [41].Nickens KP, Patierno SR, Ceryak S. Chromium genotoxicity: A double-edged sword. Chem Biol Interact. 2010;188:276–288. doi: 10.1016/j.cbi.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Azad N, Iyer AK, Wang L, Lu Y, Medan D, Castranova V, Rojanasakul Y. Nitric oxide-mediated bcl-2 stabilization potentiates malignant transformation of human lung epithelial cells. Am J Respir Cell Mol Biol. 2010;42:578–585. doi: 10.1165/rcmb.2009-0094OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Son YO, Hitron JA, Wang X, Chang Q, Pan J, Zhang Z, Liu J, Wang S, Lee JC, Shi X. Cr(VI) induces mitochondrial-mediated and caspase-dependent apoptosis through reactive oxygen species-mediated p53 activation in JB6 Cl41 cells. Toxicol Appl Pharmacol. 2010;245:226–235. doi: 10.1016/j.taap.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen ZX, Velaithan R, Pervaiz S. mitoEnergetics and cancer cell fate. Biochim Biophys Acta. 2009;1787:462–467. doi: 10.1016/j.bbabio.2008.12.009. [DOI] [PubMed] [Google Scholar]
- [45].Indran IR, Hande MP, Pervaiz S. hTERT overexpression alleviates intracellular ROS production, improves mitochondrial function, and inhibits ROS-mediated apoptosis in cancer cells. Cancer Res. 2011;71:266–276. doi: 10.1158/0008-5472.CAN-10-1588. [DOI] [PubMed] [Google Scholar]
- [46].Kulawiec M, Owens KM, Singh KK. Cancer cell mitochondria confer apoptosis resistance and promote metastasis. Cancer Biol Ther. 2009;8:1378–1385. doi: 10.4161/cbt.8.14.8751. [DOI] [PubMed] [Google Scholar]
- [47].Fang H, Shen L, Chen T, He J, Ding Z, Wei J, Qu J, Chen G, Lu J, Bai Y. Cancer type-specific modulation of mitochondrial haplogroups in breast, colorectal and thyroid cancer. BMC Cancer. 2010;10:421. doi: 10.1186/1471-2407-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.