

Abstract
MMP-9 contributes to the pathogenesis of chronic inflammatory diseases and cancer. Thus, identifying targetable components of signaling pathways that regulate MMP-9 expression may have broad therapeutic implications. Our previous studies revealed a nexus between metalloproteinases and prostanoids whereby MMP-1 and MMP-3, commonly found in inflammatory and neoplastic foci, stimulate macrophage MMP-9 expression via the release of TNF-α and subsequent induction of cyclooxygenase-2 (Cox-2), and PGE2 engagement of EP4 receptor. In the present studies, we determined whether MMP-induced Cox-2 expression was coupled to the expression of PGE synthase family members. We found that MMP-1 and MMP-3-dependent release of TNF-α induced rapid and transient expression of Egr-1 in macrophages followed by sustained elevation in mPGES-1 expression. Metalloproteinase-induced PGE2 levels and MMP-9 expression were markedly attenuated in macrophages in which mPGES-1 was silenced, thereby identifying mPGES-1 as a therapeutic target in the regulation of MMP-9 expression. Finally, the induction of mPGES-1 was regulated, in part, through a positive feedback loop dependent on PGE2 binding to EP4. Thus, in addition to inhibiting macrophage MMP-9 expression, EP4 antagonists emerge as potential therapy to reduce mPGES-1 expression and PGE2 levels in inflammatory and neoplastic settings.
Introduction
MMP family members play an essential role in tissue remodeling via their ability to degrade components of the extracellular matrix including collagens and adhesive glycoproteins (1,2). However, it is apparent that these endopeptidases do more than simply degrade extracellular matrix. For example, the biological actions of MMP-9, a type IV collagenase, result from its ability to proteolytically modify the activities of cytokines and chemokines, growth factors, and proteinase inhibitors (3,4). Evidence derived from mouse models indicates that MMP-9 participates in a number of physiologic processes (5–9), as well as the pathogenesis of airway disease (10), bullous pemphigoid (11), cancer (12,13) and both occlusive (14,15) and aneurysmal vascular diseases (16). Thus, identifying targetable components of pathways that regulate MMP-9 expression has significant therapeutic implications.
MMP-9 expression, generally absent in normal tissues, is induced by inflammatory cytokines, growth factors, LPS, and extracellular matrix components (3). Results of our recent experiments defined a complex mechanism by which MMP-1 and MMP-3, commonly found in both inflamed and neoplastic tissues, stimulate macrophage MMP-9 expression (17). Proteinase-induced MMP-9 expression is dependent on the release of TNF-α, induction of Cox-2 expression and PGE2 engagement of the EP4 prostanoid receptor.
Although it is well established that Cox activity plays a major role in the synthesis of PGE2, the isomerization of Cox-derived PGH2, an unstable endoperoxide, to PGE2 is catalyzed by a family of PGE synthases including mPGES-1, mPGES-2 and cytoplasmic PGES (cPGES/p23) (18,19). Although cPGES/p23 and mPGES-2 are constitutively expressed, they may not play a major role in PGE2 synthesis in vivo (20,21). In contrast, mPGES-1 is induced by inflammatory stimuli and appears to be functionally coupled to Cox-2 expression in a variety of pathophysiologic settings (22–25). To date, the regulatory role of MMPs on the expression of PGE synthases has not been explored.
In studies reported here, we determined whether MMP-1 and MMP-3 modulation of the Cox-2→PGE2→EP4 receptor axis was coupled to changes in the PGE synthase expression in macrophages. We found that MMP-1 and MMP-3 selectively induced mPGES-1 expression, which was dependent on the release of TNF-α and induction of Egr-1 expression. The critical role of mPGES-1 in both MMP and LPS-induced MMP-9 expression was demonstrated in macrophages transfected with mPGES-1 siRNA. Elevated mPGES-1 expression and PGE2 levels were regulated by a positive feedback loop that was dependent on the EP4 prostanoid receptor. Thus, in addition to inhibiting macrophage MMP-9 expression, EP4 antagonists emerge as potential therapeutic agents to reduce mPGES-1 expression and PGE2 levels in inflammatory or neoplastic settings.
Materials and Methods
Macrophages
Thioglycollate-elicited peritoneal macrophages were obtained from Swiss Webster mice by the method of Edelson and Cohn (26) as described previously (27). Mice were injected IP (3 ml/mouse) with 3% Brewer Thioglycollate Medium (DIFCO). Four days later, cells were harvested by lavage with cold Dulbecco's PBS. Peritoneal cells were recovered by centrifugation and resuspended in DMEM supplemented with 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml) and 4 mM glutamine, and plated into multi-well plates. Cells were allowed to adhere for 4 h and then washed free of nonadherent cells. For serum free conditions, macrophages were grown in DMEM supplemented with antibiotics, glutamine and 0.1% BSA (Sigma Aldrich) containing low levels of endotoxin (≤ 0.1 ng/mg; LE-BSA). Cellgro® DMEM and heat inactivated FBS were obtained from Mediatech, Inc. Antibiotics and glutamine were obtained from either Mediatech, Inc. or Gibco/Life Technologies. The murine macrophage cell line RAW264.7 (28) was obtained from American Type Culture Collection, and maintained as adherent cultures in DMEM-10% FBS. All animal studies described in this report have been reviewed and approved by the Weill Cornell Medical College Institutional Animal Care and Use Committee.
MMPs
Human recombinant pro-MMP-1 and an active recombinant fragment of MMP-3 were obtained from Calbiochem. Pro-MMP-1 was activated prior to use by incubation with 1 mM 4-amino-phenyl-mercuric acetate for 2 h at 37°C, and dialyzed as previously described (17). The conversion of pro-MMP-1 to its active form is associated with a loss of the pro-domain (~10 kDa), which was verified by Western blot (data not shown).
Preparation of cell lysates
Cell lysates utilized in the analysis of Cox-2 expression were prepared by adding 1× SDS sample buffer directly to washed macrophage monolayers. In experiments designed to monitor levels of phosphorylated and total MAPKerk1/2, cells were lysed in TBS containing 2 mM EDTA, 1% Triton X-100, 25 mM β-glycerophosphate, 1 mM sodium vanadate, 2 mM sodium pyrophosphate, 1 mM PMSF and 10 μg/ml aprotinin. Lysates were centrifuged (14,000 × g) for 20 min at 4°C. The supernatants were recovered, normalized for protein and mixed with SDS sample buffer with β-mercaptoethanol and boiled for 5 min. Equal amounts of cell lysates were applied to gels based on protein content.
Western blots
Cell lysates were electrophoresed in 4–15% polyacrylamide gels and proteins were transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked in 5% defatted milk in TBST, washed in TBST and incubated 18 h in blocking buffer containing affinity purified goat IgG raised against a peptide from the C-terminus of human Cox-2 (0.2 μg/ml; Santa Cruz Biotechnology, Inc.) or rabbit anti-phosphospecific p44/p42 MAPK (i.e. MAPKerk1/2) IgG (75 ng/ml; Cell Signaling Technology). Membranes were washed 2× in TBST and incubated 1 h in blocking buffer containing affinity purified rabbit anti-goat IgG conjugated to horseradish peroxidase (HRP) (0.1 μg/ml; R&D Systems) or goat anti-rabbit IgG conjugated to HRP (0.3 μg/ml; BioRad Laboratories). The membranes were washed 3× in TBST and bound HRP was visualized utilizing chemiluminescence (Pierce/Thermo Scientific). In the case of membranes probed with anti-phosphospecific MAPKerk1/2, following visualization of bound HRP, the membranes were stripped in 62.5 mM Tris buffer (pH 6.7) containing 100 mM β-mercaptoethanol and 2% SDS for 30 min at 50°C, washed and probed for total MAPKerk1/2 (Cell Signaling Technology).
Macrophage conditioned media were electrophoresed in gradient gels and proteins were transferred to a PVDF membrane. The membrane was placed in blocking buffer for 1 h, washed in TBST and incubated 18 h in blocking buffer containing rabbit anti-mouse MMP-9 IgG (0.4 μg/ml; Abcam). Membranes were washed 2× in TBST and incubated 1 h in blocking buffer containing goat anti-rabbit IgG conjugated to HRP. Densitometric analysis of scanned Western blots were determined utilizing Scion Image software (Scion Corporation) and normalized for levels of GAPDH, which served as a loading control. Data are reported as relative density units (RDU).
Gene knockdowns
mPGES-1: RAW264.7 macrophages were transfected with ONTARGETplus SMARTpool mPGES-1 siRNA or nonspecific siRNA (Dharmacon/Thermo Scientific) utilizing HiPerFect transfection reagent according to manufacturer's recommended protocols (Qiagen). Cells were plated into a 24-well (6 × 104 cells/well) plate and incubated 24 h with transfection complexes before exposure to MMPs or LPS. TNF-α: RAW264.7 macrophages were transfected with TNF-α siRNA or nonspecific siRNA (Santa Cruz) according to the manufacturer's protocol, as described previously (17). EP4: Macrophages were transfected with siGENOME SMARTpool EP4 or nonspecific (NS) siRNA (Thermo Scientific/Dharmacon) utilizing Gene Porter 3000 transfection reagent (Genlantis/Gene Therapy Systems, Inc.) according to manufacturer's instructions. Macrophages were aliquoted into a 12-well plate (2.5 × 105 cells/well) in antibiotic free DMEM-10% FBS and incubated 18 h. The next day, the transfection lipoplexes containing 50 nM siRNA were added to the cells, and incubated 4 h. After the initial 4 h incubation, an equal volume of DMEM-20% FBS was added, and cells were incubated an additional 48 h. For each gene, PCR was utilized to determine the extent of knockdown.
PCR analysis
RNA was prepared using Trizol reagent kits (Invitrogen). RNA (2 μg) was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Invitrogen) and oligo d(T)16 primer or random hexamers. The resulting cDNA was then used for amplification. The primers and conditions for PCR analysis of murine Cox-2, MMP-9, EP4 and actin have been previously described (29). Primers for murine mPGES-1, mPGES-2 and Egr-1 were designed utilizing the Primer 3 program and were ordered from Sigma. The resulting PCR products were verified by sequencing (Cornell University Sequencing Facility). The primers for mPGES-1 were: forward 5'-CCTTGAGCTGACAGCCTACC-3' (nucleotides 1484–1503) and reverse 5'-CAGCCTAATGTTCAGCGACA-3' (nucleotides 2048–2029). Primers for mPGES-2 were: forward 5'-ACTTCCACTCCCTGCCCTAT-3' (nucleotides 744–763) and reverse 5'-GCCCTCACGGACAATGTAGT-3' (nucleotides 1186–1167). The primers for Egr-1 were: forward 5'-CTCCCTCACTGCGTCTAAGG-3' (nucleotides 552–573) and reverse 5'-CAGGGTCACTTTCCAGGTGT-3' (nucleotides 826–805).
DNA (1–2μl) and primers (400 nM) were added to Platinum® PCR SuperMix (final volume 25μl; Invitrogen) in a thermocycler under the following conditions. mPGES-1: denature for 30 sec at 94°C, anneal for 30 sec at 55°C, and extend for 1 min at 72°C; repeat for 35 cycles with a final extension for 10 min at 72°C. mPGES-2: denature for 20 sec at 94°C, anneal for 20 sec at 65°C, and extend for 30 sec at 72°C; repeat for 35 cycles with a final extension for 10 min at 72°C. Egr-1: denature for 20 sec at 94°C, anneal for 20 sec at 65°C, and extend for 30 sec at 72°C; repeat for 35 cycles with a final extension for 10 min at 72°C. PCR products were electrophoresed in a 1% agarose gel with either 0.5 μg/ml ethidium bromide or 1× GelRed™ (Biotium; Hayward, CA) and photographed under UV light. PCR conditions for the target genes were optimized to ensure that experimental samples, excluding exuberant positive controls, are in the linear range when scanned. Densitometric analysis of the scanned target mRNAs were determined and normalized for levels of actin mRNA, which served as a loading control. Data are reported as RDU.
For real-time PCR analysis, the reaction volume was 20 μl and contained 5 μl cDNA, 2× SYBR Green PCR master mix, and forward and reverse primers as described previously (30). Real-time PCR primers for mPGES-1 were forward 5'- TCCCATTTGGAGCCACTTAC-3' (nucleotides 730–749) and reverse 5'-ATGGCTCTGTTGGTCAATCC- 3' (nucleotides 844–863). Primers for GAPDH were forward: 5'-AATGTGTCCGTCGTGGATCT-3' (nucleotides 759–778) and reverse: 5'- CATCGAAGGTGGAAGAGTGG-3' (nucleotides 914–933). Levels of mPGES-1 mRNA were normalized to GAPDH mRNA.
Determination of PGE2 levels in macrophage conditioned media
The concentrations of PGE2 in conditioned media were determined utilizing the PGE2 EIA kit (monoclonal) (Cayman Chemical).
Evaluation of cellular toxicity
To rule out toxic effects of the inhibitors utilized in these studies, cellular morphology and/or protein content following incubation are regularly monitored. In addition, we test for potential cytotoxicity by monitoring mitochondrial dehydrogenase activity utilizing a MTT assay kit (Sigma-Aldrich). Levels of mitochondrial dehydrogenase activity were not significantly reduced following exposure to any of the inhibitors over the time periods described in the manuscript.
Statistics
Mean RDUs for various genes and proteins, and levels of PGE2 in cellular conditioned media were compared utilizing single or two-factor analysis of variance, depending on the particular experimental design. Following the determination of significant differences between the means of a given experiment, subsequent pair-wise comparisons were performed utilizing Newman-Keuls multiple range testing (31).
Results
MMP-1 and MMP-3 induce mPGES-1 in macrophages
Cox-2-derived PGH2 is converted to PGE2 by a family of PGE synthases including mPGES-1, mPGES-2 and cPGES (19). In studies reported here, we determined whether MMP-induced Cox-2 expression in macrophages and elevated PGE2 secretion (17) were coupled to changes in expression of these PGE synthases. Incubation with LPS, a potent inducer of mPGES-1 in macrophages, served as a positive control (22,23,32). Levels of mPGES-1 and mPGES-2 mRNA were monitored utilizing semi-quantitative PCR and levels of cPGES were determined utilizing Western blot. As seen in Figure 1A, treatment of RAW264.7 macrophages with active MMP-1 and MMP-3 induced mPGES-1 approximately 3- and 6-fold (p<0.005), respectively. In contrast, levels of mPGES-2 mRNA or cPGES antigen, which were constitutively expressed in control cells, were unchanged.
Figure 1.

MMP-1 and MMP-3 induce macrophage expression of mPGES-1. [A] RAW264.7 macrophages (6-well plate; 2 × 106 cells/well) or [B] thioglycollate-elicited peritoneal macrophages (12-well plate; 7.5 × 105 cells/well) were incubated in DMEM-0.1% LE-BSA (Ctrl) or media containing 50nM MMP-1, MMP-3 or 10 ng/ml LPS. Following 18 h incubation, total RNA or cell lysates were recovered. Levels of mPGES-1 and mPGES-2 mRNAs were determined by PCR. Levels of cPGES and GAPDH in cell lysates were determined by Western blot. Data are representative blots and the mean levels (± SD) of target mRNAs or proteins, presented as RDUs, of 3 experiments.
To corroborate these findings, we examined the effect of active MMPs on the expression of PGE synthases by macrophages recovered from thioglycollate-induced peritonitis (Figure 1B). mPGES-1 mRNA levels were barely detected in the elicited macrophages. Incubation with MMP-1 and MMP-3 strongly induced macrophage expression of mPGES-1 (P<0.02); whereas levels of mPGES-2 mRNA and cPGES antigen were unchanged. These data are consistent with reports that mPGES-1 is inducible, and plays a major role in the elevation of PGE2 production observed in sites of inflammation (22–25,33).
Macrophage MMP-9 expression is dependent on inducible mPGES-1 expression
To determine whether mPGES-1 played a principal role in proteinase-induced MMP-9 expression, mPGES-1 expression was knocked down utilizing siRNA. For this purpose, we initially evaluated the ability of mPGES-1 siRNA to knockdown LPS-induced mPGES-1 expression and PGE2 synthesis by RAW264.7 macrophages. As observed in Figure 2A, LPS induced mPGES-1 expression in macrophages transfected with NS siRNA. Paralleling the increase in mPGES-1 expression, levels of PGE2 in conditioned media recovered from LPS treated cells were increased >50-fold, as compared to control cells (p<0.005). Transfection with mPGES-1 siRNA effectively blocked LPS-induced mPGES-1 expression and reduced the induction of PGE2 levels by about 70% (p<0.01). Next, we monitored MMP-induced MMP-9 expression in macrophages transfected with either NS or mPGES-1 siRNA. MMP-9 mRNA was barely detected in control macrophages transfected with either NS or mPGES-1 siRNA (Figure 2B). Likewise, MMP-9 was barely detected in conditioned media derived from control cells transfected with either NS or mPGES-1 siRNA (Figure 2C). Following incubation with MMP-1, MMP-3 or LPS, levels of MMP-9 mRNA and protein were markedly increased in macrophages transfected with NS siRNA (p<0.001; Figures 2B and C). MMP-9 in conditioned medium recovered from MMP-3 treated macrophages was in the pro (105 kDa) and active (95 kDa) forms, confirming the ability of MMP-3 to activate pro-MMP-9 (34–37). Notably, both MMP and LPS-induced MMP-9 expression was reduced ~70% in macrophages transfected with mPGES-1 siRNA (p<0.001). Likewise, MMP-9 levels in conditioned media recovered from these cells were reduced ~65% (p<0.001). Thus, mPGES-1 plays a principal role in proteinase and LPS-mediated induction of MMP-9.
Figure 2.

mPGES-1 silencing attenuates MMP-induced MMP-9 expression. [A] RAW264.7 macrophages (24-well plate; 6 × 104/well) transfected with NS or mPGES-1 siRNA were incubated 18 h with DMEM-0.1% LE-BSA (Ctrl) or media containing 10 ng/ml LPS. Total RNA and conditioned media were recovered, and levels of mPGES-1 mRNA and PGE2 were determined utilizing PCR and ELISA, respectively. PGE2 levels are the means (± SD) of 3 experiments. [B] Transfected macrophages (24-well plate; 6 × 104/well) were incubated 18 h with DMEM-0.1% LE-BSA or media containing 50 nM MMP-1, MMP-3 or 10 ng/ml LPS. Total RNA was recovered and levels of MMP-9 mRNA determined. [C] Transfected macrophages (48-well plate; 3.5 × 105/well) were incubated with DMEM-0.1% LE-BSA or media containing MMP-1, MMP-3 or LPS 18 h. Conditioned media and cell lysates were recovered, and levels MMP-9 and GAPDH were determined by Western blot. Data are representative blots and the mean levels (± SD) of MMP-9 mRNA or protein, presented as RDUs, of 4 and 3 experiments, respectively.
MMP-induced mPGES-1 expression is dependent on TNF-α
Proteinases regulate cellular functions by activating protease activated receptors (38,39), releasing tethered growth factors (40–42) and cytokines (43,44) or generating biologically active fragments of extracellular matrix (45–47). We previously reported that MMP-1 and MMP-3-induced MMP-9 expression in macrophages is dependent on the release of TNF-β, induction of Cox-2 expression and PGE2 engagement of EP4 (17). Since mPGES-1 is induced in a variety of cells by inflammatory cytokines, we determined whether MMP mediated release of TNF-β from macrophages induced the expression of mPGES-1. First, we examined the ability of recombinant murine TNF-α to induce mPGES-1 in RAW264.7 macrophages. As observed in Figure 3A, levels of mPGES-1 mRNA increased, in a dose-dependent manner, when cells were incubated with exogenous TNF-α (p<0.05), and reached levels achieved following incubation with LPS.
Figure 3.

Neutralizing anti-TNF-α IgG and TNF-α silencing blocks MMP-induced mPGES-1 expression. [A] RAW264.7 macrophages (6-well plate; 2 × 106 cells/well) were incubated 18 h in DMEM-0.1% LE-BSA containing 0 – 100 ng/ml recombinant murine TNF-α. Total RNA was recovered and levels of mPGES-1 mRNA were determined utilizing real time-PCR. [B] Macrophages (6-well plate; 2 × 106/well) were pre-incubated 2 h with non-immune IgG1 (nIgG; 20 μg/ml) or rat monoclonal anti-mouse TNF-α IgG1 (20 μg/ml), followed by 18 h incubation with 50 nM MMP-1, MMP-3 or LPS (10 ng/ml). [C] Macrophages (6-well plate; 2 × 106/well) transfected with nonspecific (NS) or TNF-α siRNA were incubated 18 h with DMEM-0.1% LE-BSA (Ctrl) or media containing MMP-1, MMP-3, or LPS. Total RNA was recovered and levels of mPGES-1 and actin mRNA were determined utilizing PCR. Data are representative blots and the mean levels (± SD) of mPGES-1mRNA, presented as RDUs, of 3 experiments.
We next utilized a neutralizing rat monoclonal anti-murine TNF-β IgG to determine whether MMP-induced release of TNF-β was responsible for stimulating mPGES-1 expression in macrophages. For this purpose, cells were pre-incubated 2 h with 20 μg/ml normal IgG or anti-TNF-β IgG prior to the addition of MMPs or LPS. As shown in Figure 3B, MMP-1 and MMP-3-mediated induction of mPGES-1 was blocked by anti-TNF-α IgG (p<0.05). Similarly, LPS-induced mPGES-1 expression was suppressed ~50% in the presence of anti-TNF-β IgG (p<0.05). To directly test the role of TNF-β in proteinase-induced mPGES-1, macrophages were transfected with either NS siRNA or TNF-β siRNA as previously reported (17). The level of TNF-β mRNA in cells transfected with TNF-β siRNA was reduced >80% when compared to cells transfected with nonspecific siRNA (data not shown). When TNF-β siRNA transfected cells were exposed to the metalloproteinases, MMP-1 and MMP-3 failed to significantly induce mPGES-1 expression (p<0.005). Moreover, consistent with data obtained utilizing anti-TNF-α IgG, TNF-β knockdown attenuated LPS-induced mPGES-1 expression ~50% (p<0.05) in transfected RAW264.7 macrophages (Figure 3C).
MMP-1 and MMP-3 induce MAPKerk1/2–dependent expression of Egr-1
The transcription factor Egr-1 plays a key role in regulating mPGES-1 gene expression by binding to GC-box in the mPGES-1 promoter (48). To determine whether MMP-induced mPGES-1 expression was associated with changes in Egr-1 expression, macrophages were treated with MMP-1 or MMP-3 and levels of Egr-1 and mPGES-1 mRNA were monitored over time utilizing PCR (Figure 4A). Levels of Egr-1 and mPGES-1 gene expression were either undetectable or very low in control RAW264.7 macrophages. Following the addition of MMPs, Egr-1 mRNA levels were strongly elevated at 1 h and returned to low or undetectable levels at 4 and 18 h. In contrast, levels of mPGES-1 mRNA were elevated at 4 h and remained elevated over the experimental period (18 h). Thus, MMP-induced Egr-1 expression preceded mPGES-1 expression. To determine whether MMP-dependent Egr-1 expression was dependent on the release of TNF-α, macrophages were exposed to MMP-1 and MMP-3 in the presence of nIgG or anti-TNF-α IgG. As seen in Figure 4B, anti-TNF-α IgG reduced MMP-induced Egr-1 expression ~65–80% in macrophages (p<0.01).
Figure 4.

MMP-induced Egr-1 is dependent on TNF-α. [A] RAW264.7 macrophages (12-well plate; 7 × 105/well) were incubated 0 – 18 h in DMEM-0.1% LE-BSA (Ctrl), or media containing 50 nM MMP-1 or MMP-3. [B] In other experiments, macrophages (6-well plate; 2 × 106/well) were pre-incubated for 2 h with non-immune IgG1 (nIgG; 20 μg/ml) or rat monoclonal anti-mouse TNF-α IgG1 (20 μg/ml), followed by 1 h incubation with 50 nM MMP-1 or MMP-3. Total RNA was isolated, and mRNA levels for Egr-1, mPGES-1 and actin were determined by PCR. Data are representative blots and the mean levels (± SD) of target mRNAs, presented as RDUs, of 3 experiments.
Since the induction of Egr-1 is regulated by MAPKerk1/2 (49–51) and exposure of macrophages to MMP-1 and MMP-3 activates MAPKerk1/2 (17), we determined whether MMP-induced Egr-1 and mPGES-1 expression were attenuated by a MEK-1 inhibitor (U0126). Pre-incubation of macrophages with U0126 blocked MMP-induced Egr-1 expression (Figure 5A) and mPGES-1 expression (Figure 5B) >90% (p<0.001). Together with results of our earlier studies (17), these data indicate that MMP-dependent release of TNF-α triggers phosphorylation of MAPKerk1/2, induction of Egr-1 and subsequently mPGES-1.
Figure 5.

MMP-induced Egr-1 and mPGES-1 expression are dependent on MAPK kinase. [A] RAW264.7 macrophages (12-well plate; 5 × 105/well) were incubated in DMEM-0.1% LE-BSA (Ctrl) or pre-incubated 30 min in media containing 10 μM U0126 (MAPK kinase inhibitor) followed by the addition of 50 nM MMP-1 or MMP-3, and incubated 1 h. [B] Macrophages (12-well plate; 7 × 105/well) were incubated in DMEM-0.1% LE-BSA or pre-incubated 30 min in media containing 10 μM U0126 followed by the addition of 50 nM MMP-1 or MMP-3, and incubated 18 h. Total RNA was isolated, and mRNA levels for Egr-1, mPGES-1 and actin were determined by PCR. Data are representative blots and the mean levels (± SD) of target mRNAs, presented as RDUs, of 3 experiments.
EP4-dependent positive feedback loop regulates mPGES-1 expression
The diverse physiological and pathophysiological effects of PGE2 are mediated by the EP family of prostanoid receptors (52,53). The EP family is comprised of four subtypes (EP1–4), which exhibit varying affinities for PGE2, and trigger distinct signaling pathways. It has been reported that PGE2 binding to EP4 triggers the activation of MAPKerk1/2 followed by induction of Egr-1 (50,51). Thus, the ability of MMP-1 and -3 to stimulate Cox-2-dependent PGE2 secretion by macrophages (17) may contribute to an EP4-dependent feedback loop that coordinately regulates mPGES-1 expression. To test this hypothesis, we first examined the ability of exogenous PGE2 to stimulate mPGES-1 expression in macrophages. As observed in Figure 6A, levels of mPGES-1 mRNA were elevated in cells incubated with 1–10 μM PGE2. Subsequently, we examined the effect of celecoxib, a selective Cox-2 inhibitor, on MMP and cytokine induced mPGES-1 expression in macrophages. Pre-treatment with celecoxib blocked PGE2 production by macrophages incubated with MMP-1, MMP-3, TNF-α or LPS (data not shown) and their ability to induce mPGES-1 expression (p<0.001; Figure 6B).
Figure 6.

MMP, TNF-α and LPS-induced mPGES-1 is dependent on Cox-2-derived PGE2. [A] RAW264.7 macrophages (12-well plate; 7 × 105/well) were incubated 18 h in DMEM-0.1% LE-BSA containing 0–10 μM PGE2 or 10 ng/ml LPS. [B] Macrophages (6-well plate; 2 × 106/well) were pre-incubated 30 min in DMEM-0.1% LE-BSA containing 5 μM celecoxib, and then incubated 18 h with media (Ctrl), 50 nM MMP-1 or MMP-3, 50 ng/ml TNF-α or 10 ng/ml LPS. Total RNA was isolated, and mRNA levels for mPGES-1 and actin were determined by PCR. Data are representative blots and the mean levels (± SD) of mPGES-1mRNA, presented as RDUs, of 3 experiments.
Next, we examined whether MMP-induced mPGES-1 expression was dependent on PGE2 engagement of EP4. In this regard, we determined whether macrophage expression of the PGE2 receptor family members (i.e. EP1–4) was regulated by MMPs. We previously reported that RAW264.7 macrophages constitutively express EP1–4 (29). Following 18 h incubation with MMP-1 and MMP-3, levels of EP2 and EP4 mRNA were elevated (Figure 7A); whereas, there were no changes in expression of EP1 and EP3 (data not shown). LPS treated macrophages serve as a positive control in these studies (54,55). In order to differentiate the roles of EP2 and EP4 in mPGES-1 expression, cells were pre-incubated 30 min with antagonists of EP2 (AH6809) or EP4 (AH23848) prior to incubation with LPS (Figure 7B). Over a range of 0.1 – 10 μM, the EP2 antagonist had no effect on LPS-induced mPGES-1 expression; whereas, LPS-induced mPGES-1 expression was attenuated by 65% and >90% with 5 μM and 10 μM EP4 antagonist, respectively (p<0.05). Likewise, pre-incubation with the EP4 antagonist reduced MMP-1 and MMP-3-induced mPGES-1 expression by >90% and 70%, respectively (p<0. 005); whereas, the EP2 antagonist had no effect (Figure 7C).
Figure 7.

EP4 antagonist blocks LPS and MMP-induced mPGES-1 expression. [A] RAW264.7 macrophages (6-well plate; 1 × 106 cells/well) were incubated in DMEM-0.1% LE-BSA alone (Ctrl), or media containing 25 nM MMP-1, MMP-3 or 10 ng/ml LPS. Following 18 h incubation, total RNA was isolated, and mRNA levels for PGE2 receptors (EP1–4) determined by PCR. [B] Macrophages were pre-incubated 30 min in DMEM-0.1% LE-BSA containing 0 – 10 μM EP2 antagonist (AH6809) or EP4 antagonist (AH23848). Following an 18 h incubation with 10 ng/ml LPS, RNA was isolated, and mRNA levels for mPGES-1 and actin were determined by PCR. [C] Macrophages were pre-incubated 30 min in DMEM-0.1% LE-BSA containing 10 μM EP2 or EP4 antagonist. Following an 18 h incubation with media alone (Ctrl), 50 nM MMP-1 or MMP-3, RNA was isolated, and mRNA levels for mPGES-1 and actin were determined by PCR. Data are representative blots and the mean levels (± SD) of mPGES-1mRNA, presented as RDUs, of 3 experiments.
To directly test whether PGE2 binding to EP4 underlies a feedback loop (EP4→MAPKerk1/2→Egr-1) that regulates mPGES-1 expression, we first examined the abilities of exogenous PGE2, EP2 agonist butaprost (56) and EP4 agonist PGE1-OH (57) to trigger phosphorylation of MAPKerk1/2 (Figure 8A). Treatment of cells with 10 μM PGE2 or PGE1-OH for 0 – 25 minutes resulted in rapid increase in phosphorylation of MAPKerk1/2 (p<0.01 and P<0.02, respectively). In contrast, incubation with butaprost had no significant effect. These data corroborate earlier reports demonstrating that PGE2→EP4 induced activation of MAPKerk1/2 in HEK-293 cells transfected with EP4 (51) and HCA-7 colon cancer cells (50). Having confirmed that PGE2→EP4→MAPKerk1/2, we next determined the ability of an EP4 antagonist to block PGE2 induced Egr-1 and mPGES-1 expression in macrophages. As observed in Figure 8B, pre-incubation of macrophages with EP4 antagonist AH23848 blocked PGE2-induced Egr-1 and mPGES-1 expression (p<0.01). Similarly, pre-incubation of macrophages with ONO-AE3-208, another EP4 antagonist, also blocked PGE2-induced Egr-1 and mPGES-1 expression (Figure 8C; p<0.01). Thus, MMP-induction of Cox-2 and subsequent PGE2 secretion contributes to an EP4-dependent feedback loop that regulates mPGES-1 expression in macrophages.
Figure 8.

PGE2 binding to EP4 triggers the activation of MAPKerk1/2 and Egr-1 and mPGES-1 expression. [A] RAW264.7 macrophages (12-well plate; 5 × 105/well) were cultured in DMEM-0.1% LE-BSA for 24 h. Cells were then incubated with 10 μM butaprost (EP2 agonist), PGE2 or PGE1-OH (EP4 agonist) for 0–25 min, and lysates were prepared. Levels of phosphorylated and total MAPKerk1/2 were determined utilizing Western blot. Data are representative blots and the mean levels (± SD) of phosphorylated MAPKerk1/2 (P~erk1/2), presented as RDUs, of 3 experiments. [B,C] Macrophages (12-well plate; 5–7 × 105/well) were pre-incubated 30 min in DMEM-0.1% LE-BSA or media containing EP4 antagonists AH23848 or ONO-AE2-208, followed by PGE2. Total RNA was collected at 30 min and 18 h, and mRNA levels for Egr-1, mPGES-1 and actin were determined utilizing PCR. Data are representative blots and the mean levels (± SD) of target mRNAs, presented as RDUs, of 3 experiments.
Finally, we utilized a genetic approach to interrogate the role of EP4 in mPGES-1 expression. For this purpose, RAW264.7 macrophages were transfected with either NS siRNA or EP4 siRNA and exposed to LPS. As expected, LPS potently induced Cox-2 and mPGES-1 expression in RAW264.7 macrophages transfected with NS siRNA (Figure 9A), and led to >900% increase in the level of PGE2 in their conditioned media (Figure 9B). In contrast, LPS-induced mPGES-1 (p<0.04) and Cox-2 (p<0.01) expression was markedly attenuated in macrophages transfected with EP4 siRNA, which resulted in approximately a 75% reduction in LPS-mediated induction of PGE2 levels.
Figure 9.

EP4 silencing reduces LPS-induced mPGES-1 and Cox-2 expression. [A] RAW264.7 macrophages transfected with nonspecific (NS) or EP4 siRNA (12-well plate; 2.5 × 105 cells/well) received DMEM-0.1% LE-BSA (Ctrl) or media containing 10 ng/ml LPS, and were incubated 18 h. Total RNA was recovered and levels of mPGES-1 and Cox-2 mRNA were determined utilizing PCR. Data are representative blots and the mean levels (± SD) of target mRNAs, presented as RDUs, of 3 experiments. [B] Levels of PGE2 in conditioned media were determined utilizing ELISA, and are the mean of duplicate samples.
EP4 antagonist attenuates mPGES-1 expression in thioglycollate-elicited macrophages
Results of experiments described here reveal that PGE2 engagement of EP4 regulates the major inducible form of PGE synthase (i.e. mPGES-1) in RAW264.7 macrophages. Thus, the administration of an EP4 antagonist to inhibit macrophage MMP-9 expression (29), would be expected to drive down levels of PGE2 by attenuating both Cox-2 and mPGES-1 expression. To test this hypothesis, macrophages recovered from thioglycollate induced peritonitis were pre-treated with AH23848 or celecoxib followed by an 18 h incubation with LPS. As shown in Figure 10A, LPS potently induced Cox-2 and mPGES-1 expression. Commensurate with the observed increase in Cox-2 and mPGES-1 expression, the levels of PGE2 in conditioned media recovered from these cells was increased approximately 5-fold (p<0.005) compared to untreated macrophages (Figure 10B). Pre-treatment with either AH23848 or celecoxib attenuated LPS-induced mPGES-1 expression similarly (~80%; p<0.001). Likewise, AH23848 and celecoxib similarly reduced LPS-induced Cox-2 expression (~50%; p<0.05) (Figure 10A). However, the effect of the EP4 antagonist and Cox-2 inhibitor on PGE2 levels in macrophage conditioned media was significantly different. Treatment with celecoxib reduced LPS-induced levels of PGE2 to those observed in untreated control cells. In contrast, although treatment with AH23848 reduced LPS-induced levels of PGE2 >60% (p<0.005), they remained elevated relative to controls (p<0.05). This reflects the ability of celecoxib to both inhibit Cox-2 activity and attenuate Cox-2 and mPGES-1 expression. Finally, both treatments were effective in blocking LPS-induced MMP-9 expression by thioglycollate-elicited macrophages (Figure 10C). Taken together, these data confirm that administration of an EP4 antagonist to inhibit macrophage MMP-9 expression, results in reduced PGE2 synthesis by attenuating both Cox-2 and mPGES-1 expression.
Figure 10.
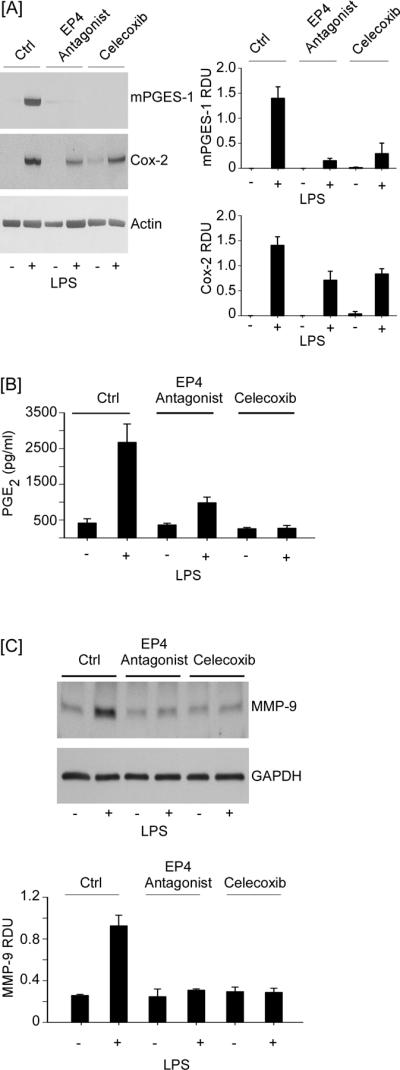
EP4 antagonist reduces LPS-induced PGE2 and MMP-9 levels in cultures of elicited peritoneal macrophages by attenuating mPGES-1 and Cox-2 expression. Thioglycollate-elicited macrophages (12-well plate; 7.5 × 105/well) were pre-incubated 30 min with DMEM-0.01% LE-BSA (Ctrl) or media containing AH23848 or celecoxib, and then incubated 18 h with 10 ng/ml LPS. [A] Total RNA was recovered and levels of mPGES-1, Cox-2 and actin mRNA were determined utilizing PCR. [B,C] Conditioned media were recovered and levels of PGE2 and MMP-9 were determined utilizing ELISA and Western blot, respectively. Data are representative blots and the mean levels (± SD) of target mRNAs and protein, presented as RDUs, of 3 experiments. PGE2 levels are presented as the means (± SD) of 3 experiments.
Discussion
MMP-9 plays a significant role in the pathogenesis of chronic inflammatory diseases and cancer (10–16). Thus, identifying targetable components of signaling pathways that regulate MMP-9 expression may have broad therapeutic implications. Earlier, we reported that MMP-1 and MMP-3 stimulate macrophage MMP-9 expression via the release of TNF-α and subsequent induction of Cox-2, and PGE2 engagement of EP4 receptor (17). While it is well established that Cox activity plays a major role in the synthesis of PGE2 under normal and pathological settings, the isomerization of Cox-derived PGH2 to PGE2 is catalyzed by a family of PGE synthases (18,19). In studies reported here, the regulatory role MMPs may have on the expression on PGE synthases was examined. We found that MMP-1 and MMP-3-dependent release of TNF-α induced rapid and transient expression of Egr-1 in macrophages followed by sustained elevation in mPGES-1 expression. Moreover, proteinase-induced MMP-9 expression was markedly attenuated in macrophages in which mPGES-1 was silenced. Thus, inhibition of mPGES-1 expression and/or activity emerges as a therapeutic target in the regulation of MMP-9 expression (Figure 11).
Figure 11.

MMP-1 and MMP-3 induce macrophage mPGES-1 expression: Role of TNF-α and EP4 receptor. Our data suggest that MMP-1 and MMP-3 selectively induce mPGES-1 expression, which is dependent on the release of TNF-α, activation of MAPKerk1/2 and induction of Egr-1 expression. Proteinase induced MMP-9 expression in macrophages was blocked by mPGES-1 silencing. Elevated mPGES-1 expression and PGE2 levels were regulated by a positive feedback loop that was dependent on the EP4 prostanoid receptor. Thus, in addition to inhibiting macrophage MMP-9 expression, EP4 antagonists emerge as potential therapeutic agents to reduce mPGES-1 expression and PGE2 levels in inflammatory or neoplastic settings.
Elevated levels of PGE2 associated with inflammation and several cancers depends on the coupling of Cox-2 and mPGES-1 expression (58–62) (23,32,63). MMP-mediated induction of Cox-2 and mPGES-1 in macrophages suggests the activation of a common signaling pathway. In this regard, we previously have shown that the activation of MAPKerk1/2 is a key determinant of MMP-induced Cox-2 expression and MMP-9 synthesis (17). The expression of Egr-1, a transcription factor that recognizes GC-rich sequences in the mPGES-1 promoter (18), is dependent on MAPKerk1/2 signaling (50,51). Therefore, we examined whether proteinase triggered activation of MAPKerk1/2 played a role in Egr-1 and mPGES-1 expression. Our studies demonstrate that MMP-induced Egr-1 gene expression was rapid and transient, and preceded mPGES-1 expression. Importantly, pre-incubation of macrophages with a MEK-1 inhibitor blocked MMP-1 and MMP-3-induction of mPGES-1. Thus, MMP-triggered activation of MAPKerk1/2 appears to be the common signaling pathway that drives MMP-induced Cox-2 and mPGES-1 expression in macrophages (Figure 11).
The diverse biologic actions of PGE2 are mediated by the EP1–4 family of prostanoid receptors (52,53). Here we report that MMP-1 and MMP-3 induce macrophage EP2 and EP4 expression. We and others have demonstrated PGE2 engagement of EP4 stimulates macrophage MMP-9 expression (29,64) and enhances Cox-2 expression via a positive feedback loop (29,65,66). It has been reported that PGE2→EP4 triggers MAPKerk1/2–dependent expression of Egr-1 (50,51,67). As reported here, PGE2 and the selective EP4 agonist PGE1-OH triggered rapid and transient phosphorylation of MAPKerk1/2 in macrophages; and PGE2-induced Egr-1 and mPGES-1 expression were blocked by the EP4 antagonists AH23848 and ONO-AE3-208. Thus, PGE2→EP4 participates in a feedback loop driving mPGES-1 expression. This pathway was shown to be critical in MMP-induced mPGES-1 expression insomuch that MMP triggered MAPKerk1/2-dependent induction of Egr-1 and mPGES-1 was blocked by when macrophages were incubated with either celecoxib or an EP4 antagonist (Figure 11).
Our studies reveal a nexus between proteinases and prostanoids whereby MMP-1 and MMP-3, proteinases commonly found in inflammatory foci, stimulate macrophage secretion of PGE2 by inducing Cox-2 and mPGES-1 expression. PGE2 is a major member of the prostanoid class of bioactive lipids with diverse immunomodulatory functions (68,69). The traditional view of PGE2 as an immunosuppressant is, in part, derived from its ability to inhibit T-cell proliferation and differentially regulate Th1 and Th2 cytokine expression (70,71), and attenuate cytokine and chemokine expression by inflammatory macrophages (72–74). Moreover, results of recent experiments suggest that PGE2 engagement of EP4 suppresses inflammation in vivo. The conditional deletion of EP4 in macrophages and/or microglia increased pro-inflammatory gene expression in brain and isolated microglia following LPS administration (75). Similarly, deletion of EP4 in bone marrow derived-cells enhanced inflammation associated with experimental models of atherosclerosis (76) and aneurysm formation (77), and administration of EP4 agonist suppressed production of pro-inflammatory cytokines and chemokines in cardiac transplants (78).
Despite the reported immunosuppressive and anti-inflammatory actions of PGE2, several lines of evidence support a critical role of mPGES-1-derived PGE2 in the pathogenesis of chronic inflammatory diseases. For example, PGE2 stimulates edema formation (24), pain (24,79) and proteinase expression (80–83). In addition, thioglycollate induced recruitment of macrophages to the peritoneal cavity and carrageenan-induced leukocyte recruitment to the pleural cavity are attenuated in mPGES-1 null mice (79,84). Finally, the deletion of mPGES-1 markedly attenuates experimentally induced arthritis (24,79), atherosclerosis (85) and aneurysm formation (86). Moreover, PGE2 acting via EP4 on T cells and dendritic cells facilitates TH1 cell differentiation and amplifies interleukin-23-mediated Th17 cell expansion in vitro (87). Administration of an EP4 antagonist decreases accumulation of both TH1 and TH17 cells in regional lymph nodes and suppresses experimental-induced autoimmune encephalomyelitis and contact hypersensentivity (87). Likewise, administration of EP4 antagonists inhibit inflammation in experimental models of skin injury (88) and arthritis (89). Similarly, experimentally-induced arthritis was reduced in EP4 null mice (90).
Clearly, understanding the environmental context that determines whether PGE2 acts as an immunosuppressive/anti-inflammatory or immunostimulatory/pro-inflammatory capacity remains a critical goal. We have reported that MMP-1 and MMP-3 stimulate macrophage MMP-9 expression by releasing TNF-α, which up-regulates key components of the PGE2 biosynthetic and signaling pathways (i.e. COX-2, mPGES-1 and EP4). Proteinase-induced PGE2 synthesis and EP4 expression likely contribute to the inflammatory response, since PGE2 engagement of EP4 stimulates MMP-9 expression, which is causally associated with the pathogenesis of several inflammatory diseases. Selective targeting of EP4 blocks PGE2-dependent MMP-9 expression, and attenuates PGE2 synthesis by eliminating a feedback loop attenuating expression of Cox-2 and mPGES-1.
Acknowledgements
The authors wish to thank Dr. Takayuki Maruyama (ONO Pharmaceutical Company, Osaka, Japan) for supplying the EP4 antagonist ONO-AE3-208.
1These studies were supported by research grant HL093331 (to D.J. Falcone) from the National Institutes of Health, and grants from the New York Crohn's Foundation and Flight Attendants Medical Research Institute (to A.J. Dannenberg).
Abbreviations used in this paper
- cPGES
cytoplasmic prostaglandin sythase
- Cox-2
cyclooxygenase-2
- Egr-1
early growth response protein 1
- EIA
enzyme immunoassay
- HRP
horseradish peroxidase
- LE
low levels of endotoxin
- MMP
matrix metalloproteinase
- mPGES
microsomal prostaglandin synthase
- NS
nonspecific
- PVDF
polyvinylidene fluoride
- RDU
relative density unit
- siRNA
small interfering RNA
Reference List
- 1.Nagase H, Okada Y. Proteinases and Matrix Degradation. In: Kelly WN, Harris ED Jr., Ruddy S, Sledge CB, editors. The Textbook of Rheumatology. 5 ed. W.B.Saunders; Philadelphia: 1996. pp. 323–341. [Google Scholar]
- 2.Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007;8:221–233. doi: 10.1038/nrm2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Opdenakker G, Van den Steen PE, Van Damme J. Gelatinase B: a tuner and amplifier of immune functions. Trends in Immunology. 2001;22:571–579. doi: 10.1016/s1471-4906(01)02023-3. [DOI] [PubMed] [Google Scholar]
- 4.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nature Reviews Immunology. 2004;4:617–629. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 5.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colnot C, Thompson Z, Miclau T, Werb Z, Helms JA. Altered fracture repair in the absence of MMP9. Development. 2003;130:4123–4133. doi: 10.1242/dev.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MAS, Werb Z, Rafii S. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of Kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corry DB, Kiss A, Song LZ, Song L, Xu J, Lee SH, Werb Z, Kheradmand F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB Journal. 2004;18:995–997. doi: 10.1096/fj.03-1412fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lelongt B, Bengatta S, Delauche M, Lund LR, Werb Z, Ronco PM. Matrix metalloproteinase 9 protects mice from anti-glomerular basement membrane nephritis through its fibrinolytic activity. Journal of Experimental Medicine. 2001;193:793–802. doi: 10.1084/jem.193.7.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez FG, Campbell LG, Liu W, Shipley JM, Itohara S, Patterson GA, Senior RM, Mohanakumar T, Jaramillo A. Inhibition of obliterative airway disease development in murine tracheal allografts by matrix metalloproteinase-9 deficiency. Am. J. Transplant. 2005;5:671–683. doi: 10.1111/j.1600-6143.2005.00751.x. [DOI] [PubMed] [Google Scholar]
- 11.Liu Z, Zhou XY, Shapiro SD, Shipley JM, Twining SS, Diaz LA, Senior RM, Werb Z. The serpin alpha 1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. 2000;102:647–655. doi: 10.1016/s0092-8674(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 12.Chantrain CF, Shimada H, Jodele S, Groshen S, Ye W, Shalinsky DR, Werb Z, Coussens LM, DeClerck YA. Stromal matrix metalloproteinase-9 regulates the vascular architecture in neuroblastoma by promoting pericyte recruitment. Cancer Research. 2004;64:1675–1686. doi: 10.1158/0008-5472.can-03-0160. [DOI] [PubMed] [Google Scholar]
- 13.Masson V, de la Ballina LR, Munaut C, Wielockx B, Jost M, Maillard C, Blacher S, Bajou K, Itoh T, Itohara S, Werb Z, Libert C, Foidart JM, Noel A. Contribution of host MMP-2 and MMP-9 to promote tumor vascularization and invasion of malignant keratinocytes. FASEB J. 2005;19:234–236. doi: 10.1096/fj.04-2140fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lessner SM, Martinson DE, Galis ZS. Compensatory vascular remodeling during atherosclerotic lesion growth depends on matrix metalloproteinase-9 activity. Arteriosclerosis Thrombosis and Vascular Biology. 2004;24:2123–2129. doi: 10.1161/01.ATV.0000141840.27300.fd. [DOI] [PubMed] [Google Scholar]
- 15.Choi ET, Collins ET, Marine LA, Uberti MG, Uchida H, Leidenfrost JE, Khan MF, Boc KP, Abendschein DR, Parks WC. Matrix metalloproteinase-9 modulation by resident arterial cells is responsible for injury-induced accelerated atherosclerotic plaque development in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1020–1025. doi: 10.1161/01.ATV.0000161275.82687.f6. [DOI] [PubMed] [Google Scholar]
- 16.Ikonomidis JS, Barbour JR, Amani Z, Stroud RE, Herron AR, McClister DM, Jr., Camens SE, Lindsey ML, Mukherjee R, Spinale FG. Effects of deletion of the matrix metalloproteinase 9 gene on development of murine thoracic aortic aneurysms. Circulation. 2005;112:I242–I248. doi: 10.1161/CIRCULATIONAHA.104.526152. [DOI] [PubMed] [Google Scholar]
- 17.Steenport M, Khan KM, Du B, Barnhard SE, Dannenberg AJ, Falcone DJ. Matrix metalloproteinase (MMP)-1 and MMP-3 induce macrophage MMP-9: evidence for the role of TNF-alpha and cyclooxygenase-2. J. Immunol. 2009;183:8119–8127. doi: 10.4049/jimmunol.0901925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fahmi H. mPGES-1 as a novel target for arthritis. Curr. Opin. Rheumatol. 2004;16:623–627. doi: 10.1097/01.bor.0000129664.81052.8e. [DOI] [PubMed] [Google Scholar]
- 19.Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin. Immunol. 2006;119:229–240. doi: 10.1016/j.clim.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 20.Jania LA, Chandrasekharan S, Backlund MG, Foley NA, Snouwaert J, Wang IM, Clark P, Audoly LP, Koller BH. Microsomal prostaglandin E synthase-2 is not essential for in vivo prostaglandin E2 biosynthesis. Prostaglandins Other Lipid Mediat. 2009;88:73–81. doi: 10.1016/j.prostaglandins.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lovgren AK, Kovarova M, Koller BH. cPGES/p23 is required for glucocorticoid receptor function and embryonic growth but not prostaglandin E2 synthesis. Mol. Cell Biol. 2007;27:4416–4430. doi: 10.1128/MCB.02314-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uematsu S, Matsumoto M, Takeda K, Akira S. Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J. Immunol. 2002;168:5811–5816. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- 23.Bezugla Y, Kolada A, Kamionka S, Bernard B, Scheibe R, Dieter P. COX-1 and COX-2 contribute differentially to the LPS-induced release of PGE(2) and TxA(2) in liver macrophages. Prostaglandins & Other Lipid Mediators. 2006;79:93–100. doi: 10.1016/j.prostaglandins.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc. Natl. Acad. Sci. U. S. A. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westman M, Korotkova M, af Klint E, Stark A, Audoly LP, Klareskog L, Ulfgren AK, Jakobsson P. Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis and Rheumatism. 2004;50:1774–1780. doi: 10.1002/art.20286. [DOI] [PubMed] [Google Scholar]
- 26.Edelson PJ, Cohn ZA. Purification and cultivation of monocytes and macrophages. In: Bloom BR, David JR, editors. In vitro methods in cell mediated tumor immunity. Academic Press; New York: 1976. pp. 333–340. [Google Scholar]
- 27.Falcone DJ, Ferenc MJ. Acetyl-LDL stimulates macrophage-dependent plasminogen activation and degradation of extracellular matrix. J. Cellular Physiol. 1988;135:387–396. doi: 10.1002/jcp.1041350305. [DOI] [PubMed] [Google Scholar]
- 28.Raschke WC, Baird S, Ralph P, Nakoinz I. Functional macrophage cell lines transformed by Abelson leukemia virus. Cell. 1978;15:261–267. doi: 10.1016/0092-8674(78)90101-0. [DOI] [PubMed] [Google Scholar]
- 29.Pavlovic S, Du B, Sakamoto K, Khan KMF, Natarajan C, Breyer RM, Dannenberg AJ, Falcone DJ. Targeting prostaglandin E-2 receptors as an alternative strategy to block cyclooxygenase-2-dependent extracellular matrix-induced matrix metalloproteinase-9 expression by macrophages. Journal of Biological Chemistry. 2006;281:3321–3328. doi: 10.1074/jbc.M506846200. [DOI] [PubMed] [Google Scholar]
- 30.Hou Z, Falcone DJ, Subbaramaiah K, Dannenberg AJ. Macrophages induce COX-2 expression in breast cancer cells: role of IL-1beta autoamplification. Carcinogenesis. 2011;32:695–702. doi: 10.1093/carcin/bgr027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zar JH. Biostatistical Analysis. Second Edition Prentice Hall; Englewood Cliffs, N.J.: 1984. [Google Scholar]
- 32.Ikeda-Matsuo Y, Ikegaya Y, Matsuki N, Uematsu S, Akira S, Sasaki Y. Microglia-specific expression of microsomal prostaglandin E2 synthase-1 contributes to lipopolysaccharide-induced prostaglandin E2 production. J. Neurochem. 2005;94:1546–1558. doi: 10.1111/j.1471-4159.2005.03302.x. [DOI] [PubMed] [Google Scholar]
- 33.Subbaramaiah K, Yoshimatsu K, Scherl E, Das KM, Glazier KD, Golijanin D, Soslow RA, Tanabe T, Naraba H, Dannenberg AJ. Microsomal prostaglandin E synthase-1 is overexpressed in inflammatory bowel disease. Evidence for involvement of the transcription factor Egr-1. J Biol Chem. 2004;279:12647–12658. doi: 10.1074/jbc.M312972200. [DOI] [PubMed] [Google Scholar]
- 34.Ogata Y, Itoh Y, Nagase H. Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases- 1 complex by 4-aminophenylmercuric acetate and proteinases. J. Biol. Chem. 1995;270:18506–18511. doi: 10.1074/jbc.270.31.18506. [DOI] [PubMed] [Google Scholar]
- 35.Goldberg GI, Strongin A, Collier IE, Genrich LT, Marmer BL. Interaction of 92-Kda Type-Iv Collagenase with the Tissue Inhibitor of Metalloproteinases Prevents Dimerization, Complex-Formation with Interstitial Collagenase, and Activation of the Proenzyme with Stromelysin. Journal of Biological Chemistry. 1992;267:4583–4591. [PubMed] [Google Scholar]
- 36.Dreier R, Wallace S, Fuchs S, Bruckner P, Grassel S. Paracrine interactions of chondrocytes and macrophages in cartilage degradation: articular chondrocytes provide factors that activate macrophage-derived pro-gelatinase B (pro-MMP-9) J Cell Sci. 2001;114:3812–3822. doi: 10.1242/jcs.114.21.3813. [DOI] [PubMed] [Google Scholar]
- 37.Ramos-DeSimone N, Hahn-Dantona E, Sipley J, Nagase H, French DL, Quigley JP. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J. Biol. Chem. 1999;274:13066–13076. doi: 10.1074/jbc.274.19.13066. [DOI] [PubMed] [Google Scholar]
- 38.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. Journal of Thrombosis and Haemostasis. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 39.Boire A, Covic L, Agarwal A, Jacques S, Sherifl S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 40.Saksela O, Rifkin DB. Release of basic fibroblasssst growth factor-Heparan sulfate complexes from endothelial cells by plasminogen activator- mediated proteolytic activity. J. Cell Biol. 1990;110:767–775. doi: 10.1083/jcb.110.3.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol. 2004;164:769–779. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falcone DJ, McCaffrey TA, Haimovitz-Friedman A, Vergilio J-A, Nicholson AC. Macrophage and foam cell release of matrix bound growth factors: Role of plasminogen activation. J. Biol. Chem. 1993;268:11951–11958. [PubMed] [Google Scholar]
- 43.Gearing AJ, Beckett P, Christodoulou M, Churchill M, Clements J, Davidson AH, Drummond AH, Galloway WA, Gilbert R, Gordon JL. Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature. 1994;370:555–557. doi: 10.1038/370555a0. [DOI] [PubMed] [Google Scholar]
- 44.Mohan MJ, Seaton T, Mitchell J, Howe A, Blackburn K, Burkhart W, Moyer M, Patel I, Waitt GM, Becherer JD, Moss ML, Milla ME. The tumor necrosis factor-alpha converting enzyme (TACE): a unique metalloproteinase with highly defined substrate selectivity. Biochemistry. 2002;41:9462–9469. doi: 10.1021/bi0260132. [DOI] [PubMed] [Google Scholar]
- 45.Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–228. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- 46.Khan KMF, Laurie GW, McCaffrey TA, Falcone DJ. Exposure of cryptic domains in the alpha1-chain of laminin-1 by elastase stimulates macrophages urokinase and matrix metalloproteinase-9 expression. J. Biol. Chem. 2002;277:13778–13786. doi: 10.1074/jbc.M111290200. [DOI] [PubMed] [Google Scholar]
- 47.Adair-Kirk TL, Atkinson JJ, Kelley DG, Arch RH, Miner JH, Senior RM. A chemotactic peptide from laminin alpha 5 functions as a regulator of inflammatory immune responses via TNF alpha-mediated signaling. Journal of Immunology. 2005;174:1621–1629. doi: 10.4049/jimmunol.174.3.1621. [DOI] [PubMed] [Google Scholar]
- 48.Naraba H, Yokoyama C, Tago N, Murakami M, Kudo I, Fueki M, Oh-ishi S, Tanabe T. Transcriptional regulation of the membrane-associated prostaglandin E2 synthase gene. Essential role of the transcription factor Egr-1. J. Biol. Chem. 2002;277:28601–28608. doi: 10.1074/jbc.M203618200. [DOI] [PubMed] [Google Scholar]
- 49.Guha M, O'Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, Stern D, Mackman N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood. 2001;98:1429–1439. doi: 10.1182/blood.v98.5.1429. [DOI] [PubMed] [Google Scholar]
- 50.Cherukuri DP, Chen XB, Goulet AC, Young RN, Han Y, Heimark RL, Regan JW, Meuillet E, Nelson MA. The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Exp. Cell Res. 2007;313:2969–2979. doi: 10.1016/j.yexcr.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujino H, Xu W, Regan JW. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J. Biol. Chem. 2003;278:12151–12156. doi: 10.1074/jbc.M212665200. [DOI] [PubMed] [Google Scholar]
- 52.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu. Rev. Pharmacol. Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 53.Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol. Ther. 2004;103:147–166. doi: 10.1016/j.pharmthera.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 54.Hubbard NE, Lee S, Lim D, Erickson KL. Differential mRNA expression of prostaglandin receptor subtypes in macrophage activation. Prostaglandins Leukot. Essent. Fatty Acids. 2001;65:287–294. doi: 10.1054/plef.2001.0327. [DOI] [PubMed] [Google Scholar]
- 55.Ikegami R, Sugimoto Y, Segi E, Katsuyama M, Karahashi H, Amano F, Maruyama T, Yamane H, Tsuchiya S, Ichikawa A. The expression of prostaglandin E receptors EP2 and EP4 and their different regulation by lipopolysaccharide in C3H/HeN peritoneal macrophages. J. Immunol. 2001;166:4689–4696. doi: 10.4049/jimmunol.166.7.4689. [DOI] [PubMed] [Google Scholar]
- 56.Tani K, Naganawa A, Ishida A, Egashira H, Sagawa K, Harada H, Ogawa M, Maruyama T, Ohuchida S, Nakai H, Kondo K, Toda M. Design and synthesis of a highly selective EP2-receptor agonist. Bioorg. Med. Chem. Lett. 2001;11:2025–2028. doi: 10.1016/s0960-894x(01)00359-6. [DOI] [PubMed] [Google Scholar]
- 57.Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsumoto H, Naraba H, Murakami M, Kudo I, Yamaki K, Ueno A, Ohishi S. Concordant induction of prostaglandin E(2) synthase with cyclooxygenase-2 leads to preferred production of prostaglandin E(2) over thromboxane and prostaglandin D-2 in lipopolysaccharide-stimulated rat peritoneal macrophages. Biochemical and Biophysical Research Communications. 1997;230:110–114. doi: 10.1006/bbrc.1996.5894. [DOI] [PubMed] [Google Scholar]
- 59.Yoshimatsu K, Golijanin D, Paty PB, Soslow RA, Jakobsson PJ, DeLellis RA, Subbaramaiah K, Dannenberg AJ. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 2001;7:3971–3976. [PubMed] [Google Scholar]
- 60.Yoshimatsu K, Altorki NK, Golijanin D, Zhang F, Jakobsson PJ, Dannenberg AJ, Subbaramaiah K. Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin. Cancer Res. 2001;7:2669–2674. [PubMed] [Google Scholar]
- 61.Golijanin D, Tan JY, Kazior A, Cohen EG, Russo P, Dalbagni G, Auborn KJ, Subbaramaiah K, Dannenberg AJ. Cyclooxygenase-2 and microsomal prostaglandin E synthase-1 are overexpressed in squamous cell carcinoma of the penis. Clin. Cancer Res. 2004;10:1024–1031. doi: 10.1158/1078-0432.ccr-1032-3. [DOI] [PubMed] [Google Scholar]
- 62.Boulet L, Ouellet M, Bateman KP, Ethier D, Percival MD, Riendeau D, Mancini JA, Methot N. Deletion of microsomal prostaglandin E-2 (PGE(2)) synthase-1 reduces inducible and basal PGE(2) production and alters the gastric prostanoid profile. Journal of Biological Chemistry. 2004;279:23229–23237. doi: 10.1074/jbc.M400443200. [DOI] [PubMed] [Google Scholar]
- 63.Inada M, Matsumoto C, Uematsu S, Akira S, Miyaura C. Membrane-bound prostaglandin E synthase-1-mediated prostaglandin E-2 production by osteoblast plays a critical role in lipopolysaccharide-induced bone loss associated with inflammation. Journal of Immunology. 2006;177:1879–1885. doi: 10.4049/jimmunol.177.3.1879. [DOI] [PubMed] [Google Scholar]
- 64.Cipollone F, Fazia ML, Iezzi A, Cuccurullo C, De Cesare D, Ucchino S, Spigonardo F, Marchetti A, Buttitta F, Paloscia L, Mascellanti M, Cuccurullo F, Mezzetti A. Association between prostaglandin E receptor subtype EP4 overexpression and unstable phenotype in atherosclerotic plaques in human. Arteriosclerosis Thrombosis and Vascular Biology. 2005;25:1925–1931. doi: 10.1161/01.ATV.0000177814.41505.41. [DOI] [PubMed] [Google Scholar]
- 65.Faour WH, He YL, He QW, de Ladurantaye M, Quintero M, Mancini A, Di Battista JA. Prostaglandin E-2 regulates the level and stability of cyclooxygenase-2 mRNA through activation of p38 mitogen-activated protein kinase in interleukin-1 beta-treated human synovial fibroblasts. Journal of Biological Chemistry. 2001;276:31720–31731. doi: 10.1074/jbc.M104036200. [DOI] [PubMed] [Google Scholar]
- 66.Wang D, Buchanan FG, Wang H, Dey SK, DuBois RN. Prostaglandin E2 enhances intestinal adenoma growth via activation of the Rasmitogen-activated protein kinase cascade. Cancer Res. 2005;65:1822–1829. doi: 10.1158/0008-5472.CAN-04-3671. [DOI] [PubMed] [Google Scholar]
- 67.Li T, Qi J, Cowley EA. Activation of the EP(4) prostanoid receptor induces prostaglandin E(2) and pro-inflammatory cytokine production in human airway epithelial cells. Pulm. Pharmacol. Ther. 2011;24:42–48. doi: 10.1016/j.pupt.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 68.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends Immunol. 2002;23:144–150. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 69.Sakata D, Yao C, Narumiya S. Prostaglandin E2, an immunoactivator. J. Pharmacol. Sci. 2010;112:1–5. doi: 10.1254/jphs.09r03cp. [DOI] [PubMed] [Google Scholar]
- 70.Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J. Immunol. 1991;146:108–113. [PubMed] [Google Scholar]
- 71.Hilkens CM, Vermeulen H, van Neerven RJ, Snijdewint FG, Wierenga EA, Kapsenberg ML. Differential modulation of T helper type 1 (Th1) and T helper type 2 (Th2) cytokine secretion by prostaglandin E2 critically depends on interleukin-2. Eur. J. Immunol. 1995;25:59–63. doi: 10.1002/eji.1830250112. [DOI] [PubMed] [Google Scholar]
- 72.Shinomiya S, Naraba H, Ueno A, Utsunomiya I, Maruyama T, Ohuchida S, Ushikubi F, Yuki K, Narumiya S, Sugimoto Y, Ichikawa A, Oh-ishi S. Regulation of TNFalpha and interleukin-10 production by prostaglandins I(2) and E(2): studies with prostaglandin receptor-deficient mice and prostaglandin E-receptor subtype-selective synthetic agonists. Biochem. Pharmacol. 2001;61:1153–1160. doi: 10.1016/s0006-2952(01)00586-x. [DOI] [PubMed] [Google Scholar]
- 73.Takayama K, Garcia-Cardena G, Sukhova GK, Comander J, Gimbrone MA, Jr., Libby P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J. Biol. Chem. 2002;277:44147–44154. doi: 10.1074/jbc.M204810200. [DOI] [PubMed] [Google Scholar]
- 74.Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC, Liu J, Driver A, Bao XR, Sternweis PC, Simon MI, Fraser ID. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKA-AKAP95-p105. Sci. Signal. 2009;2:ra28. doi: 10.1126/scisignal.2000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K. The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J. Immunol. 2010;184:7207–7218. doi: 10.4049/jimmunol.0903487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tang EH, Shimizu K, Christen T, Rocha VZ, Shvartz E, Tesmenitsky Y, Sukhova G, Shi GP, Libby P. Lack of EP4 receptors on bone marrow-derived cells enhances inflammation in atherosclerotic lesions. Cardiovasc. Res. 2011;89:234–243. doi: 10.1093/cvr/cvq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang EH, Shvartz E, Shimizu K, Rocha VZ, Zheng C, Fukuda D, Shi GP, Sukhova G, Libby P. Deletion of EP4 on Bone Marrow-Derived Cells Enhances Inflammation and Angiotensin II-Induced Abdominal Aortic Aneurysm Formation. Arterioscler. Thromb. Vasc. Biol. 2011;31:261–269. doi: 10.1161/ATVBAHA.110.216580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ogawa M, Suzuki J, Kosuge H, Takayama K, Nagai R, Isobe M. The mechanism of anti-inflammatory effects of prostaglandin E2 receptor 4 activation in murine cardiac transplantation. Transplantation. 2009;87:1645–1653. doi: 10.1097/TP.0b013e3181a5c84c. [DOI] [PubMed] [Google Scholar]
- 79.Kamei D, Yamakawa K, Takegoshi Y, Mikami-Nakanishi M, Nakatani Y, Oh-ishi S, Yasui H, Azuma Y, Hirasawa N, Ohuchi K, Kawaguchi H, Ishikawa Y, Ishii T, Uematsu S, Akira S, Murakami M, Kudo I. Reduced pain hypersensitivity and inflammation in mice lacking microsomal prostaglandin E synthase-1. Journal of Biological Chemistry. 2004;279:33684–33695. doi: 10.1074/jbc.M400199200. [DOI] [PubMed] [Google Scholar]
- 80.Shankavaram UT, DeWitt DL, Funk SE, Sage EH, Wahl LM. Regulation of human monocyte matrix metalloproteinases by SPARC. Journal of Cellular Physiology. 1997;173:327–334. doi: 10.1002/(SICI)1097-4652(199712)173:3<327::AID-JCP4>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 81.Corcoran ML, Kibbey MC, Kleinman HK, Wahl LM. Laminin SIKVAV peptide induction of monocyte/macrophage prostaglandin E2 and matrix metalloproteinases. J. Biol. Chem. 1995;270:10365–10368. doi: 10.1074/jbc.270.18.10365. [DOI] [PubMed] [Google Scholar]
- 82.Ottino P, Bazan HEP. Corneal stimulation of MMP-1, -9 and uPA by platelet-activating factor is mediated by cyclooxygenase-2 metabolites. Current Eye Research. 2001;23:77–85. doi: 10.1076/ceyr.23.2.77.5471. [DOI] [PubMed] [Google Scholar]
- 83.Khan KMF, Howe LR, Falcone DJ. Extracellular matrix-induced cyclooxygenase-2 regulates macrophage proteinase expression. Journal of Biological Chemistry. 2004;279:22039–22046. doi: 10.1074/jbc.M312735200. [DOI] [PubMed] [Google Scholar]
- 84.Hara S, Kamei D, Sasaki Y, Tanemoto A, Nakatani Y, Murakami M. Prostaglandin E synthases: Understanding their pathophysiological roles through mouse genetic models. Biochimie. 2010;92:651–659. doi: 10.1016/j.biochi.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 85.Wang M, Zukas AM, Hui YQ, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang M, Lee E, Song W, Ricciotti E, Rader DJ, Lawson JA, Pure E, FitzGerald GA. Microsomal prostaglandin E synthase-1 deletion suppresses oxidative stress and angiotensin II-induced abdominal aortic aneurysm formation. Circulation. 2008;117:1302–1309. doi: 10.1161/CIRCULATIONAHA.107.731398. [DOI] [PubMed] [Google Scholar]
- 87.Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, Sugimoto Y, Narumiya S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat. Med. 2009;15:633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 88.Murase A, Okumura T, Sakakibara A, Tonai-Kachi H, Nakao K, Takada T. Effect of prostanoid EP4 receptor antagonist, CJ-042,794, in rat models of pain and inflammation. European Journal of Pharmacology. 2008;580:116–121. doi: 10.1016/j.ejphar.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 89.Clark P, Rowland SE, Denis D, Mathieu MC, Stocco R, Poirier H, Burch J, Han Y, Audoly L, Therien AG, Xu D. MF498 [N-{[4-(5,9-Diethoxy-6-oxo-6,8-dihydro-7H-pyrrolo[3,4-g]quinolin-7-yl)-3-m ethylbenzyl]sulfonyl}-2-(2-methoxyphenyl)acetamide], a selective E prostanoid receptor 4 antagonist, relieves joint inflammation and pain in rodent models of rheumatoid and osteoarthritis. J Pharmacol Exp. Ther. 2008;325:425–434. doi: 10.1124/jpet.107.134510. [DOI] [PubMed] [Google Scholar]
- 90.McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J. Clin. Invest. 2002;110:651–658. doi: 10.1172/JCI15528. [DOI] [PMC free article] [PubMed] [Google Scholar]