

Abstract
Dystrophin is an actin-binding protein thought to stabilize cardiac and skeletal muscle cell membranes during contraction. Here, we investigated the contributions of each dystrophin domain to actin binding function. Cosedimentation assays and pyrene-actin fluorescence experiments confirmed that a fragment spanning two-thirds of the dystrophin molecule (from N-terminal ABD1 through ABD2) bound actin filaments with high affinity and protected filaments from forced depolymerization, but was less effective in both assays compared to full-length dystrophin. While a construct encoding the C-terminal third of dystrophin displayed no specific actin binding activity or competition with full-length dystrophin, our data show that it confers an unexpected regulation of actin binding by the N-terminal two-thirds of dystrophin when present in cis. Time-resolved phosphorescence anisotropy experiments demonstrated that the presence of the C-terminal third of dystrophin in cis also influences actin interaction in terms of restricting actin’s rotational amplitude. We propose that the C-terminal region of dystrophin allosterically stabilizes an optimal actin binding conformation of dystrophin.
Keywords: Dystrophin, actin binding protein, actin dynamics, cooperative binding, and muscular dystrophy
Introduction
Dystrophin is an essential component of the dystrophin-glycoprotein complex (DGC) that functions to protect the muscle cell membrane from contraction-induced injury 1. The DGC is localized to a lattice-like structure in muscle called the costamere that is thought to transmit force radially from the z-disc to neighboring muscle fibers 2–5. Dystrophin is a multidomain protein with two globular domains at its N- and C-termini that are connected by a large rod domain consisting of 24 spectrin-like repeats with 4 interspersed hinge regions 6; 7. The C-terminal domain of dystrophin encodes a WW domain, a ZZ domain and two EF hand domains that interact with the cytoplasmic tail of β-dystroglycan 8–12. Dystrophin interacts with actin filaments at two sites; ABD1 at its N-terminus composed of a tandem calponin homology domain 13; 14 and ABD2 within spectrin-like repeats 11–17, which interacts with actin filaments via electrostatic attraction 15. Through the concerted action of these two distinct binding interactions, dystrophin is thought to physically anchor the sarcolemma to the costameric actin cytoskeleton thereby stabilizing the membrane against mechanical damage during muscle use. Recent biophysical characterization of the dystrophin-actin interaction showed that binding of full length dystrophin restricts actin microsecond rotational amplitude but increases the rate 16. This paradoxical combination is novel to dystrophin and utrophin 16, and is hypothesized to effect a strong but resilient dystrophin-actin interaction to prevent contraction-induced damage.
Here, we have systematically measured the specific contributions of each dystrophin domain to its actin binding function, through actin cosedimentation, depolymerization, and time-resolved phosphorescence anisotropy (TPA) assays. While the extent of actin binding to dystrophin constructs can be evaluated through actin cosedimentation assays and depolymerization assays, these measurements provide no information about the physical properties of the bound complex. TPA provides a direct measure of the structural dynamics of the bound complex 16. We have established through previous TPA studies that full-length dystrophin restricts actin rotational amplitude but increases the rate, and we proposed that these effects are important for the stability and resilience of the dystrophin-actin complex at the subsarcolemmal region 16. Our binding assays demonstrate that large fragments containing a single ABD tested showed similar affinities for actin as isolated ABDs. Most interestingly, while the C-terminal third of dystrophin displayed no measurable affinity for actin, or competition with full-length dystrophin binding to actin, our data suggest a regulatory function for the C-terminal domain in the stabilization of a dystrophin conformation that binds actin with maximal affinity. In addition, we demonstrate through TPA assays that the C-terminal domain can impact the regulation of actin rotational dynamics (i.e. flexibility) by the actin-binding domains of dystrophin.
Results
Dystrophin domain contributions
Several previous studies from our laboratory reported the affinities of the dystrophin N-terminal ABD1 17, middle rod ABD2 18, ABD1 plus spectrin repeats 1–10 19, or both ABD1 and ABD2 linked by spectrin repeats 1–10 19. Here we directly compared the affinities of two previously studied constructs (DysN-R10, Dys N-R17) as well as ABD2 in context of the DP260 isoform of dystrophin to determine whether the small, unique N-terminus of DP260, or whether the presumed actin non-binding C-terminal third of dystrophin influences actin binding affinity (Fig. 1). We also measured the actin binding affinity of a therapeutically relevant miniaturized dystrophin protein 20 that retains ABD1 and the C-terminus but lacks ABD2 (Dys ΔH2-R19). Because the dystrophin constructs exhibited variable degrees of concentration-dependent self-pelleting, we varied the concentration of actin in our cosedimentation assay around a fixed concentration of dystrophin construct at 0.5 μM. Varying the actin concentration is advantageous because any self-pelleting protein can be easily subtracted from the protein which cosediments with actin filaments 21. DP260 bound actin filaments with a Kd of 6.85 +/− 4.62 μM (Fig. 2a), which is in good agreement with previously published data (7.3 μM) for isolated ABD2 18. These results suggest that neither the unique N-terminus of DP260 nor the C-terminal region influences the actin-binding affinity of ABD2. Dys ΔH2-R19 and DysN-R10 bound to actin filaments with approximately an order of magnitude lower affinity compared to the full-length protein with a Kd of 3.53 +/− 1.73 μM for Dys ΔH2-R19 (Fig. 2c) and 3.92 +/− 2.18 μM for DysN-R10 (Fig. 2b) versus a Kd of 0.11 +/− 0.0082 μM for dystrophin (Fig. 2e). These data confirm that two ABDs are necessary in cis to achieve the sub-micromolar binding affinity measured for full-length dystrophin 15; 19.
Fig. 1. Domain map of proteins assayed.

Globular N- and C-terminal domains are represented by rectangles. Hinge regions are marked by tilted rectangles. Spectrin-like repeats are represented by ovals and colored blue for actin binding repeats. Non-actin binding spectrin-like repeats are yellow.
Fig. 2. Analysis of dystrophin actin binding domains.

Binding isotherms from actin cosedimentation assays with increasing actin concentration and a constant concentration of DP260 (a), DysN-R10 (b), Dys ΔH2-R19 (c), DysN-R17 (d), FL-Dystrophin (e) and DysR18-CT (f). Plots display the results from at least three independent experiments on each graph. Binding curves were fit using regression analysis to determine Kd and Bmax (inset). The color code for domain diagrams is the same as in figure 1.
Interestingly, comparison of data for DysN-R17 (Fig. 2d) with dystrophin (Fig, 2e) suggested that the C-terminal third of dystrophin may contribute to enhanced actin binding affinity. By fitting the aggregate data from three independent binding experiments to a hyperbola, we measured a Kd of 0.6 +/− 0.24 μM and Bmax of 0.066 +/− 0.0087 for DysN-R17 (Fig. 2d) compared to a Kd of 0.11 +/−.0082 μM and Bmax of 0.065 +/− 0.006 for dystrophin (Fig. 2e). To determine if the actin binding affinities of dystrophin and DysN-R17 were significantly different, Kd’s were determined from curve fits to each individual binding experiment and values averaged for each construct. By this method, the mean Kd for DysN-R17 was 0.553 +/− 0.118 (SEM) μM. The mean Kd for dystrophin was 0.152 μM +/− 0.124 μM, which was significantly different from that of DysN-R17 by Student’s T-test (p= 0.0063). DysN-R17 and full-length dystrophin were previously shown to bind with apparent Kd’s of 0.76 μM and 0.3 μM, respectively, but using an assay design where their concentrations were varied around a fixed actin concentration 19. While the relative difference in actin binding affinities between DysN-R17 and dystrophin are consistent between our current and previous study 19, we suggest that self association at high DysN-R17 concentrations increased error around the measurements previously reported to obscure significance.
Although their similar actin binding stoichiometries suggest that DysN-R17 encodes the full actin binding domain of dystrophin, the significantly lower affinity of DysN-R17 compared with full-length dystrophin suggests that the C-terminal third of dystrophin may play a role in actin binding. However, an isolated C-terminal fragment encoding spectrin repeat 18 though the C-terminus of dystrophin (DysR18-CT), showed only non-specific binding to actin filaments up to a concentration of 25 μM actin (Fig. 2f).
To further address whether DysN-R17 binds actin filaments with lower affinity than full-length dystrophin, we measured the ability of each protein to protect actin filaments from forced depolymerization 19. Consistent with previous studies 22; 23, none of the large dystrophin fragments containing only ABD1 or ABD2 (DP260, DysN-R10, and Dys ΔH2-R19) was able to protect actin filaments from forced depolymerization, confirming that both ABD1 and ABD2 must be present in cis to afford protection (Fig. 3a–c). DysN-R17 significantly protected actin filaments from depolymerization, albeit with reduced capacity compared to the full-length protein, which further suggests that the C-terminal region enhances actin binding activity (Fig. 3d).
Fig. 3. Actin depolymerization protection analysis of dystrophin fragments.

Actin depolymerization was monitored by measuring the decay of pyrene actin fluorescence after dilution into low salt buffer below critical concentration. Actin was measured alone or in the presence of molar ratios of 1:6, 1:12, 1:24 (Dys:actin) DysN-R10 (a), DP260 (b), Dys- ΔH2-R19 (c) and DysN-R17 (d). (a–d) Error bars represent standard error of the mean (SEM). Black curve in (d) represents values from full-length dystrophin measured previously 21.
Effect of the C-terminal region of dystrophin on actin binding
Although Dys-R18-CT exhibited no measureable actin-binding activity (Fig. 2f) DysN-R17 exhibited a significantly lower affinity compared to full length dystrophin. Therefore, we hypothesized that the actin non-binding C-terminal third of dystrophin may facilitate a cooperative interaction between adjacent dystrophin molecules docked on an actin filament analogous to the cooperative model of actin binding in tropomyosin 24. To test if DysR18-CT may participate in actin binding through a head to tail association with a second dystrophin molecule, we assayed the ability of Dys-R18-CT to inhibit the binding of full-length dystrophin to actin filaments. However, increasing concentrations of DysR18-CT had no measureable effect on dystrophin’s affinity for actin, as measured in both the high-speed cosedimentation (Fig. 4a and b) and forced depolymerization assays (Fig. 4c and d).
Fig. 4. Dystrophin C-terminus competition.
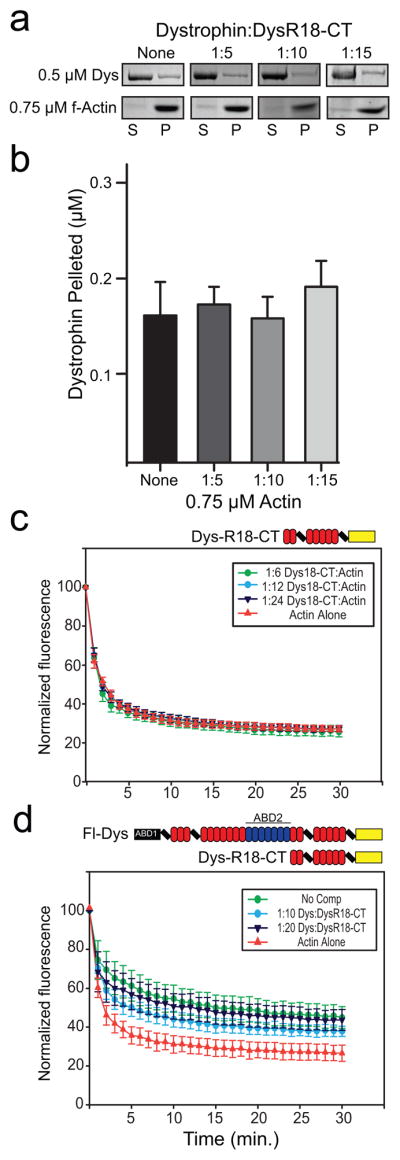
(a) A single point dystrophin actin cosedimentation assay with a fixed concentration of actin of 0.75 μM and a concentration of 0.5 μM dystrophin. DysR18-CT was added at molar ratios of 1:5, 1:10, and 1:15 (Dystrophin:DysR18-CT) to compete for full-length dystrophin binding. Boxed images are coomassie stained polyacrylamide gels that display the amount of dystrophin and actin in the supernatant and pellet for each ratio of DysR18-CT added. (b) Bar graph depicting the quantitation of the cosedimentation assay in (a). No significant difference in actin binding activity is observed with increasing amounts of DysR18-CT. (c) Actin depolymerization protection control experiment at molar ratios of 1:6, 1:12, and 1:24 for DysR18-CT:actin. (d) Actin depolymerization protection assay with full-length dystrophin alone at a molar ratio of 1:12 (Dystrophin:actin) and DysR18-CT added as a competitor at molar ratios of 1:10 and 1:20 (Dystrophin:DysR18-CT). No significant competition was observed for the amounts of DysR18-CT tested. (b–d) Error bars represent SEM.
Dystrophin domain contributions to actin dynamics
We recently demonstrated that dystrophin binding affects the torsional flexibility of actin filaments 16. To test possible effects of the dystrophin C-terminal region on actin torsional flexibility, we performed TPA to compare the effects of dystrophin constructs with and without the C-terminal region on actin dynamics. TPA was performed with erythrosine-iodoacetamide (ErIA) coupled to Cys-374 of each actin protomer, and the degree of filament anisotropy (orientational order) was measured as a function of time after excitation with a pulsed laser. From the calculated anisotropy decays, the amplitudes and rates corresponding to filament bending and twisting motions of actin (Fig. 5a) were calculated 25. We previously demonstrated that the dystrophin-actin complex is characterized by a paradoxical combination of a restriction in actin rotational amplitude but increase in rate 16. Additionally, our previous TPA studies 16 showed a cooperative restriction of actin rotational amplitude and an increase in rotational rate when full length dystrophin was bound (Fig. 5b and c, red). In contrast, DysN-R17 lost functional cooperativity, as its titration caused a linear decrease in the angular amplitude of actin rotational motion (Fig. 5b, blue), corresponding to a drop in the degree of cooperativity (n) from 9.6 to 1.3 when compared with full-length dystrophin (Fig. 5d and e, blue). Loss of the C-terminal region from DysN-R17 also ablated the effect on rate observed with full-length dystrophin (Fig. 5c, blue). These results suggest that the decreased protection of DysN-R17 on actin depolymerization was also the result of an altered mode of interaction between the ABDs and actin.
Fig. 5. TPA shows that the C-terminal region of dystrophin contributes to cooperative restriction of actin rotational amplitude.

(a) Diagram of actin rotational dynamics evaluated by time-resolved phosphorescence anisotropy (TPA) when bound to full length dystrophin (red), DysN-R17 (blue) or DP260 (green). Effects on amplitudes (b) and rates (c) of actin rotational motion are plotted against the fraction of bound actin protomers (Eq 5). Full length dystrophin (red) restricts the amplitude and increases the rate of actin rotational motion 16. Deletion of the C-terminal region of dystrophin in DysN-R17 (blue) shows similar restriction of amplitude (b) at higher titrations compared with full length dystrophin, but there is loss in the cooperativity of the effect. DP260 (green), containing the C-terminal tail regains the cooperative effect on restricting actin rotational amplitude. However, either loss of the C-terminal or N-terminal regions in DysN-R17 or DP260 fails to produce any increase on the rate of rotational motion in actin (c). The degree of cooperativity (n) was determined by fitting to the equilibrium binding constant (Eq. 5) (d) and summarized in (e). The results from the fits in (d) show that DysN-R17 loses considerable cooperativity in its effect on rotational amplitude of actin filaments compared with dystrophin. On the other hand, DP260 with the C-terminal region intact restores the degree of cooperativity seen in full length dystrophin in restricting the angular amplitude of rotational motion.
Despite its lower affinity and lack of protection in depolymerization assays, DP260 showed a dramatic rescue of the cooperative effect in restricting actin rotational amplitude in TPA (Fig. 5b, green), increasing the degree of cooperativity to that observed in full length dystrophin at 9.8 (Fig. 5d and e, green). However, DP260 failed to increase actin rotational rate (Fig. 5c, green). These results suggest that the C-terminal region allosterically influences ABD2 to restrict actin’s rotational amplitude, but both ABDs and the C-terminal region appear to be necessary to increase the rotational rate.
Despite the high sensitivity of the TPA assay to detect changes in actin dynamics, a large range of Dys-R18-CT (0.3uM to 13.5um) to ErIA-actin (1μM) concentrations had no significant effect on actin rotational motion (Fig. 6a and b). The effect on actin rotational amplitude (Fig. 6a) or rate (Fig. 6b) was minimal and mostly within range of ErIA-actin alone. The occasional outlier showed at most a 15% change in actin rotational amplitude and a 6% change in rate, which are quite minimal compared to the average 26% and 80% changes, respectively, observed when dystrophin was bound to actin (red dotted lines in Fig. 5a and b). The TPA results are congruent with both the co-sedimentation and actin-depolymerization data. We conclude that the C-terminal third of dystrophin neither directly participates in actin binding nor enhances binding through a head-to-tail interaction with neighboring dystrophin molecules (analogous to the tropomyosin model 24), but its presence in cis with ABD1 and ABD2 enhances the ability of dystrophin to bind and regulate actin filament dynamics.
Fig. 6. Lack of specific interaction of the C-terminal region (Dys-R18-CT) with actin.

Dys-R18-CT has negligible effect in restricting the amplitude (a) or increasing the rate (b). The ranges of values from actin only (dash lines, black) and when 40% of the actin is decorated with dystrophin (dotted line, red) are shown.
Discussion
It is well established by multiple studies that dystrophin binds actin filaments with sub-micromolar affinity through the concerted action of ABD1 and ABD2 15; 18; 19. While previous studies have measured the actin binding properties of ABD1 and ABD2 in isolation and reported ~10-fold lower affinities than measured for full-length dystrophin, ours is the first to directly compare the properties of ABD1, ABD2 and the C-terminal third of dystrophin with full-length protein (Fig. 2). We found that large constructs containing only one ABD (DP260, DysN-R10 and Dys ΔH2-R19) exhibited actin-binding affinities that matched well with those previously reported for isolated ABD1 and ABD2, and none of these single ABD-containing fragments were able to protect actin filaments against forced depolymerization (Fig. 3). Ablation of one ABD also weakened the effect of dystrophin on actin filament dynamics (Fig. 5, green). Loss of either the N-terminal region in DP260 or the C-terminal region in DysN-R17 eliminated their effect on actin rotational rate (Fig. 5c), although their restriction of rotational amplitude was preserved at higher titrations (Fig. 5b).
Our binding data demonstrate that Dys ΔH2-R19 and DP260 have similar affinities for actin filaments in vitro and we previously demonstrated that both Dys ΔH2-R19 and DP260 can restore the link between the sarcolemma and costameric cytoskeleton when transgenically overexpressed in mdx muscle 26. However, based on differences between Dys ΔH2-R19 and DP260 in rescuing other dystrophic phenotypes 27; 28, it appears that the N-terminal ABD1 may perform an additional function in addition to binding actin filaments. Dys ΔH2-R19 fully rescued the dystrophic phenotypes of the mdx mouse when overexpressed 27. In contrast, while DP260 expression prevented contraction induced injury, it failed to rescue the muscle weakness, necrosis and regeneration associated with dystrophin deficiency 28. In support of an actin-independent function for ABD1, the highly homologous ABD of plectin has been shown to differentially bind to actin filaments or integrin cytoplasmic domain dependent on its open versus closed conformation 29. Moreover, dystrophin associates with cytokeratins through a direct interaction of keratin 19 with ABD1 30–32.
While DysN-R17, containing both ABD1 and ABD2, bound actin filaments with similar stoichiometry as full-length dystrophin and significantly protected filaments from depolymerization (Fig. 3d), it bound with significantly lower affinity than full-length dystrophin (Fig. 2d). These new data suggest a role for the C-terminal region of dystrophin in increasing actin affinity. However, the isolated C-terminal region (Dys-R18-CT) exhibited no measureable specific actin-binding activity in both cosedimentation (Fig. 2f) and forced polymerization assays (Fig. 4c).
We hypothesize that the dystrophin C-terminal region must be in cis with both ABDs to allosterically influence their binding to actin. We initially thought that the dystrophin actin interaction may exhibit a head-to-tail cooperative binding similar to the coiled-coil domains of tropomyosin 24. However, we measured no specific actin-binding activity in Dys-R18-CT (Fig. 2f), and competition experiments showed it had no effect on full-length dystrophin binding to actin (Fig. 4). Nevertheless, we cannot rule out a head-to-tail cooperative binding activity since it may be dependent on the presence of the N-terminal domain.
Additionally, our TPA experiments demonstrate that dystrophin fragments cooperatively restrict actin rotational amplitude only when the C-terminal region is present in the construct (Fig. 5b). The results in Fig. 2f and Fig. 4 indicate that dystrophin does not convey cooperative binding similar to that observed in tropomyosin. However, our TPA results indicate that actin rotational amplitude responds cooperatively to dystrophin binding, that this cooperativity is largely dependent on the presence of the C-terminal region, and that the full-length protein is required for increased rotational rate (see table in Fig. 5). The degree of cooperativity (n) quantifies actin’s remarkable allosteric properties in response to different binding partners, which can be as high as several hundred 33; 34. The high degree of cooperativity observed in the full length dystrophin-actin complex (n = 9.6, Fig. 5) could explain why a low amount of expressed dystrophin (29–57%) in humans can prevent the characteristic muscle weakness in muscular dystrophy 35. It also may explain why expression of 20% dystrophin in transgenic mdx mice (dys null) was sufficient to rescue dystrophic phenotype and that a threshold of only ~5% is enough to partially restore muscle function in Dko mice (dys−/utr−)36–38.
Multiple approaches to treat DMD rely on the deletion of dystrophin domains. Recently, we demonstrated that internal truncation compromises the stability of dystrophin 20. Our new finding that the C-terminal region of dystrophin influences both actin binding and actin dynamics adds yet another layer of complexity into the design of DMD therapies, but also highlights the potential to biophysically optimize gene therapy constructs prior to extensive testing in animals. For example, inclusion of specific C-terminal spectrin-like repeats in next generation gene therapy constructs may improve efficacy through enhanced actin binding activity.
Recently, we demonstrated that the isolated C-terminal region (Dys-R18-CT) exhibits markedly greater thermal stability compared to N-terminal constructs (Dys-N-R17) or even full-length dystrophin 20. Since the N- and C-terminal regions of dystrophin have drastically different thermal stabilities (50 °C and 70 °C respectively), one would expect to observe two melting transitions but instead only a single transition exactly halfway in between each is observed (60 °C). We propose that when present in cis, the highly stable C-terminal third of dystrophin allosterically influences the N-terminal region of the protein to effect increased thermal stability and orient or stabilize a conformation required for optimal actin binding. Alternatively, the presence of the N-terminal region in cis with the C-terminal domain may stabilize an actin binding conformation in the C-terminus. Further experiments will be needed to understand how the C-terminal region of dystrophin influences actin binding activity.
Materials and Methods
Cloning, protein expression and purification
All constructs used to express protein in this manuscript were previously generated for other studies 20. Each cDNA was N-terminally Flag tagged using PCR and cloned into either pFastbac1 or pFastbac duel downstream of the polyhedrin promoter. Once in pFastbac, bacmids were generated using the Bac-to-bac system from Invitrogen. High titer virus was used to infect Sf21 insect cells at a density of between 1.0 and 1.5 × 106 cells/ml with a volume of 250 milliliters. Proteins were purified by Flag affinity chromatography and dialyzed against two changes of phosphate buffered saline pH 7.5 to remove excess flag peptide. Proteins were concentrated in Millipore Amicon Ultra centrifuge-based concentrators with cutoffs of either 100 kDa or 10 kDa depending on protein molecular weight.
Actin cosedimentation assays
In actin co-sedimentation assays the concentration of actin was varied instead of dystrophin identically to Henderson et al 2010 21. Each reaction was incubated at room temperature for thirty minutes and centrifuged at 100,000 × g for 30 minutes. Resulting supernatant and pellet fraction we subjected to SDS-PAGE and stained with Coomassie blue. Supernatant and pellet fractions were determined by densitometry with UVP analysis software. The concentration of actin was varied between 0.1 and 25 μM and the concentration of dystrophin or dystrophin fragment was fixed at 0.5 μM. Additionally, the fraction of dystrophin protein found to pellet without actin was quantitated by densitometry from Coomassie stained gels and the self-pelleting fraction was subtracted from subsequent reactions where actin was present. Three independent experiments were performed for each dystrophin protein and the aggregate data was plotted and fit using non-linear regression analysis identically to Henderson et al 2010 and Rybakova et al 2006 19; 21. A single ligand binding site equation was used in Sigma Plot (Systat Software) for non-linear regression.
Actin depolymerization assays
Depolymerization protection assays were performed with pyrene labeled actin from Cytoskeleton as previously described 21. In short, pyrene labeled actin at 2 μM was incubated with dystrophin or dystrophin fragments at ratios of 1:6, 1:12 or 1:24 (dystrophin:actin). Actin was forced to depolymerize by dilution below critical concentration with g-buffer (10 mM Tris pH 8.0 and 2 mM CaCl2). Samples were immediately read on a Gemini (Molecular Devices) fluorescence plate reader and every minute for 30 minutes.
C-terminal competition experiments
Dystrophin actin-binding was performed as described above at a near Kd concentration of 0.75 μM actin and incubated with increasing concentrations of DysR18-CT (1:5, 1:10 and 1:15). Results were compiled by analyzing the scanned Coomassie stained gel by densitometry and plotted as a bar graph. Student’s t-test was used to determine significance. Actin depolymerization protection assays were performed as described above except that DysR18-CT was added at ratios of 1:10 and 1:20 (dystrophin:DysR18-CT) to a reaction containing full-length dystrophin at a ratio of 1 dystrophin per 12 actin monomers.
Time-resolved phosphorescence anisotropy
Actin preparation and labeling with phosphorescent erythrosine-iodoacetamide (ErIA) (Anaspec) was as described in 16. Phalloidin-stabilized ErIA-actin was diluted in U/D buffer (100 mM NaCl2, 2 mM MgCl2, 0.2 mM ATP, 1 mM DTT, 10 mM Tris pH 7.5) to 1uM. Increasing concentrations of dystrophin and fragments (Fig. 1) were added to 1 μuM ErIA-actin. An oxygen removing system containing glucose oxidase (55ug/ml), catalase (36ug/ml), and glucose (45ug/ml) was added to the sample prior to each experiment to maximize the phosphorescence signal 39; 40. The phosphorescent dye was excited at 532nm with a vertically polarized 1.2 ns laser pulse from a FDSS 532-150 laser (CryLas) with a 100 Hz repetition rate. Emission was detected through a 670 nm glass cutoff filter (Corion) using a photomultiplier (R928; Hamamatus) and transient digitizer (CompuScope 14100; GaGe) with a resolution of 1 μs per channel. Time-resolved anisotropy is defined by
| Eq 1 |
Where Iv(t) and Ih(t) is defined by the vertical and horizontal components of the detected phosphorescent emission, using a single detector at 90° and a rotating sheet polarizer alternating between the two orientations every 500 laser pulses. The instrument response function G was calibrated by detection of the signal with horizontally polarized excitation pulse, and correcting so that the anisotropy is zero. All time-resolved anisotropy experiments were recorded with 30 cycles, with 500 pulses each in the horizontal and vertical planes.
The anisotropy decays were analyzed by fitting to the function:
| Eq 2 |
Where rotational correlation times φ1 (slow) and φ2 (fast) and amplitudes r1, r2 and r∞ were varied and using a least-squares minimization procedure 41. Results were validated by comparison of residuals and chi-squares of the fits at one, two and three exponential terms. Increase in final anisotropy r∞ indicates a decrease in the amplitudes of microsecond rotational dynamics, which is attributed to a decrease in actin filament flexibility. A maximally flexible molecule (isotropic) would exhibit a final anisotropy value of 0. The slower motions (φ1, r1) represent mainly the actin filament bending, while the faster motions (φ2, r2) represent mainly the actin filament twisting. The combined rates of the filament twisting and bending motions were evaluated by φAve which is calculated by a weighted average of φ1 and φ2. The overall angular amplitudes of the microsecond motions in actin were calculated using the wobble-in-a-cone model 25
| Eq 3 |
This reflects the combined amplitude of microsecond filament bending and twisting motions depicted in Fig. 6a.
Rates of the corresponding angular amplitudes were calculated by:
| Eq 4 |
Graphs of amplitude and rate of actin rotation dynamics are plotted against the fractional saturation of protein-decorated actin (Fraction bound, actin) 16. This is different from the cosedimentation plots since the goal of this assay is not to evaluate the degree of binding but to measure the structural effects of the bound state. Cosedimentation assays using a fixed concentration of ErIA (6μM) binding to varied concentrations of dystrophin constructs were performed to obtain values of Kd and Ymax. These values were then used to calculate the fraction of actin bound to dystrophin constructs in our TPA assays using the quadratic equation:
| Eq 5 |
Where y is the fractional saturation of actin by bound dystrophin construct (mol/mol actin) and x is the added concentration of dystrophin construct. Pt is defined as the concentration of available binding sites for dystrophin construct, which is [actin, μM]*Ymax. The fraction of bound actin is then calculated by y/Ymax.
The degree of cooperativity is determined by fitting the plot of angular amplitudes θ∞ (Eq 2) verses fraction of actin decorated with dystrophin or dystrophin constructs to the equilibrium binding equations 34.
| Eq 6 |
Where θC(OBS) is the observed amplitude, θa is the amplitude of actin only and θ∞ is the amplitude of the actin bound to dystrophin or related constructs listed in Fig. 1. The parameter v is the fraction of actin decorated with dystrophin or related constructs and n is the degree of cooperativity in the system, i.e. the number of unbound actin monomers affected by a single monomer binding to dystrophin.
Acknowledgments
We thank Bin Li for excellent technical support. This work was supported by the NIH Training Program in Muscle Research [AR007612], [NIH RO1 AR042423] to JME, [NIH R01 AG026160] to DDT, and [NIH F30 AG034033] to AYL. TPA experiments were performed in the Biophysical Spectroscopy Facility supported by [NIH P30 AR057220].
Abbreviations used are
- DGC
Dystrophin glycoprotein complex
- ABD1 and ABD2
Actin binding domain 1 or 2
- TPA
time-resolved phosphorescence anisotropy
- ErIA
erythrosine-iodoacetamide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ervasti JM, Sonnemann KJ. Biology of the striated muscle dystrophin-glycoprotein complex. Int Rev Cytol. 2008;265:191–225. doi: 10.1016/S0074-7696(07)65005-0. [DOI] [PubMed] [Google Scholar]
- 2.Porter GA, Dmytrenko GM, Winkelmann JC, Bloch RJ. Dystrophin colocalizes with beta-spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J Cell Biol. 1992;117:997–1005. doi: 10.1083/jcb.117.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Straub V, Bittner RE, Leger JJ, Voit T. Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J Cell Biol. 1992;119:1183–1191. doi: 10.1083/jcb.119.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ervasti JM. Costameres: the Achilles’ heel of Herculean muscle. J Biol Chem. 2003;278:13591–13594. doi: 10.1074/jbc.R200021200. [DOI] [PubMed] [Google Scholar]
- 5.Zubrzycka-Gaarn EE, Bulman DE, Karpati G, Burghes AH, Belfall B, Klamut HJ, Talbot J, Hodges RS, Ray PN, Worton RG. The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature. 1988;333:466–469. doi: 10.1038/333466a0. [DOI] [PubMed] [Google Scholar]
- 6.Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell. 1988;53:219–228. doi: 10.1016/0092-8674(88)90383-2. [DOI] [PubMed] [Google Scholar]
- 7.Koenig M, Kunkel LM. Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem. 1990;265:4560–4566. [PubMed] [Google Scholar]
- 8.Ishikawa-Sakurai M, Yoshida M, Imamura M, Davies KE, Ozawa E. ZZ domain is essentially required for the physiological binding of dystrophin and utrophin to beta-dystroglycan. Hum Mol Genet. 2004;13:693–702. doi: 10.1093/hmg/ddh087. [DOI] [PubMed] [Google Scholar]
- 9.Ponting CP, Blake DJ, Davies KE, Kendrick-Jones J, Winder SJ. ZZ and TAZ: new putative zinc fingers in dystrophin and other proteins. Trends Biochem Sci. 1996;21:11–13. [PubMed] [Google Scholar]
- 10.Hnia K, Zouiten D, Cantel S, Chazalette D, Hugon G, Fehrentz JA, Masmoudi A, Diment A, Bramham J, Mornet D, Winder SJ. ZZ domain of dystrophin and utrophin: topology and mapping of a beta-dystroglycan interaction site. Biochem J. 2007;401:667–77. doi: 10.1042/BJ20061051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bork P, Sudol M. The WW domain: a signalling site in dystrophin? Trends Biochem Sci. 1994;19:531–533. doi: 10.1016/0968-0004(94)90053-1. [DOI] [PubMed] [Google Scholar]
- 12.Rentschler S, Linn H, Deininger K, Bedford MT, Espanel X, Sudol M. The WW domain of dystrophin requires EF-hands region to interact with beta-dystroglycan. Biol Chem. 1999;380:431–42. doi: 10.1515/BC.1999.057. [DOI] [PubMed] [Google Scholar]
- 13.Levine BA, Moir AJ, Patchell VB, Perry SV. The interaction of actin with dystrophin. FEBS Lett. 1990;263:159–162. doi: 10.1016/0014-5793(90)80728-2. [DOI] [PubMed] [Google Scholar]
- 14.Way M, Pope B, Cross RA, Kendrick-Jones J, Weeds AG. Expression of the N-terminal domain of dystrophin in E. coli and demonstration of binding to F-actin. FEBS Lett. 1992;301:243–5. doi: 10.1016/0014-5793(92)80249-g. [DOI] [PubMed] [Google Scholar]
- 15.Amann KJ, Renley BA, Ervasti JM. A cluster of basic repeats in the dystrophin rod domain binds F-actin through an electrostatic interaction. J Biol Chem. 1998;273:28419–28423. doi: 10.1074/jbc.273.43.28419. [DOI] [PubMed] [Google Scholar]
- 16.Prochniewicz E, Henderson D, Ervasti JM, Thomas DD. Dystrophin and utrophin have distinct effects on the structural dynamics of actin. Proc Natl Acad Sci U S A. 2009;106:7822–7. doi: 10.1073/pnas.0812007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Renley BA, Rybakova IN, Amann KJ, Ervasti JM. Dystrophin binding to nonmuscle actin. Cell Motil Cytoskelet. 1998;41:264–270. doi: 10.1002/(SICI)1097-0169(1998)41:3<264::AID-CM7>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 18.Amann KJ, Guo AW, Ervasti JM. Utrophin lacks the rod domain actin binding activity of dystrophin. J Biol Chem. 1999;274:35375–35380. doi: 10.1074/jbc.274.50.35375. [DOI] [PubMed] [Google Scholar]
- 19.Rybakova IN, Humston JL, Sonnemann KJ, Ervasti JM. Dystrophin and utrophin bind actin through distinct modes of contact. J Biol Chem. 2006;281:9996–10001. doi: 10.1074/jbc.M513121200. [DOI] [PubMed] [Google Scholar]
- 20.Henderson DM, Belanto JJ, Li B, Heun-Johnson H, Ervasti JM. Internal deletion compromises the stability of dystrophin. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Henderson DM, Lee A, Ervasti JM. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc Natl Acad Sci U S A. 2010;107:9632–7. doi: 10.1073/pnas.1001517107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rybakova IN, Amann KJ, Ervasti JM. A new model for the interaction of dystrophin with F-actin. J Cell Biol. 1996;135:661–672. doi: 10.1083/jcb.135.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rybakova IN, Ervasti JM. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J Biol Chem. 1997;272:28771–28778. doi: 10.1074/jbc.272.45.28771. [DOI] [PubMed] [Google Scholar]
- 24.Tobacman LS. Cooperative binding of tropomyosin to actin. Adv Exp Med Biol. 2008;644:85–94. doi: 10.1007/978-0-387-85766-4_7. [DOI] [PubMed] [Google Scholar]
- 25.Prochniewicz E, Zhang Q, Howard EC, Thomas DD. Microsecond rotational dynamics of actin: spectroscopic detection and theoretical simulation. J Mol Biol. 1996;255:446–57. doi: 10.1006/jmbi.1996.0037. [DOI] [PubMed] [Google Scholar]
- 26.Hanft LM, Rybakova IN, Patel JR, Rafael-Fortney JA, Ervasti JM. Cytoplasmic gamma-actin contributes to a compensatory remodeling response in dystrophin-deficient muscle. Proc Natl Acad Sci U S A. 2006;103:5385–5390. doi: 10.1073/pnas.0600980103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, Chamberlain JS. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 28.Warner LE, DelloRusso C, Crawford RW, Rybakova IN, Patel JR, Ervasti JM, Chamberlain JS. Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin-glycoprotein complex and partially prevents dystrophy. Hum Mol Genet. 2002;11:1095–105. doi: 10.1093/hmg/11.9.1095. [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Alvarez B, Bobkov A, Sonnenberg A, de Pereda JM. Structural and functional analysis of the actin binding domain of plectin suggests alternative mechanisms for binding to F-actin and integrin beta4. Structure. 2003;11:615–25. doi: 10.1016/s0969-2126(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 30.O’Neill A, Williams MW, Resneck WG, Milner DJ, Capetanaki Y, Bloch RJ. Sarcolemmal organization in skeletal muscle lacking desmin: evidence for cytokeratins associated with the membrane skeleton at costameres. Mol Biol Cell. 2002;13:2347–59. doi: 10.1091/mbc.01-12-0576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ursitti JA, Lee PC, Resneck WG, McNally MM, Bowman AL, O’Neill A, Stone MR, Bloch RJ. Cloning and characterization of cytokeratins 8 and 19 in adult rat striated muscle. Interaction with the dystrophin glycoprotein complex. J Biol Chem. 2004;279:41830–8. doi: 10.1074/jbc.M400128200. [DOI] [PubMed] [Google Scholar]
- 32.Stone MR, O’Neill A, Catino D, Bloch RJ. Specific interaction of the actin-binding domain of dystrophin with intermediate filaments containing keratin 19. Mol Biol Cell. 2005;16:4280–93. doi: 10.1091/mbc.E05-02-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prochniewicz E, Zhang Q, Janmey PA, Thomas DD. Cooperativity in F-actin: binding of gelsolin at the barbed end affects structure and dynamics of the whole filament. J Mol Biol. 1996;260:756–66. doi: 10.1006/jmbi.1996.0435. [DOI] [PubMed] [Google Scholar]
- 34.Prochniewicz E, Janson N, Thomas DD, De la Cruz EM. Cofilin increases the torsional flexibility and dynamics of actin filaments. J Mol Biol. 2005;353:990–1000. doi: 10.1016/j.jmb.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 35.Neri M, Torelli S, Brown S, Ugo I, Sabatelli P, Merlini L, Spitali P, Rimessi P, Gualandi F, Sewry C, Ferlini A, Muntoni F. Dystrophin levels as low as 30% are sufficient to avoid muscular dystrophy in the human. Neuromuscular Disorders. 2007;17:913–918. doi: 10.1016/j.nmd.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 36.Wells DJ, Wells KE, Asante EA, Turner G, Sunada Y, Campbell KP, Walsh FS, Dickson G. Expression of human full-length and minidystrophin in transgenic mdx mice: implications for gene therapy of Duchenne muscular dystrophy. Hum Mol Genet. 1995;4:1245–50. doi: 10.1093/hmg/4.8.1245. [DOI] [PubMed] [Google Scholar]
- 37.Phelps SF, Hauser MA, Cole NM, Rafael JA, Hinkle RT, Faulkner JA, Chamberlain JS. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum Mol Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- 38.Li D, Yue Y, Duan D. Marginal level dystrophin expression improves clinical outcome in a strain of dystrophin/utrophin double knockout mice. PLoS One. 2010;5:e15286. doi: 10.1371/journal.pone.0015286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prochniewicz E, Walseth TF, Thomas DD. Structural dynamics of actin during active interaction with myosin: different effects of weakly and strongly bound myosin heads. Biochemistry. 2004;43:10642–52. doi: 10.1021/bi049914e. [DOI] [PubMed] [Google Scholar]
- 40.Eads TM, Thomas DD, Austin RH. Microsecond rotational motions of eosin-labeled myosin measured by time-resolved anisotropy of absorption and phosphorescence. J Mol Biol. 1984;179:55–81. doi: 10.1016/0022-2836(84)90306-1. [DOI] [PubMed] [Google Scholar]
- 41.Prochniewicz E, Thomas DD. Perturbations of functional interactions with myosin induce long-range allosteric and cooperative structural changes in actin. Biochemistry. 1997;36:12845–53. doi: 10.1021/bi971201r. [DOI] [PubMed] [Google Scholar]