

Abstract
Membranous adenylyl cyclases (mACs) constitute a family of nine isoforms with different expression patterns. Studies with mAC gene knockout mice provide evidence for the notion that AC isoforms play distinct (patho)physiological roles. Consequently, there is substantial interest in the development of isoform-selective mAC inhibitors. Here, we review the current literature on mAC inhibitors. Structurally diverse inhibitors targeting the catalytic site and allosteric sites (e.g. the diterpene site) have been identified. The catalytic site of mACs accommodates both purine and pyrimidine nucleotides, with a hydrophobic pocket constituting a major affinity-conferring domain for substituents at the 2′- and 3′-O-ribosyl position of nucleotides. BODIPY-forskolin stimulates ACs 1 and 5 but inhibits AC2. However, so far, no inhibitor has been examined at all mAC isoforms, and data obtained with mAC inhibitors in intact cells have not always been interpreted cautiously enough. Future strategies for the development of the mAC inhibitor field are discussed critically.
mACs
ACs catalyze the conversion of ATP into the second messenger cAMP. cAMP plays a crucial role in the regulation of numerous cell functions. Mammals express nine mAC isoforms and a sAC. mACs consist of two transmembrane domains with six predicted helices each, and two cytosolic domains, referred to as C1 and C2, respectively [1-3]. The C1- and C2-domains of mACs show considerable homology with each other and together constitute the catalytic core of the enzyme. The C1- and C2 domains are pseudosymmetrically arranged and, at their interface, form a pair of ligand-binding sites, i.e. the catalytic site and the regulatory diterpene site.
The diterpene forskolin (FS) comes from the Indian plant Coleus forskohlii [4] and activates mACs 1-8, but not mAC9 [1,2]. It has been postulated that in polycystic kidney disease, an endogenous FS-like molecule occurs in the cysts [5], but these studies need to be confirmed. FS possesses some structural similarity with α-D-glucose [4]. However, the interactions of the diterpene site of mACs with sugars have still to be examined. All mAC isoforms are activated by the G-protein Gs being stimulated following binding of hormones and neuotransmitters to their cognate G protein-coupled receptors (GPCRs) [1-3].
mAC isoforms are differentially expressed in cells and organs, suggesting specific (patho)physiological functions of each isoform [1-3]. This notion is supported by unique phenotypes of transgenic animals overexpressing defined AC isoforms or knock-out animals missing a single AC isoform. For example, Ca2+/calmodulin-stimulated AC1 plays a role in learning, memory formation, neurotoxicity, and pain responses, and AC5 provides protection from heart failure and enhances life span [3,6,7]. Deletion of AC5 in mice provides protection from heart failure and enhances life span, and AC1 is involved in neurotoxicity and pain responses [3,6-8]. These findings have evoked considerable enthusiasm in the research community that selective AC5 inhibitors could constitute innovative drugs for treatment of heart failure and ageing and that AC1 inhibitors could be used in the treatment of diseases associated with neuronal damage and chronic pain.
The aim of this review is to critically discuss the challenges in the field of mAC inhibitor development, recent progress on mAC inhibitors and future directions. Table 1 presents the specific properties and limitations of representative mAC inhibitors, and Table 2 provides a summary of selected patents in the mAC inhibitor field. Potential clinical indications for mAC inhibitors covered in patents include ageing, cardiovascular diseases, gastrointestinal infections, vascular diseases and neurological disorders.
Table 1. Overview on publications on mAC inhibitors.
| Inhibitor name and structure | Pharmacological key data | Comments | |||
|---|---|---|---|---|---|
MANT-GTP
|
Ki values in AC/GC assay (+ Mn2+): AC1: 90 nM; AC2: 610 nM; AC5: 53 nM; AC6: 91 nM; sAC: > 100 μM; sGC: 710 nM; EF: 1.7 μM; CyaA: 6.5 μM; mouse heart AC: 21 nM. [22,28,29]. Competitive AC inhibition. Binds to the catalytic site. | MANT-GTP is the reference AC inhibitor for the group of (M)ANT-NTPs. MANT-GTPγS and MANT-GppNHp are hydrolysis-resistant versions of MANT-GTP. MANT-GTPγS and MANT-GppNHp were originally used as G-protein probes, but they possess higher affinities for mACs than for G-proteins [9,22]. MANT-GTPγS has been used as AC5 inhibitor in electrophysiological experiments [60]. MANT-GTP has been widely used for fluorescence studies with purified VC1:IIC2 to characterize both the catalytic and the diterpene site [23-25,28,40]. MANT-GTP has been used in crystallographic studies (PDB:1TL7) [23]. Note the lack of selectivity of MANT-GTP for AC5 relative to AC6. MANT-GTP is commercially available as an experimental tool. | |||
MANT-ITP
|
Ki values in AC assay (+ Mn2+): AC1: 2.8 nM; AC2: 14 nM; AC5: 1.2 nM; mouse heart AC: 4 nM. [28,29]. Competitive AC inhibition. Binds to the catalytic site. | MANT-ITP is the most potent competitive mAC inhibitor known so far. Based on previous data obtained with ITPγS and MANT-ITPγS [22], the exceptional potency of MANT-ITP at mACs was predicted. MANT-ITPγS has a higher mAC-selectivity relative to G-proteins than MANT-GTPγS [22]. The high affinity of MANT-ITP for mACs is explained by a tight interaction of the triphosphate chain with the protein [25]. However, in electrophysiological experiments, MANT-ITP exhibits off-target effects that are independent of AC inhibition (PDB:3G82) [30], despite its high affinity. MANT-ITP has been used in crystallographic and fluorescence spectroscopy studies [25,28]. The base hypoxanthine is generic and can bind to both adenine- and guanine nucleotide-binding proteins with substantial affinity. MANT-ITP should also be a potent sGC inhibitor. | |||
MANT-ATP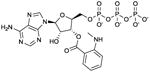
|
Ki values in AC/GC assay (+ Mn2+): AC1: 150 nM; AC2: 330 nM; AC5: 100 nM; AC6: 280 nM; sAC: 5.6 μM; sGC: 430 nM; EF: 230 nM; CyaA: 5.4 μM; mouse heart AC: 64 nM. [22,28,29]. Competitive AC inhibition. Binds to the catalytic site. | On first glance, it was quite unexpected to find that the adenine nucleotide MANT-ATP was not a more potent mAC inhibitor than the guanine nucleotide MANT-GTP [22]. Along the same line, sGC has no preference for guanine nucleotide-based inhibitors. However, with only few mutations substrate- and inhibitor-specificities of ACs and GCs can be switched [36,61]. MANT-ATP gives rise to smaller fluorescence signals with VC1:IIC2 than MANT-GTP, probably due to less favorable positioning of the MANT-group in the hydrophobic pocket [24]. MANT-ATP has been used in crystallographic studies (PDB:2GVZ) [24]. Note the lack of selectivity of MANT-ATP for AC5 relative to AC6. MANT-ATP is commercially available as experimental tool. In electrophysiological studies, MANT-ATP exerts paradoxical stimulatory effects that cannot be explained by AC5 inhibition, but must be due to hitherto unexplained off-target effects [30]. | |||
MANT-UTP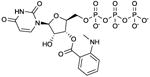
|
Ki values in AC assay (+ Mn2+): AC1: 46 nM; AC2: 460 nM; AC5: 32 nM; mouse heart AC: 12 nM [28,29]. Competitive AC inhibition. Binds to the catalytic site. | MANT-UTP is a prototypical representative of the pyrimidine-based mAC inhibitors, surpassing the affinities of MANT-ATP. The high-affinity inhibition of mACs by MANT-UTP reflects the broad base-specificity of the catalytic site of these enzymes [24]. MANT-UTP yields distinct fluorescence responses with VC1:IIC2 compared to other MANT-NTPs, reflecting unique positioning of the nucleotide in the catalytic site [28]. MANT-UTP could also be a potent sGC inhibitor. The broad base-specificity of mACs and sGC with a slight preference for the pyrimidine uracil relative to adenine is puzzling. The situation is reminiscent to the identification of UTP-binding P2Y-receptors two decades ago [62]. These UTP-binding GPCRs are now being firmly established. | |||
TNP-ATP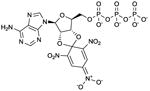
|
Ki values in AC/GC assay (+ Mn2+): AC1: 9.0 nM; AC2: 99 nM; AC5: 3.7 nM; sAC: 710 nM; sGC: 7.3 nM [27]. Competitive AC inhibition. Binds to the catalytic site. | TNP-ATP is the reference AC inhibitor for the group of TNP-NTPs [27].Overall, TNP-NTPs have been less extensively studied than (M)ANT-NTP-based AC inhibitors. TNP-AMP is effectively phosphorylated to TNP-ATP in AC reaction mixtures by the myokinase/pyruvate kinase-based NTP-regenerating system. Phosphorylation can also occur with MANT-NDPs [22]. This must be considered when studying mono- and diphosphates. TNP-NTPs give rise to large basal fluorescence signals upon interaction with purified VC1:IIC2. In contrast to the data obtained with (M)ANT-NTPs, FS reduces fluorescence signals with TNP-NTPs [27,28]. TNP-ATP has been used in crystallographic studies (PDB:2GVD) [24]. Notably, TNP-NTPs are also very potent sGC inhibitors [27]. One could envisage that TNP-NTPs (and possibly MANT-NTPs) are useful tools for achieving an elusive goal in nucleotidyl cyclase research, namely the crystallization of sGC. TNP-ATP binds to numerous types of nucleotide-binding proteins and is not specific for mACs or sGC [27,31]. TNP-ATP is commercially available as experimental tool. | |||
Bis-Cl-ANT-ATP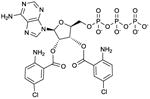
|
Ki values in AC assay (+ Mn2+): AC1: 1.7 μM; AC2: 2.4 μM; AC5: 1.6 μM; CyaA: 16 nM; EF: 220 nM [26,63]. Competitive AC inhibition. Binds to the catalytic site. | Bis-Cl-ANT-ATP serves as reference AC inhibitor for the group of bis-(M)ANT-NTPs. Bis-Cl-ANT-ATP constitutes a very potent CyaA inhibitor with substantial selectivity relative to mACs, indicating that in principle, by targeting the catalytic site, AC isoform-selectivity can be obtained. The catalytic site of EF is less spacious than the catalytic site of CyaA. Hence Bis-Cl-ANT-ATP is a less potent inhibitor of EF than of CyaA [63]. Bis-(M)ANT-nucleotides are also characterized by low basal fluorescence and high signal-to noise ratio in studies with purified CyaA. At mACs, these nucleotides have not yet been examined in fluorescence studied [26,63]. The affinity of mACs for bis-(M)ANT-nucleotides may be too low for fluorescence spectroscopy studies, but the high signal-to-noise ratio of the nucleotides may compensate for this disadvantage The introduction of two (M)ANT groups into an inhibitor increases substantially the number of possible chemical substitutions. This property may facilitate identification of mAC-selective inhibitors. However, spatial constraints in mACs are of concern. | |||
|
UTPγS |
Ki values in AC assay (+ Mn2+): VC1:IIC2: 8.5 μM; AC1: 53 μM; AC2: > 100 μM; AC5: 18 μM; sGC: 4.1 μM; sAC: > 100 μM [22]. Competitive AC inhibition. Binds to the catalytic site. | Originally, UTPγS was described as relatively potent Gs activator, giving rise to AC stimulation [22]. However, at concentrations above 1 μM, UTPγS (like ITPγS) causes strong AC inhibition. UTPγS and ITPγS have been very important tools for unmasking the broad base-specificity of mACs [22,64]. Notably, UTPγS is also a quite potent sGC inhibitor [22]. Based thereon, we assume that uracil nucleotide-based nucleotides can be developed into highly potent sGC inhibitors. For sGC crystallography, such compounds may be most useful. UTPγS is commercially available as experimental tool. | |||
|
2′,5′-dd-3′-ATP |
IC50 values in AC assay (+ Mn2+): AC1: 170 nM; AC2: 280 nM; AC6: 150 nM; AC7: 90 nM; AC8: 150 nM [15]. VC1:IIC2: 38 nM; AC1: 37 nM; AC2: 220 nM; AC5: 37 nM; sAC: 690 nM [22]. Non-competitive/un-competitive AC inhibition. Binds to the catalytic site. | 2′,5′-dd-3′-ATP is a prototypical polyphosphate-containing potent P-site inhibitor of mAC that has also been used in crystallographic studies (PDB:1CUL) [38]. The compound illustrates the difficulties in obtaining AC isoform-selective P-site inhibitors [15]. Notably, the compound is a relatively potent sAC inhibitor [22]. 2′,5′-dd-3′-ATP is commercially available as experimental tool. | |||
AraAde
|
IC50 values in AC assay (+ Mg2+): AC2: 700 μM; AC2: 380 μM; AC5: 9.8 μM [18]. Non-competitive AC inhibition. Probably binds to the catalytic site. | The compound is a virustatic drug [17] and constitutes a prototypical low-affinity P-site inhibitor. Clinically, potential cancerogenic, embryotoxic and antiproliferative effects of AraAde and structurally related compounds must be taken into consideration. Compounds structurally related to AraAde are supposed to act as selective AC5 inhibitors [17], but AC isoform has not yet been studied in sufficient detail. In addition, the low potency is of concern, increasing the probability of interactions with other targets and toxic effects when used in animals or humans. Also, it is difficult to obtain fully saturated concentration/response curves even in in vitro experiments. AraAde is commercially available as experimental tool. | |||
PMC6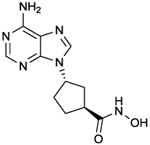
|
IC50 values in AC assay (+ Mg2+): AC2: 85 μM; AC2: 11 μM; AC5: 320 nM [18]. Non-competitive AC inhibition. Probably binds to the catalytic site. | The compound exhibits substantial selectivity for AC5 relative to ACs 2 and 3, but from molecular modelling studies (Fig. 3C), it remained unclear what the molecular basis for this selectivity may be. Formally, it cannot be excluded that PMC6 also binds to another site than the catalytic site. Similar considerations also apply to AraAde, NKY80 and NB001. Crystallographic studies would be required to answer the question although identification of a binding site beyond the crystallized VC1:IIC2 domains, e.g. in the transmembrane domains, is very challenging. The selectivity of PMC6 for AC6 and other targets (except for ACs 2 and 3) has not yet been examined. In cardiomyocytes, PMC6 exhibits beneficial effects on apoptosis, supposedly mediated via AC5 inhibition [18]. | |||
SQ 22,536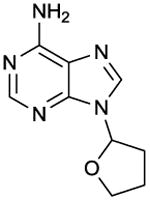
|
IC50 values in AC assay (+ Mg2+): AC1: 120 μM; AC2: 670 μM; AC6: 360 μM; AC8: 120 μM [15]. AC2: 290 μM; AC3: 100 μM; AC5: 2.2 μM [14]. Note the differences in IC50 values between ACs 5 and 6 between the two studies. The difference is difficult to interpret because a direct comparison of both AC isoforms has not yet been performed. Non-competitive AC inhibition. Probably binds to the catalytic site. | SQ 22,536 is one of the first mAC inhibitors developed and was introduced into experimental pharmacology [13]. Considering the structural similarity between ACs 5 and 6, the supposed AC5-selectivity of SQ 22,536 is quite amazing, but a direct side-by-side comparison of SQ 22,526 at both ACs 5 and 6 has not yet been presented. A problem in the analysis of SQ 22,536 is its rather low potency so that it is difficult to obtain fully saturated inhibition curves for all mAC isoforms. SQ 22,536 is probably the most-widely used mAC inhibitor, specifically with respect to intact cell studies. A pubmed research on September 15, 2011, revealed 377 entries with the key word “SQ 22,536”. In many studies the specificity of SQ 22,536 for mACs is taken for granted without considering off-target effects. However, considering the rather low potency of SQ 22,536, such effects cannot be excluded. Conversely, in some studies, SQ 22,536 was used at rather low concentrations (1 μM) [65] that may be insufficient to inhibit AC and prevent cAMP formation. SQ 22,536 is commercially available as experimental tool. | |||
NKY80
|
IC50 values in AC assay (+ Mg2+): AC2: 2.6 mM; AC3: 230 μM; AC5: 15 μM [18]. AC2: 1.7 mM; AC3: 130 μM; AC5: 8.3 μM [14]. Non-competitive AC inhibition. Probably binds to the catalytic site. | The compound constitutes a prototypical low-affinity P-site inhibitor and was identified in a virtual screen of > 850,000 compounds [14]. The compound is marketed as “selective AC5 inhibitor” for experimental purposes, but AC isoform has not yet been studied in sufficient detail. In fact, there are doubts whether NKY80 is selective for AC5 relative to AC6 [20]. In addition, the low potency of NKY80 for AC5 is of concern, increasing the probability of interactions with other targets. The lack of effects of NKY80 in a typical AC5 system has also been noted [18]. Due to the low affinity of NKY80 for mACs, it is difficult to obtain fully saturated concentration/response curves for precise calculation of IC50 values [14]. NKY80 is commercially available as experimental tool. | |||
NB001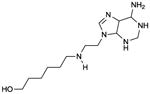
|
IC50 values in cAMP accumulation assay in intact transfected HEK293 cells: AC1: 10 μM; AC5: 210 μM; AC6: 170 μM; AC7: 190 μM; AC8: 140 μM [8]. Molecular mechanism undefined. Probably binds to the catalytic site. | The molecular mechanism of AC inhibition by NB001 has not yet been demonstrated because AC activity studies with membranes have not yet been performed. Only studies with intact cells have been performed. It cannot be taken for granted that cAMP accumulation assays in intact cells reflect AC activity even if phosphodiesterases are blocked. Specifically, intracellularly formed cAMP may be exported from cells via multidrug resistance proteins [66], introducing bias into the experimental setting. This specific experimental design renders comparison with other AC inhibitors that are routinely tested in the broken cell AC assay difficult. Based on our modelling (Fig. 3C), NB001 presumably acts as a non-competitive P-site inhibitor. However, our modelling failed to reveal the molecular basis for the AC1-selectivity. NB001 is a low-affinity mAC inhibitor, raising questions whether in addition to AC1, the compound also interacts with other targets in mammalian cells. The paper by Wang et al. [8] does not provide information on structure/activity relationships for AC inhibition by NB001 and related compounds. | |||
MDL 12330A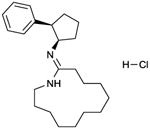
|
MDL 12330 inhibits histamine-stimulated AC activity in the guines pig ventricle according to a biphasic concentration/response curve. IC50-1 ∼20 μM; IC50-2 ∼ 300 μM [67]. MDL 12330 (100 μM) shows modest inhibition of ACs 2 and 3, but not of AC5 [14]. However, complete concentration-response curves have not yet been presented. | MDL 12330A, like SQ 22,536, is one of the most widely used mAC inhibitor for intact cell studies. A pubmed research on September 15, 2011, revealed 166 entries with the key word “MDL 12330A”.MDL 12330A is a non-nucleoside-based mAC inhibitor. It was introduced into experimental pharmacology into the early 1980s [67]. It is very well documented that in addition to AC inhibition, the compound exhibits numerous pleiotropic effects including inhibition of Na+/K+-ATPase and phosphodiesterases [16,67,68]. Nonetheless, in several studies using MDL 12330A, specificity for mAC inhibition is assumed although exceedingly high concentrations (up to 10 mM) are used [69]. Overall, MDL 12330A has not yet been thoroughly examined at mAC isoforms. In some studies, MDL 12330A exerts also stimulatory effects on AC [70], a possible indication for isoform-specific effects like with BODIPY-FS [11,34]. MDL 12330A is commercially available as experimental tool. | |||
BODIPY-FS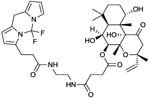
|
EC50 values in AC assay (+ Mn2+): AC1: 2.9 μM; AC5: 2.3 μM. IC50 value in AC assay (+ Mn2+): AC2: 1.2 μM [34]. The effects of BODIPY-FS strongly depend on the experimental conditions, the type of divalent cation being a critical determinant. In a follow-up study [11], the following data were obtained: EC50 values in AC assay (+ Mn2+): AC1: 1.2 μM; AC5: 2.7 μM. IC50 value in AC assay (+ Mn2+): AC2: 170 nM. EC50 values in AC assay (+ Mg2+): AC1: 900 nM; AC5: 24 μM. IC50 value in AC assay (+ Mg2+): AC2: 500 nM. Binds to the diterpene site. | BODIPY-FS is a partial agonist (compared to FS) in terms of AC1- and AC5 activation, but reduces the catalytic activity of AC2 in the presence of GTPγS [11,34]. The potencies and efficacies of BODIPY-FS are strongly determined by the divalent cation. Most notably, in the presence of Mg2+, the inhibitory effect of BODIPY-FS is much more pronounced than in the presence of Mn2+ [11]. BODIPY-FS is only weakly effective at activating VC1:IIC2 but inhibits the stimulatory effect of FS (3 μM) with an IC50 of 800 nM [34]. BODIPY-FS is a fluorescent molecule and has been studied to a very limited extent to localize mACs in cells [41]. BODIPY-FS could also be used as fluorescence probe for VC1:IIC2, but so far, no data have been published. BODIPY-FS was withdrawn from the market because of lack of demand, but because of the renewed interest in the compound, it is now again commercially available as experimental tool. Other fluorescent groups than BODIPY have not yet been assessed, but linkage of the FS core to other fluorophores is technically feasible. Thus, BODIPY-FS serves as a promising starting point for extensive structure/activity relationship studies. Based on the experimental data [11,34] and molecular modelling (Figs. 3A and B), the development of mAC isoform-selective diterpenes is anticipated. The BODIPY substituent increases the affinity of FS for mACs substantially. As control reagent for BODIPY-FS, the free dye, BODIPY was used [11,34]. This compound is devoid of effects on AC activity. | |||
1,9dd-FS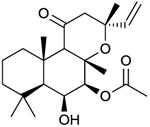
|
No activation of VC1:IIC2, AC1, AC2 or AC5 but apparently non-competitive inhibition of FS-stimulated catalysis of mACs [34,40]. Binds to the diterpene site. A recent study [41] suggests 1,9dd-FS can also exert stimulatory effects on as yet unidentified mAC isoforms | Historically, the compound was considered not to interact with mACs [4,39]. However, fluorescence studies with MANT-GTP at VC1:IIC2 clearly demonstrated that 1,9dd-FS (and other “inactive” 1d-FS derivatives) bind to the diterpene site without activating catalysis [34,40]. The non-competitive inhibition is probably due to the slow exchange of FS and 1,9dd-FS. Concentration-response curves for the inhibitory effects of 1,9dd-FS have not yet been performed. Only a fixed 1,9-dd-FS concentration of 100 μM was studied. 1,9dd-FS is very lipophilic so that very high concentrations of dimethyl sulfoxide (up to 6%, v/v) have to be used in experiments. Fortunately, mACs expressed in Sf9 cells and VC1:IIC2 are very dimethyl sulfoxide-resistant. In fact, we recommend to use high dimethyl sulfoxide concentrations in experiments with 1,9dd-FS in particular and with diterpenes in general because otherwise, incomplete dissolution of compounds introduces substantial experimental errors. The poor water-solubility of 1,9dd-FS limits the use of this compound in intact cell studies. 1,9dd-FS is commercially available as experimental tool. | |||
6A7DA-FS
|
AC assay in the presence of Mg2+:AC1: EC50, 6.5 μM. AC2: IC50, 1.8 μM; EC50, 61 μM. AC5: EC50, 52 μM [11]. The inhibitory effect of 6A7DA on AC2 in the presence of Mg2+ is relatively small and is not observed in the presence of Mn2+. Binds to the diterpene site. | 6A7DA is also referred to as iso-forskolin because the acetyl group is switched from the 7-position to the 6-position of the diterpene ring. In the presence of Mn2+, FS and 6A7DA-FS are similarly potent activators of ACs 1, 2 and 5, but there are differences in efficacy. In the presence of Mg2+, FS and 6A7DA-FS are similarly potent activators of AC1, but at AC5, 6A7DA is about ten-fold less potent than FS [11]. Most notably, in the presence of Mg2+, 6A7DA-FS exerts high-potency inhibitory effects and low-potency stimulatory effects on AC2 [11]. These data corroborate the unique position of AC2 among mACs in terms of inhibition. Biphasic effects on mACs were also observed for calmidazolium [12]. | |||
Calmidazolium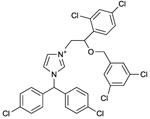
|
IC50 values in AC assay (+ Mg2+): mouse AC9: ∼6 μM; human AC9: ∼15 μM; rat AC2, ∼30 μM; soluble C1-C2 fusion protein from AC9: 8 μM [12]. Binds to a still undefined site. The IC50 of calmidazolium for calmodulin in a fluorescence binding assay is 2-3 nM [71]. | Calmidazolium is a well-known high-affinity calmodulin antagonist [71]. However, calmidazolium also binds to other proteins exhibiting hydrophobic sites, i.e. many effects of calmidazolium are actually calmodulin-independent [72]. The precise molecular mechanism by which calmidazolim inhibits mAC activity has not yet been determined [12], but it is not dependent on calmodulin. Notably, at a concentration of ∼5 μM, calmidazolium stimulates human AC9 by up to ∼60%. At a concentration of ∼2-3 μM, calmidazolium stimulates mouse AC9 by up to ∼70%. At a concentration of 10 μM, calmidazolium increases AC2 activity even by ∼220%. At higher concentrations, very steep inhibition isotherms are observed for all mACs studied so far, indicative for cooperative inhibitor binding. The peculiar (biphasic and steep) concentration-response curves for calmidazolium could point to the existence of multiple ligand binding sites or to a single binding site adopting two affinity states and displaying positive cooperativity. To our knowledge, this is the first study in which species-dependent sensitivity of a mAC to activators and inhibitors has been observed. Recently, we observed biphasic stimulatory and inhibitory effects of 6A7DA-FS on AC2 [11]. Binding of calmidazolium to hydrophobic sites in proteins other than calmodulin has been repeatedly reported [72]. Hence, again, target-specificity of a mAC inhibitor is of concern. The concentrations required to inhibit mACs are considerably higher than these needed to block calmodulin function [71]. Calmidazolim is commercially available as experimental tool. Calmidazolium may represent an archetype of allosteric mAC inhibitor targeting as yet unexplored sites that may encompass the poorly studied transmembrane domains. | |||
Tyrphostin A25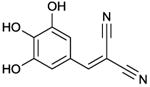
|
IC50 value in AC/GC assay (+ Mn2+): GC-C: 5.8 μM; sGC: 34 μM; AC-HEK cells: 120 μM; C1-C2 fusion protein (C1 from AC1 and C2 from AC2): 16 μM [59]. Binds to a still undefined site. | The precise molecular mechanism by which tyrphostin A25 inhibits mAC and GC activity has not yet been determined [59]. Tyrphostin A25 has been suggested to bind to hydrophobic residues close to the catalytic site [59]. An interesting aspect of [59] concerns the fact that this is, to our knowledge, the only report reporting on inhibition of a particulate GC in comparison to mACs. The area of particulate GC inhibition needs to be developed much more intensively. Tyrphostin A25 is commercially available as experimental tool. Tyrphostin may represent an archetype of allosteric mAC inhibitor targeting as yet unexplored sites that may encompass the poorly studies transmembrane domains. | |||
In the Table, representative mAC inhibitors from various chemical classes are listed. Structures and pharmacological key data are shown, and specific compound properties and problems are discussed. Please, note that due to space limitations, the list of inhibitors is not comprehensive. This table focuses only on these inhibitors that have been examined at various ACs (and GCs). Inhibition data of (M)ANT- and TNP-nucleotides for VC1:IIC2 are listed in Fig. 2F [22,24,28]. It should be noted that the potencies of mAC inhibitors differ vastly from each other. For example, MANT-ITP inhibits AC5 with a Ki of a bout 1 nM, whereas the “AC5-selective inhibitor” NKY80 inhibits AC5 with an IC50 of about 10 μM, i.e. the potency difference amounts to about 10,000-fold. Even the highly potent MANT-ITP exhibits effects unrelated to AC5 [30], rendering it likely that much less potent mAC inhibitors such as NKY80 and NB001 also exhibit off-target effects. The possibility of off-target effects of mAC inhibitors has not yet been comprehensively studied and must be carefully examined in future studies. For competitive inhibitors, Ki values are given; for non-competitive inhibitors, IC50 values are given.
Table 2. Overview on patent activities in the AC field with emphasis on mAC inhibitors.
| Indication | Patent Titles | Key Patents and Applications | Proposed Use of AC-Inhibitors | AC inhibitors/compounds (name listed as written in patent) |
|---|---|---|---|---|
| Senescence | Signals and molecular species involved in senescence | WO2009139511 US7482134 |
|
|
|
| ||||
| Heart Disease | Treatment of cardiac disease including heart attack, myocardial apoptosis, and heart failure, comprises administering type 5 adenylyl cyclase inhibiting compound | WO2009099676 |
|
|
| New 9-substituted adenine derivatives are adenylyl cyclase inhibitors used to treat cardiac function and performance and as cytostatics. | WO2002004475 US7045309 |
|||
|
| ||||
| Intestinal fluid loss | Treatment of intestinal fluid loss e.g. diarrhea or condition associated with increased 3′-5′-adenosine monophosphate levels involves administering composition comprising e.g. cycloalkenone derivatives, and tricyclic and bicyclic derivatives | WO2001094369 US20020188016 US20020032228 EP1353664 |
|
|
|
| ||||
| Fibropro-liferative vasculopathy | New adenine derivatives, used to treat e.g. chronic allograft rejection, vascular restenosis, congestive heart failure, psoriasis, tumor growth, diabetic retinopathy and arteriosclerosis, are adenyl cyclase inhibitors | WO2002040481 US6887880 |
|
|
|
| ||||
| Circadian Rhythm | Use of composition comprising inhibitor of adenylyl cyclase (e.g. purine site inhibitor) for elongation of circadian rhythm and for treating e.g. jet lag, familial advanced sleep phase syndrome and shift lag | WO2007135387 |
|
|
|
| ||||
| Wound healing | Pharmaceutical composition useful for promoting wound healing in mammals, preferably humans, comprises adenylyl cyclase inhibitor and lysophosphatidic acid inhibitor | WO2010126260 |
|
|
We performed a keyword search in the Thompson Innovation patent literature databases including the Derwent World Patents Index (http://www.thomsoninnovation.com/ti/contentsets/patents/). The search term was: CTB=((adenylyl or adenylate) cyclase inhibitor*) OR CTB=(ADCY* inhibitor *) and the date of search was June 5th, 2011. The initial 247 hits were individually reviewed, the patents dealing with mAC inhibitors were identified, and the information contained therein was condensed and compared according to the criteria of the Table. The usefulness of the patents will be critically determined by the selectivity of the compounds for mACs or specific mAC isoforms relative to other targets. As is outlined in the text and Table 1, specificity of nucleoside-based inhibitors among AC isoforms and off-target effects are a great concern. One prominent example in this regard is SQ 22,536 (also referred to as 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (THFA or 9-THF-Ade)), mentioned in several patents. The different nomenclature of this compound in various publications [13-16,65] may have caused some confusion in the literature regarding compound identity.
Challenges to isoform-specific mAC inhibitors
AC inhibitors are divided into four classes: i), inhibitors competing with the substrate ATP at the catalytic site [9]; ii), non-competitive/un-competitive inhibitors mimicking the cAMP·PPi transition state (P-site inhibitors) [10]; iii), allosteric non-competitive inhibitors targeting the diterpene site [11]; and iv), allosteric non-competitive inhibitors targeting as yet undefined sites [12]. Both the catalytic and diterpene site are highly conserved among mAC isoforms (Figure 1). Thus, from a structural perspective, the development of mAC isoform-selective inhibitors is very challenging.
Fig. 1. Multiple sequence alignment of the C1 and C2 subunits of mAC isoforms and of sGC α1- and β1 subunits.




Sequences are identified with human (hAC), murine (muAC) or human soluble GCS (hsGC) and a unique GenBank identification number. The full sequence of AC1 is shown as reference sequence. A, C1 domain of ACs and α1-subunit of hsGC; B, C2 domain of ACs and β1-subunit of hsGC. The conserved residues correspond to the C1 and C2 subunits of other AC isoforms and hsGC subunits and are indicated as “.”; amino acid differences are indicated in the one-letter code, and sequence gaps are indicated as “-”. The secondary structures correspond to the C1 from AC5 (VC1) and C2 from AC2 (IIC2) constructs that were determined by X-ray crystallography and are shown above the alignment; arrows represent β strands, and the cylinders represent α helices. Other structural elements, such as random coils and turns, are represented by a solid line. The functional residues in C1 and C2 subunits are indicated: bold, substrate binding; bold/italic, FS binding. The underlined residues indicate the hydrophobic region important for binding to (M)ANT- und TNP groups of 2′,3′-O-ribosyl-substituted nucleotides.
Historically, research on mAC inhibitors has focused on the catalytic site. The first mAC inhibitors available were nucleoside-based compounds such as SQ 22,536 [9-(tetrahydro-2-furanyl)-9H-purin-6-amine; also known as THFA or 9-THF-Ade] that inhibit mACs non-competitively [13]. Although these compounds are sufficiently lipophilic to penetrate the plasma membrane so that they can be used in intact cell studies, the generally low potency of these compounds is of concern [14,15]. Given the fact that high concentrations (often above 100 μM) are required to elicit effects [3], limited solubility and off-target effects cannot be dismissed. In intact cell studies it is often assumed that AC inhibitors reduce cAMP concentrations, but cAMP concentrations are actually not determined [16]. Moreover, the low potency of compounds renders it very difficult to achieve full saturation in concentration/response curves so that IC50 values cannot be precisely calculated [14].
Investigators who use AC inhibitors as pharmacological tools in their specific fields of research may not be sufficiently aware of potential off-target effects. One typical P-site inhibitor, AraAde [9-β-D-arabinosyladenine (vidarabine)], is also used as virustatic drug [17], and it is likely that such nucleoside-based AC inhibitors also interfere with purine metabolism and DNA synthesis and exhibit long-term cytotoxic effects. However, a systematic analysis of the off-target effects of P-site inhibitors has not yet been performed. So far, no mAC inhibitor has been systematically examined at all mAC isoforms with a complete profile of IC50 values/Ki values. Hence, claims about mAC isoform-specificity of a given compound are generally not sufficiently supported by experimental data. “Selective AC5 inhibitors” are a particular problem in this respect [14,18-21]. Such compounds should discriminate between ACs 5 and 6, but there is little compelling evidence that currently available inhibitors do so when they are compared side-by-side [22].
MANT [2′(3′)-O-(N-methylanthraniloyl]- and TNP [2′,3′-O-(2,4,6)-trinitrophenyl)]-substituted nucleotides are much more potent mAC inhibitors than P-site inhibitors [9,14,15,22-29]. However, MANT/TNP-nucleotides are too hydrophilic to penetrate the plasma membrane and are, therefore, not useful for intact cell studies. In an electrophysiological study, MANT-nucleotides were delivered into the cytosol of cardiomyocytes via the patch pipette to inhibit AC5-dependent activity of voltage-dependent calcium channels [30]. Strikingly, even the most potent mAC inhibitor known so far, MANT-ITP, exhibits unexpected paradoxical stimulatory effects on calcium channel activity that cannot be explained by AC5 inhibition [30]. MANT/TNP-nucleotides can bind with considerable affinity to purinergic receptors, G-proteins and protein kinases, thereby modulating signal transduction processes [9,27,30,31].
A less intuitive target for the development of mAC inhibitors is the diterpene site because traditionally, this site has been associated with AC activation [4,32]. Substantial efforts were devoted towards the development of mAC isoform-selective diterpenes, but with only modest success [11,14,33,34]. The serendipitous identification of BODIPY-FS as an AC2 inhibitor [11,34], as opposed to being an activator of ACs 1 and 5, showed that the diterpene site is a valuable target for mAC inhibitors. However, as is the case for nucleoside/nucleotide-based inhibitors targeting the catalytic site [9,30,31], diterpenes bind to, and influence the activity of, multiple other targets such as glucose transporters [4,34,35].
Exploring the catalytic site of mACs with MANT- and TNP-nucleotides
MANT-nucleotides nucleotides have provided valuable structural information on the properties of the catalytic site of mACs. Four crystal structures of the purified catalytic mAC subunits VC1:IIC2 in complex with various MANT- and TNP-nucleotides have been resolved (Figures 2A and C) [23-25]. This information provides an excellent basis for future development of non-nucleoside/nucleotide-based mAC inhibitors. The inhibitory potencies of MANT-nucleotides are catalysis-dependent, (i.e. the higher the AC activity is, the more potent are the MANT-nucleotides) [9,22-29]. This property may be of therapeutic relevance because activity-dependent AC inhibition could result in selective inhibition of pathologically increased cAMP formation while leaving normal AC activity unaffected. Catalysis-dependent AC inhibition has also been observed for P-site inhibitors [10,15,18].
Fig. 2. Crystal structures and general pharmacophore model of VC1·IIC2 in complex with MANT- or TNP-nucleotides or P-site inhibitors.



All data presented in this Figure refer to VC1:IIC2. A, Detailed views of the substrate binding site in the complex of Gαs–activated mAC with the competitive inhibitors MANT-GTP (PDB:1TL7) [23], MANT-ATP (PDB:2GVZ) [24], MANT-ITP (PDB:3G82) [24] and TNP-ATP (PDB:2GVD) [25], and two Mn2+ ions. Structures of inhibitors as bound to their respective complexes with Gαs·VC1:IIC2 are superimposed. The molecular surface was calculated using PYMOL (DeLano Scientific, San Carlos, CA, USA), based on the atomic coordinates of the Gαs·VC1:IIC2·TNP-ATP complex. Ligands are shown as stick models. Carbon atoms are colored magenta for MANT-GTP, cyan for MANT-ATP, yellow for MANT-ITP, and gray for TNP-ATP, nitrogens blue, oxygens red, sulfur yellow, and phosphorous green; the two Mn2+ ions are shown as metallic orange spheres. The secondary structures of VC1 and IIC2 domains are shown in tan and mauve, respectively. Ligands and two metal ions occupy the interdomain cleft between the C1 and C2 domains. Inhibitors prevent transition of the enzyme from the catalytically inactive open conformation to the catalytically active closed conformation because the MANT- and TNP-groups act like rigid body movement-impairing wedges. The MANT- and TNP groups insert into a hydrophobic pocket close to the catalytic site, providing substantial binding energy and giving rise to hydrophobicity-dependent fluorescence increases. Substitution of the 3′-hydroxyl group in MANT- and TNP-nucleotides prevent the 3′:5′-ATP cyclization reaction. B, Comparative views of substrate binding site of the Gαs-activated mAC complex with the prototypical non-competitive/uncompetitive P-site inhibitor, 2′,5′-dideoxy-3′-ATP (PDB:1CUL) [38]. Atoms are colored according to panel A. The Mg2+ ions are shown as metallic limeyellow spheres. Note that 2′,5′-dideoxy-ATP, while occupying the catalytic site, in contrast to MANT- and TNP-nucleotides, does not exploit the hydrophobic pocket. Right-most panels show views of the binding pocket for ribose substitutes of inhibitors and are rotated ∼80° relative to the view shown on the left-most panels. The ribose substituents of inhibitor molecules are positioned between the α4 helix of IIC2 and α1- α2 helices of VC1. C, Structures of MANT-ATP, MANT-GTP, MANT-ITP, and TNP-ATP, as bound to their respective complexes with Gαs·VC1:IIC2 are superimposed and colored as above. Average Ki values are indicated, corresponding to contributions from each type of functional group, derived from singular value decomposition analysis (SVD) D, SVD analysis of the κi values for independent components from a set of AC inhibitors. SVD analysis was performed as described [16] using published Ki values as basis [22,24,28]. P, monophosphate; PP diphosphate; PPP for triphosphate; PPSP, [γ-thio]triphosphate; PPNP, [β,γ-imido]triphosphate.
MANT-nucleotides and the structurally related TNP-nucleotides inhibit ACs 1, 5 and 6 with similar potencies, whereas AC2 is inhibited only ∼10-fold less potently [22,26-29]. Hence, as for the classic P-site inhibitors, no compelling isoform-specificity was observed [15]. Part of the moderate preference of MANT-nucleotides for ACs 1 and 5 relative to AC2 is due to an alanine→proline exchange (position 409, VC1 numbering) in AC2, hindering the movement of an α-helix that is important for hydrophobic interactions of the MANT-group with mACs in a closed conformation [23]. MANT- and TNP-nucleotides also potently inhibit nitric oxide (NO)-stimulated soluble guanylyl cyclase (sGC) [22,27]. Considering the structural similarities of mACs with sGC in the catalytic core (Figure 1) [1,36], the overlap in inhibitor pharmacology is not unexpected. By contrast, the structurally distinct sAC is much less sensitive to inhibition by MANT- and TNP-nucleotides than mACs and sGC [22,27]. Certain bis-MANT-nucleotides, bearing a MANT-group both at the 2′- and 3′-O-ribosyl position, inhibit the Bordetella pertussis AC toxin CyaA much more potently than mACs [26], opening the door for the development of potent CyaA inhibitors for treatment of whooping cough. Like protein kinases [37], mACs possess a tripartite catalytic site with binding regions for the base, the ribosyl moiety and the triphosphate chain (Figure 2C) [24]. Protein kinases and ACs exhibit a hydrophobic pocket adjacent to the ribosyl moiety that is not exploited by the substrate ATP, but is occupied by hydrophobic 2′- and 3′-O substitutents of inhibitors. For the development of potent and isoform-selective protein kinase inhibitors, this hydrophobic pocket is crucial [37].
In the crystal structures of VC1:IIC2 in complex with MANT-GTP, MANT-ITP, MANT-ATP and TNP-ATP, the base, the ribosyl substituent and the triphosphate chain align very similarly (Figure 2C) [23-25]. The triphosphate chain of MANT-ITP tightly associates with Mn2+, resulting in an exceptionally high affinity of VC1:IIC2 for MANT-ITP [25]. The (M)ANT- and TNP groups provide the largest contribution to inhibitor affinity (Figures 2C and D) [24]. Among the ribosyl substituents, the order of preference is MANT > ANT > TNP. The dominant contribution of these substituents to inhibitor affinity is afforded by the aforementioned hydrophobic pocket adjacent to the catalytic site of AC [24]. By analogy to the situation with protein kinase inhibitors [379], this pocket is of high importance for the development of high-affinity non-nucleoside/nucleotide-based inhibitors. The length and nature of the phosphate chain provide the second-largest contribution to inhibitor affinity; γ-phosphate > βγ-imidophosphate > γ-thiophosphate (Figures 2C and D). Among the bases, hypoxanthine is optimal, although it forms fewer hydrogen bonds with mAC than guanine or adenine. Probably, the smaller molecular volume of hypoxanthine relative to guanine allows for a better fit of the MANT-group into the hydrophobic pocket [25]. mACs exhibit preference for uracil relative to adenine, guanine and cytosine (Figure 2D) [24,27,28]. The broad base-specificity of mACs provides ample opportunities for structural variations in future inhibitor development.
Recent developments on P-site inhibitors
Although highly potent P-site inhibitors such as 2′,5′-dideoxy-3′-ATP have been described and mAC crystal structures with this ligand resolved (Figure 2B) [10,15,38], a major problem has been the lack of isoform-specificity of these compounds [10,15]. More recently, non-nucleoside P-site inhibitors with supposedly higher mAC isoform-specificity have been reported (Table 1). Patent activity on P-site inhibitors, in contrast to competitive inhibitors or diterpenes, has been substantial (Table 2), but documentation of the AC isoform-selectivity of the disclosed compounds such as SQ 22,536 is incomplete, most notably with respect to a side-by-side comparison of ACs 5 and 6 [14,15].
PMC6 [1R,4R-3-(6-aminopurin-9-yl)-cyclopentanecarboxylic acid hydroxyamide], AraAde and NKY80 [2-amino-7-(2-furanyl)-7,8-dihydro-5(6H)-quinazolinone] exhibit selectivity for AC5 relative to ACs 2 and 3 [14,18]. Although these AC isoforms belong to different families [1,2], true AC5-selectivity can only be claimed when all mACs are examined and off-target effects ruled out. We conducted molecular modelling studies with PMC6, AraAde and NKY80 using VC1:IIC2 as template (Figure 3C). Our in silico studies did not reveal a structural basis for selectivity at the AC5 catalytic site. Publications on PMC6, AraAde and NKY80 did not provide structural information for the supposed AC5-selectivity either [14,18]. It is possible that novel P-site inhibitors bind to an as yet unidentified allosteric mAC site that is not the catalytic site.
Fig. 3. Model of the interactions of BODIPY-FS and various P-site inhibitors with mAC.
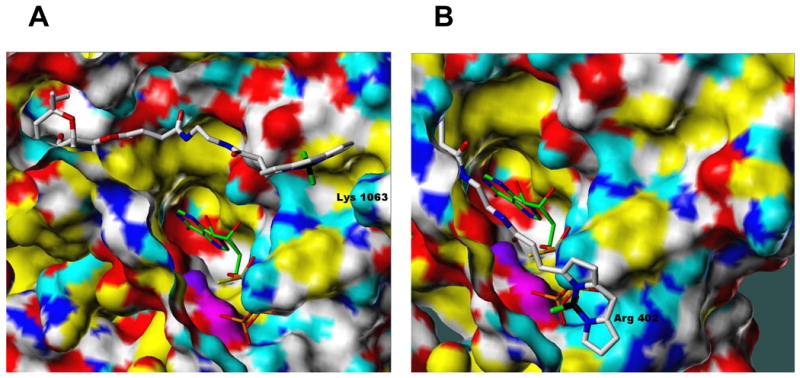

Molecular modelling studies were performed using the Surflex module in SYBYL 8.1 (Tripos Associates, St. Louis MO, 2010). To dock BODIPY-FS (in A, B), the Surflex protomol file was defined by residues within 5.0 Å of the known diterpene site plus any cationic residues within 15.0 Å of the diterpene site. To dock non-nucleoside P-site inhibitors (in C), the protomol file was defined by residues within 5.0 Å of the catalytic site. Atoms of each ligand are represented as sticks according to standard CPK coloring except for boron (black; present only in A and B) and carbon atoms (specified below). A, Interaction of BODIPY-FS (white C atoms) with AC5 showing the relative position of co-crystallized ATPαS (green C atoms). B, Interaction of BODIPY-FS (white C atoms) with AC2 showing the relative position of co-crystallized ATPαS (green C atoms). C, Interactions of ATPαS (white carbons), PMC6 (green carbons), AraAde (black carbons), NKY80 (pink carbons) and NB001 (brown carbons) with VC1:IIC2. In all cases above, the receptor surface is represented as a solvent-accessible Connolly surface, colored as follows: lipophilic regions are yellow, polar oxygens are red, polar nitrogens are blue, donatable protons are cyan, and polarized alkyl or aryl moieties are white. Approximate locations of the α-carbon atoms of key residues are labeled for reference. Note that interactions of the P-site inhibitors shown in C with sites in mACs other than the catalytic site cannot be excluded, particularly in light of the fact that our modeling does not explain the reported mAC isoform-specificity of the compounds (see Table 1). Moreover, crystal structures of VC1:IIC2 with the above-mentioned P-site inhibitors, in contrast to MANT-nucleotides, TNP-nucleotides and 2′,5′-dd-3′-ATP (see Fig. 2 and Table 1) are not yet available.
An advantage of PMC6, AraAde and NKY80 relative to (M)ANT- and TNP-nucleotides is that they can penetrate the plasma membrane [14,18,30] (Table 1). A disadvantage, however, is their low potency relative to (M)ANT- and TNP-nucleotides, raising concerns regarding target-specificity. As an example, NKY80, distributed as a “selective AC5 inhibitor” for experimental purposes, inhibits renin release from juxtaglomerular cells, so that is has been suggested that AC5 mediates protease secretion [19]. However, studies with knockout mice unequivocally revealed that both ACs 5 and 6 are involved in the activation of renin release [20]. These data highlight the importance of discriminating between effects on ACs 5 and 6 and the need for control experiments with AC knockout mice. In another study, NKY80 was designated as a dual AC5/6 inhibitor [21], although AC6 data are not yet available. NKY80 failed to inhibit cAMP accumulation in mouse cardiomyocytes [18]. Considering the structure of NKY80 (Table 1) and the fact that NKY80 shows effects in other intact cell systems [19], it is unlikely that the lack of effect of the compound in cardiomyocytes [18] is attributable to inability of NKY80 to penetrate membranes. Perhaps, inhibition of ACs 5 and 6 is rescued by as yet unidentified NKY80-insensitive ACs expressed in heart [29]. Alternatively, NKY80 could be converted to an as yet unknown inactive metabolite.
Selective AC1 inhibitors may be useful for neuroprotection because AC1 is activated by Ca2+/calmodulin following stimulation of various [Ca2+]i-increasing receptors, most notably ionotropic glutamate receptors [7]. Moreover, AC1 inhibition may be beneficial for treatment of chronic pain [3,8]. (M)ANT- and TNP-nucleotides are not selective for AC1 relative to AC5 [22-29]. Recently, NB001 [6-((2-(6-amino-9H-purin-9-yl)ethyl)amino)hexan-1-ol] has been suggested to be a selective AC1 inhibitor [8]. In silico studies indicate that like PMC6, AraAde and NKY80, NB001 can bind to the catalytic site of mACs, and the alcohol tail stretches to the phosphate-binding region (Figure 3C). Unfortunately, it is not clear why NB001 exhibits AC1-selectivity, and other binding sites of NB001 on mACs cannot be excluded. Moreover, the molecular mechanism of action of NB001 (competitive versus non-competitive) has not yet been examined, and a comprehensive analysis of all mAC isoforms has not yet been presented. Like NKY80, AraAde and PMC6, NB001 is not a potent AC1 inhibitor [8,14,18]. Despite these shortcomings, NB001 exhibits beneficial effects in animal models of chronic pain, supporting the notion that AC1 inhibition may be a valuable therapeutic principle [8].
The diterpene site as target for mAC inhibitors
Traditionally, diterpenes have been associated with stimulatory effects on mACs [4,32]. Early studies showed that ACs 1, 2 and 5 interact differently with diterpenes [33], and later studies documented different activation patterns of these ACs by diterpenes [11,34]. Fluorescence studies with MANT-GTP at VC1:IIC2 revealed that, in contrast to earlier assumptions [39], 1-deoxy-forskolin (1d-FS) and 1,9-dideoxy-forskolin (1,9dd-FS) bind to AC [40]. Unlike FS and 9-deoxy-forskolin (9d-FS), 1d-FS and 1,9dd-FS stabilize a catalytically inactive conformation of mAC. Formation of a hydrogen bond between the 1-OH group of diterpenes and Val-506 (VC1 numbering) is crucial for catalytic activation [40]. 1d-FS and 1,9dd-FS inhibit FS-stimulate cAMP formation both at VC1:IIC2 and intact mACs [34,40]. Intriguingly, a recent study on TRPC6 channel regulation by cAMP-dependent protein kinase suggests that 1,9dd-FS can also exert stimulatory effects on certain as yet unidentified mAC isoforms [41]. Thus, a careful analysis of the effects of both FS and 1,9dd-FS on all mAC isoforms is warranted.
BODIPY-FS was synthesized as a fluorescence probe to study AC localization in intact cells, but it has not yet been widely used for this purpose. BODIPY-FS is a partial activator of VC1:IIC1 and ACs 1 and 5 [11,34]. Unexpectedly, BODIPY-FS substantially reduces Gs-stimulated activity of AC2 [11,34]. A specific requirement of Gs for diterpene interaction with AC2 was noted previously [33]. An explanation for this observation is provided by molecular modelling studies (Figures 3A and B). In the case of AC2, the negatively charged BODIPY group is attracted to the positively charged Arg-402 and Mn2+ ion, whereas in AC5, BODIPY is attracted to Lys-1063 (an Asp residue in AC2 and the VC1:IIC2 complex). If one docks the ATPαS substrate mimic (protein data bank (PDB):1CJK) into these models, BODIPY and the tether may interfere with binding of the γ-phosphate in AC2. An arginine corresponding to Arg-402 in AC2 is also found in ACs 4 and 7, and in AC9, there is a lysine. Thus, we predict that BODIPY-FS also reduces the basal activity of ACs 4, 7 and 9. In ACs 5 and 6, nucleophilic amino acids are at this position, a glycine in AC1, and a glutamine in AC3. Thus, ACs 1, 3, 5 and 6 are expected not be inhibited by BODIPY-FS. In fact, BODIPY-FS partially activates ACs 1 and 5 [11,34]. Introduction of the bulky BODIPY group into ligands of the diterpene site increased affinity [11,34], indicating productive interactions of BODIPY with mACs. Intriguingly, 6-acetyl-7-deacetyl-forskolin (6A7DA-FS) constitutes a dual high-potency inhibitor/low-potency activator of AC2 in the presence of Mg2+ [11], indicating that the diterpene site can exist in different affinity states. [3H] Forskolin binding studies in rat brain membrane revealed high- and low-affinity binding sites, too, but it remained unclear whether these sites are attributable to mACs only [39].
Remaining questions
There is great interest in obtaining selective AC5 inhibitors as potential drugs for treatment of heart failure and ageing (Table 2) [6]. However, based on the high amino acid sequence similarities of the C1- and C2-domains, respectively, of mAC isoforms (Figure 1), pharmacological discrimination between ACs 5 and 6 is challenging, at least when the catalytic site is targeted [15]. Even if selective AC5 inhibitors can be developed, neurotoxicity constitutes a serious issue. Specifically, AC5 knockout leads to extrapyramidal motor disorders, alcohol addiction and stress-related behavioural problems [42-44]. Thus, achievement of organ-selectivity of AC5 inhibitors is crucial. Originally, ACs 5 and 6 were assumed to play opposite roles in heart function [2,3], but recent data have raised doubts about this notion [45]. Thus, future studies will have to answer the question whether it is necessary to discriminate between ACs 5 and 6 [6,22]. Perhaps, dual AC5/6 inhibitors are more useful than currently appreciated. At least, the development of dual AC5/6 inhibitors is a more realistic goal than the development of selective AC5 inhibitors.
There are significant differences in basal (constitutive) activity among various recombinantly expressed AC isoforms, with AC2 exhibiting particularly high constitutive catalytic activity [46]. However, whether mACs also exhibit different basal activities in physiological systems is unknown. Most AC studies have focused on activation mechanisms, and therefore, basal catalysis has been mostly considered as “noise”, serving to define the zero point in a normalized stimulation experiment. Demonstration of physiologically significant constitutive activity by certain mAC isoforms may justify a search for specific inhibitors of basal catalytic activity that, by analogy to the two-state model of GPCR activation, could be classified as inverse agonists [47].
A general problem in the field is that no single inhibitor has been examined at all mAC isoforms, sAC and guanylyl cyclases (GCs). Although our group has almost two decades of experience in expressing mACs in Spodoptera frugiperda Sf9 cells [48], nonetheless, we have encountered great difficulties in functionally expressing AC isoforms other than ACs 1, 2 and 5 in this system. Other leading groups also focus their efforts on these three ACs [33,49] that express very robustly. ACs 1, 2 and 5 represent prototypical members of various AC families [1-3]. It is reasonable to assume that within a given mAC family, pharmacological similarities are greater than among different mAC families. Co-expression with pharmacological chaperones may be a useful strategy to improve expression of “difficult” AC isoforms. In case of GPCRs, this strategy has been successful [48]. Efforts to stably express all mAC isoforms in mammalian expression systems are necessary, too. An advantage of this strategy may be that, unlike in insect cells, relevant mAC-interacting proteins are endogenously expressed [50].
Unfortunately, there is also a paucity of isoform-selective mAC antibodies [29,51]. Such antibodies must be tested against all mAC isoforms and in tissues of mAC knockout animals. The rigorous quality criteria established for GPCR antibodies must also be met for mAC antibodies [52], specifically in view of the fact that in native cells, mACs, are expressed only at low levels, increasing the risk of non-specific immunoreactions. Only with meticulous immunological studies will we be able to observe the true expression patterns of ACs at the protein level. To partially compensate for the lack of high-quality mAC antibodies, we have used a combination of pharmacological activators and inhibitors to characterize AC isoforms in organs [29,53]. However, this approach is not perfect. With few exceptions [54], previous studies on the expression of AC isoforms at the mRNA level did not allow quantitative comparison of AC isoform expression patterns. Only real-time polymerase chain reaction (PCR) studies with appropriate calibrations are suitable for comparison of relative expression levels of AC isoforms.
In the GPCR field, striking species differences in pharmacological properties have been observed [55]. However, with respect to AC inhibition, the species issue has been largely ignored so far, although there is evidence for species-specific inhibition of AC9 [12,56]. There is only a limited number of amino acid differences in the C1- and C2 domains, respectively, between human and mouse AC9 (Figure 1) so that site-directed mutagenesis can be used to unmask the molecular basis for the different pharmacological properties between AC9 species orthologs. Thus, mammalian expression plasmids and/or baculoviruses for all mAC isoforms from several species, at least from human and mouse, are needed.
Future directions
Future studies aiming at the development of inhibitors targeting the catalytic site of ACs, by analogy to protein kinases [37], should consider non-nucleoside/nucleotide-based compounds. Such studies entail high-throughput screening and are a task for the pharmaceutical industry. mAC inhibitors, particularly if they specifically explore the hydrophobic pocket in the catalytic site (Figure 2A), should possess better membrane-permeability than nucleosides/nucleotides.
In principle, it is possible to deliver nucleotide-based mAC inhibitors into cells as mononucleoside phosphate prodrugs [57]. However, this approach is more complicated than it appears on first glance. For example, certain types of phosphate-protecting groups work well, whereas others, for unknown reasons, do not. Moreover, in some cases, very substantial inhibitor concentrations may build up in cells so that off-target effects can become an issue [57]. Furthermore, the complement of cells with the enzymes required for prodrug conversion to the pharmacologically active compound, in addition to the cellular complement of mAC isoforms, may determine inhibitor effects.
Much more effort should be devoted towards the exploration of the diterpene site as target for mAC inhibitors. The diterpene site tolerates large outward-projecting substituents (Figures 3A and B) [11,34], providing a large and almost completely unchartered pharmacological territory. Starting from BODIPY-FS, we anticipate that variations on this theme will yield isoform-selective mAC inhibitors. The structure/activity relationships of diterpenes and glucose transporters are different [35], indicating that in principle, it is possible to avoid off-target effects of compounds targeting the mAC diterpene site.
A substantial limitation of the field constitutes the fact that crystal structures with a functional catalytic site and diterpene site are available only for the VC1:IIC2 heterodimer [23-25,38]. Therefore, it would be most desirable to obtain crystal structures from the catalytic cores of other mAC isoforms, but yield and stability of the respective proteins are formidable problems. Even more challenging is the crystallization of holo-mACs. However, considering the recent progress in the techniques for crystallization of membrane proteins and the increasing number of high-resolution GPCR structures [58], holo-mACs may ultimately be crystallized. This would be very important since mAC isoforms differ substantially from each other in the as yet poorly understood transmembrane domains, offering multiple opportunities for the development of isoform-selective mAC inhibitors. Calmidazolium and tyrphostin may represent (low-potency) archetypes of such inhibitors targeting novel allosteric sites [12,59].
mAC inhibitor studies also require an in-depth understanding of the precise (patho)physiological function of any given mAC isoform. To this end, most pathophysiological studies on mACs have focused on mAC knock-out and overexpression models [2,3]. However, we also need to understand the specific roles of individual mAC isoforms in disease models where all mAC isoforms are present, since compensatory changes in mAC function and expression as a result of a gene knock-out cannot be excluded. Lastly, the effects of inhibitors targeting the catalytic site and the diterpene site are substantially affected by the type of divalent cation present (Mg2+ or Mn2+) [11]. It is assumed that Mg2+ is the physiological cation for mACs [1], but proof for this assumption is missing. The resolution of this question will also provide guidance for future mAC inhibitor development.
Concluding remarks
By combining methods from biochemistry, pharmacology, biophysics and medicinal chemistry, substantial progress has been made towards understanding the molecular basis of the interactions of inhibitors with the catalytic and diterpene sites of mACs. However, none of the AC inhibitors presently available has been comprehensively evaluated for isoform-specificity and selectivity against other proteins. Targeting the diterpene site rather than the catalytic site may be a more promising strategy to obtain isoform-selective AC inhibitors. Furthermore, we need to express all mAC isoforms and analyze constitutive AC activity before we can make a judgement whether we need isoform-specific inhibitors or more non-specific inhibitors to abrogate pathologically increased constitutive or stimulated cAMP formation. Finally, the transmembrane domains and flanking regulatory domains of mAC isoforms, in contrast to their catalytic and diterpene sites, show considerable structural differences. Thus, the development of ligands for these as yet poorly understood domains may be another promising avenue towards isoform-selective mAC inhibitors.
Acknowledgments
We thank Drs. Anshuman Dixit, Sara Dizayee, Michael B. Doughty, Stefan Dove, Michael Egger, Miriam Erdorf, Jens Geduhn, Martin Göttle, Jian-Xin Guo, Stefan Herzig, Klaus Höcherl, Roger A. Johnson, Melanie Hübner, Volkhard Kaever, Burkhard König, Prantik Maity, Jan Matthes, Cibele Pinto, Mark Richter, Dennis Rottländer, Michael Schäferling, Jennifer Schmidt, Yuequan Shen, Christian Spangler, Corinna Spangler, Philip Steindel, Srividya Suryanarayana, Hesham Taha, Stephen F. Vatner, Wei-Jen Tang, Jenna Wang, Jay J.-Q. Wu and Qi-Yuan Zhang for collaboration on various aspects of the adenylyl cyclase project. We thank Jörg Bräunig and Annette Stanke for critical reading of the manuscript. We are also appreciative of the constructive critique of the reviewers. The mAC inhibitor project was supported by grants of the American Heart Association (AHA 005140Z, AHA 0450120Z) and Deutsche Forschungsgemeinschaft (Se 529/5-1, Se529/5-2) to R.S. and The National Institutes of Health (DK46371) to S.R.S.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signalling. Mol Interv. 2002;2:168–184. doi: 10.1124/mi.2.3.168. [DOI] [PubMed] [Google Scholar]
- 2.Sadana R, Dessauer CW. Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals. 2009;17:5–22. doi: 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pierre S, et al. Capturing adenylyl cyclases as potential drug targets. Nat Rev Drug Discov. 2009;8:321–335. doi: 10.1038/nrd2827. [DOI] [PubMed] [Google Scholar]
- 4.Laurenza A, et al. Forskolin: a specific stimulator of adenylyl cyclase or a diterpene with multiple sites of action? Trends Pharmacol Sci. 1989;10:442–447. doi: 10.1016/S0165-6147(89)80008-2. [DOI] [PubMed] [Google Scholar]
- 5.Putnam W, et al. Identification of a forskolin-like molecule in human renal cysts. J Am Soc Nephrol. 2007;18:934–943. doi: 10.1681/ASN.2006111218. [DOI] [PubMed] [Google Scholar]
- 6.Ho D, et al. Modulation of (β-adrenergic receptor signalling in heart failure and longevity targeting adenylyl cyclase type 5. Heart Fail Rev. 2010;15:495–512. doi: 10.1007/s10741-010-9183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Watts VJ, et al. Adenylyl cyclase isoforms as novel therapeutic targets: an exciting example of excitotoxicity neuroprotection. Mol Interv. 2007;7:70–73. doi: 10.1124/mi.7.2.6. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, et al. Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci Transl Med. 2011;3:65ra3. doi: 10.1126/scitranslmed.3001269. [DOI] [PubMed] [Google Scholar]
- 9.Gille A, Seifert R. 2′(3′)-O-(N-methylanthraniloyl)-substituted GTP analogs: a novel class of potent competitive adenylyl cyclase inhibitors. J Biol Chem. 2003;278:12672–12679. doi: 10.1074/jbc.M211292200. [DOI] [PubMed] [Google Scholar]
- 10.Dessauer CW, et al. The interactions of adenylate cyclases with P-site inhibitors. Trends Pharmacol Sci. 1999;20:205–210. doi: 10.1016/s0165-6147(99)01310-3. [DOI] [PubMed] [Google Scholar]
- 11.Erdorf M, Seifert R. Impact of divalent cations on regulation of adenylyl cyclase isoforms by forskolin analogs. Biochem Pharmacol. 2011 doi: 10.1016/j.bcp.2011.07.099. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haunsø A, et al. Small ligands modulating the activity of mammalian adenylyl cyclases: a novel mode of inhibition by calmidazolium. Mol Pharmacol. 2003;63:624–631. doi: 10.1124/mol.63.3.624. [DOI] [PubMed] [Google Scholar]
- 13.Haslam RJ, et al. Inhibition of adenylate cyclase by adenosine analogues in preparations of broken and intact platelets. Biochem J. 1978;176:83–95. doi: 10.1042/bj1760083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onda T, et al. Type-specific regulation of adenylyl cyclase. Selective pharmacological stimulation and inhibition of adenylyl cyclase isoforms. J Biol Chem. 2001;276:47785–47793. doi: 10.1074/jbc.M107233200. [DOI] [PubMed] [Google Scholar]
- 15.Johnson RA, et al. Isozyme-dependent sensitivity of adenylyl cyclase to P-site-mediated inhibition by adenine nucleosides and nucleoside 3′-polyphosphates. J Biol Chem. 1997;272:8962–8966. doi: 10.1074/jbc.272.14.8962. [DOI] [PubMed] [Google Scholar]
- 16.Lippe C, Ardizzone C. Actions of vasopressin and isoprenaline on the ionic transport across the isolated frog skin in the presence and the absence of adenylyl cyclase inhibitors MDL12330A and SQ22536. Comp Biochem Physiol C. 1991;99:209–211. doi: 10.1016/0742-8413(91)90101-x. [DOI] [PubMed] [Google Scholar]
- 17.Whitley R, et al. Vidarabine: a preliminary review of its pharmacological properties and therapeutic use. Drugs. 1980;20:267–282. doi: 10.2165/00003495-198020040-00002. [DOI] [PubMed] [Google Scholar]
- 18.Iwatsubo K, et al. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J Biol Chem. 2004;279:40938–40945. doi: 10.1074/jbc.M314238200. [DOI] [PubMed] [Google Scholar]
- 19.Ortiz-Capisano MC, et al. Adenylyl cyclase isoform V mediates renin release from juxtaglomerular cells. Hypertension. 2007;49:618–624. doi: 10.1161/01.HYP.0000255172.84842.d2. [DOI] [PubMed] [Google Scholar]
- 20.Aldehni F, et al. Stimulation of renin secretion by catecholamines is dependent on adenylyl cyclases 5 and 6. Hypertension. 2011;57:460–468. doi: 10.1161/HYPERTENSIONAHA.110.167130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rangel-Barajas C, et al. L-DOPA-induced dyskinesia in hemiparkinsonian rats is associated with up-regulation of adenylyl cyclase type V/VI and increased GABA release in the substantia nigra reticulata. Neurobiol Dis. 2011;41:51–61. doi: 10.1016/j.nbd.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 22.Gille A, et al. Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides. J Biol Chem. 2004;279:19955–19969. doi: 10.1074/jbc.M312560200. [DOI] [PubMed] [Google Scholar]
- 23.Mou TC, et al. Structural basis for the inhibition of mammalian membrane adenylyl cyclase by 2′(3′)-O-(N-methylanthraniloyl)-guanosine 5′-triphosphate. J Biol Chem. 2005;280:7253–7261. doi: 10.1074/jbc.M409076200. [DOI] [PubMed] [Google Scholar]
- 24.Mou TC, et al. Broad specificity of mammalian adenylyl cyclase for interaction with 2′,3′-substituted purine and pyrimidine nucleotide inhibitors. Mol Pharmacol. 2006;70:878–886. doi: 10.1124/mol.106.026427. [DOI] [PubMed] [Google Scholar]
- 25.Hübner M, et al. Structural basis for the high-affinity inhibition of mammalian membranous adenylyl cyclase by 2′,3′-O-(N-methylanthraniloyl)-inosine 5′-triphosphate. Mol Pharmacol. 2011;80:87–96. doi: 10.1124/mol.111.071894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geduhn J, et al. Bis-halogen-anthraniloyl-substituted nucleoside 5′-triphosphates as potent and selective inhibitors of Bordetella pertussis adenylyl cyclase toxin. J Pharmacol Exp Ther. 2011;336:104–115. doi: 10.1124/jpet.110.174219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suryanarayana S, et al. Differential inhibition of various adenylyl cyclase isoforms and soluble guanylyl cyclase by 2′,3′-O-(2,4,6-trinitrophenyl)-substituted nucleoside 5′-triphosphates. J Pharmacol Exp Ther. 2009;330:687–695. doi: 10.1124/jpet.109.155432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pinto C, et al. Structure-activity relationships for the interactions of 2′- and 3′-(O)-(N-methyl)anthraniloyl-substituted purine and pyrimidine nucleotides with mammalian adenylyl cyclases. Biochem Pharmacol. 2011;82:358–370. doi: 10.1016/j.bcp.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Göttle M, et al. Characterization of mouse heart adenylyl cyclase. J Pharmacol Exp Ther. 2009;329:1156–1165. doi: 10.1124/jpet.109.150953. [DOI] [PubMed] [Google Scholar]
- 30.Hübner M, et al. Effect of MANT-nucleotides on L-type calcium currents in murine cardiomyocytes. Naunyn-Schmiedeberg's Arch Pharmacol. 2011;383:573–583. doi: 10.1007/s00210-011-0626-x. [DOI] [PubMed] [Google Scholar]
- 31.Jameson DM, Watts JF. Fluorescent nucleotide analogs: synthesis and applications. Methods Enzymol. 1997;278:363–390. doi: 10.1016/s0076-6879(97)78020-0. [DOI] [PubMed] [Google Scholar]
- 32.Seamon KB, et al. Forskolin: Unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci USA. 1981;78:3363–3367. doi: 10.1073/pnas.78.6.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McHugh E, et al. Regulation of forskolin interactions with type I, II, V, and VI adenylyl cyclases by Gsα. Biochemistry. 1994;33:12852–12859. doi: 10.1021/bi00209a017. [DOI] [PubMed] [Google Scholar]
- 34.Pinto C, et al. Activation and inhibition of adenylyl cyclase isoforms by forskolin analogs. J Pharmacol Exp Ther. 2008;325:27–36. doi: 10.1124/jpet.107.131904. [DOI] [PubMed] [Google Scholar]
- 35.Appel NM, et al. [125I]-Labeled forskolin analogs which discriminate adenylyl cyclase and a gluose transporter: Pharmacological characterization and localization of binding sites in rat brain by in vitro receptor autoradiography. J Pharmacol Exp Ther. 1992;263:1415–1423. [PubMed] [Google Scholar]
- 36.Sunahara RK, et al. Exchange of substrate and inhibitor specificities between adenylyl and guanylyl cyclases. J Biol Chem. 1998;273:16332–16338. doi: 10.1074/jbc.273.26.16332. [DOI] [PubMed] [Google Scholar]
- 37.Fabbro D, et al. Protein kinases as targets for anticancer agents: from inhibitors to useful drugs. Pharmacol Ther. 2002;93:79–98. doi: 10.1016/s0163-7258(02)00179-1. [DOI] [PubMed] [Google Scholar]
- 38.Tesmer JJ, et al. Molecular basis for P-site inhibition of adenylyl cyclase. Biochemistry. 2000;39:14464–14471. doi: 10.1021/bi0015562. [DOI] [PubMed] [Google Scholar]
- 39.Seamon KB, et al. Binding of [3H]forskolin to rat brain membranes. Proc Natl Acad Sci USA. 1984;81:5081–5085. doi: 10.1073/pnas.81.16.5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pinto C, et al. Differential interactions of the catalytic subunits of adenylyl cyclase with forskolin analogs. Biochem Pharmacol. 2009;78:62–69. doi: 10.1016/j.bcp.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horinouchi T, et al. Adenylate cyclase-cAMP-protein kinase A signaling pathway inhibits endothelin type A receptor-operated Ca2+ entry mediated via TRPC6 channels. J Pharmaol Exp Ther. 2011 doi: 10.1124/jpet.111.187500. in press. [DOI] [PubMed] [Google Scholar]
- 42.Kim KS, Han PL. Mice lacking adenylyl cyclase-5 cope badly with repeated restraint stress. J Neurosci Res. 2009;87:2983–2993. doi: 10.1002/jnr.22119. [DOI] [PubMed] [Google Scholar]
- 43.Kim KS, et al. Mice lacking adenylyl cyclase type 5 (AC5) show increased ethanol consumption and reduced ethanol sensitivity. Psychopharmacology (Berl) 2011;215:391–398. doi: 10.1007/s00213-010-2143-x. [DOI] [PubMed] [Google Scholar]
- 44.Iwamoto T, et al. Motor dysfunction in type 5 adenylyl cyclase-null mice. J Biol Chem. 2003;278:16936–16940. doi: 10.1074/jbc.C300075200. [DOI] [PubMed] [Google Scholar]
- 45.Tang T, et al. Adenylyl cyclase 6 deletion reduces left ventricular hypertrophy, dilation, dysfunction, and fibrosis in pressure-overloaded female mice. J Am Coll Cardiol. 2010;55:1476–1486. doi: 10.1016/j.jacc.2009.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pieroni JP, et al. Distinct characteristics of the basal activities of adenylyl cyclases 2 and 6. J Biol Chem. 1995;270:21368–21373. doi: 10.1074/jbc.270.36.21368. [DOI] [PubMed] [Google Scholar]
- 47.Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn-Scmiedeberg's Arch Pharmacol. 2002;366:381–416. doi: 10.1007/s00210-002-0588-0. [DOI] [PubMed] [Google Scholar]
- 48.Schneider E, Seifert R. Sf9 cells: a versatile model system to investigate the pharmacological properties of G protein-coupled receptors. Pharmacol Ther. 2010;128:387–418. doi: 10.1016/j.pharmthera.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 49.Shen Y, et al. Selective inhibition of anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc Natl Acad Sci USA. 2004;101:3242–3247. doi: 10.1073/pnas.0306552101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dessauer CW. Adenylyl cyclase-A-kinase anchoring protein complexes: the next dimension of cAMP signalling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu CL, et al. Adenylyl cyclase type 5 protein expression during cardiac development. Am J Physiol Heart Circ Physiol. 2009;297:H1776–H1782. doi: 10.1152/ajpheart.00050.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michel MC, et al. How reliable are G-protein-coupled receptor antibodies? Naunyn-Schmiedeberg's Arch Pharmacol. 2009;379:385–388. doi: 10.1007/s00210-009-0395-y. [DOI] [PubMed] [Google Scholar]
- 53.Erdorf M, Seifert R. Pharmacological characterization of adenylyl cyclase isoforms in rabbit kidney membranes. Naunyn-Schmiedeberg's Arch Pharmacol. 2011;383:357–372. doi: 10.1007/s00210-011-0600-7. [DOI] [PubMed] [Google Scholar]
- 54.Wang T, Brown MJ. Differential expression of adenylyl cyclase subtypes in human cardiovascular system. Mol Cell Endocrinol. 2004;223:55–62. doi: 10.1016/j.mce.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 55.Schnell D, et al. Expression and functional properties of canine, rat, and murine histamine H4 receptors in Sf9 insect cells. Naunyn-Schmiedeberg's Arch Pharmacol. 2011;383:457–470. doi: 10.1007/s00210-011-0612-3. [DOI] [PubMed] [Google Scholar]
- 56.Hacker BM, et al. Cloning, chromosomal mapping, and regulatory properties of the human type 9 adenylyl cyclase (ADCY9) Genomics. 1998;50:97–104. doi: 10.1006/geno.1998.5293. [DOI] [PubMed] [Google Scholar]
- 57.Laux WH, et al. Pro-nucleotide inhibitors of adenylyl cyclases in intact cells. J Biol Chem. 2004;279:13317–13332. doi: 10.1074/jbc.M309535200. [DOI] [PubMed] [Google Scholar]
- 58.Kobilka BK. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol Sci. 2011;32:213–218. doi: 10.1016/j.tips.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaleel M, et al. Tyrphostins are inhibitors of guanylyl and adenylyl cyclases. Biochemistry. 2004;43:8247–8255. doi: 10.1021/bi036234n. [DOI] [PubMed] [Google Scholar]
- 60.Rottländer D, et al. Functional adenylyl cyclase inhibition in murine cardiomyocytes by 2′(3′)-O-(N-methylanthraniloyl)-guanosine 5′-[γ-thio]triphosphate. J Pharmacol Exp Ther. 2007;321:608–615. doi: 10.1124/jpet.106.118422. [DOI] [PubMed] [Google Scholar]
- 61.Tucker CL, et al. Two amino acid substitutions convert a guanylyl cyclase, RetGC-1, into an adenylyl cyclase. Proc Natl Acad Sci USA. 1998;95:5993–5997. doi: 10.1073/pnas.95.11.5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seifert R, Schultz G. Involvement of pyrimidinoceptors in the regulation of cell functions by uridine and uracil nucleotides. Trends Pharmacol Sci. 1989;10:365–369. doi: 10.1016/0165-6147(89)90009-6. [DOI] [PubMed] [Google Scholar]
- 63.Taha H, et al. Inhibition of the adenylyl cyclase toxin, edema factor, from Bacillus anthracis by a series of 18 mono- and bis-(M)ANT-substituted nucleoside 5′-triphosphates. Naunyn-Schmiedeberg's Arch Pharmacol. 2011 doi: 10.1007/s00210-011-0688-9. in press. [DOI] [PubMed] [Google Scholar]
- 64.Gille A, et al. Differential interactions of G-proteins and adenylyl cyclase with nucleoside 5′-triphosphates, nucleoside 5′-[γ-thio]triphosphates and nucleoside 5′-[β,γ-imido]triphosphates. Biochem Pharmacol. 2005;71:89–97. doi: 10.1016/j.bcp.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 65.Li F, et al. Role of adenylate and guanylate cyclases in β1, β2-, and β3-adrenoceptor-mediated relaxation of internal anal sphincter smooth muscle. J Pharmacol Exp Ther. 2004;308:1111–1120. doi: 10.1124/jpet.103.060145. [DOI] [PubMed] [Google Scholar]
- 66.Copsel S, et al. Mutidrug resistance protein 4 (MRP4/ABCC4) regulates cAMP cellular levels and controls human leukemia cell proliferation and differentiation. J Biol Chem. 2011;286:6979–6988. doi: 10.1074/jbc.M110.166868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grupp G, et al. Effects of RMI 12330A, a new inhibitor of adenylate cyclase on mycocardial function and subcellular activity. Br J Pharmacol. 1980;70:429–442. doi: 10.1111/j.1476-5381.1980.tb08721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gadea A, et al. The adenylate cyclase inhibitor MDL-12330A has a non-specific effect on glycine transport in Müller cells from the retina. Brain Res. 1999;838:200–204. doi: 10.1016/s0006-8993(99)01683-2. [DOI] [PubMed] [Google Scholar]
- 69.Spehr M, et al. Particulate adenylate cyclase plays a key role in human sperm olfactory receptor-mediated chemotaxis. J Biol Chem. 2004;279:40194–40203. doi: 10.1074/jbc.M403913200. [DOI] [PubMed] [Google Scholar]
- 70.Ferretti ME, et al. MDL 12330 inhibits the non-neuronal adenylyl cyclase from the freshwater snail Planorbarius corneus, but the neuronal enzyme is activated by this compound. Neurosci Lett. 1996;207:191–194. doi: 10.1016/0304-3940(96)12528-3. [DOI] [PubMed] [Google Scholar]
- 71.Johnson JD, Wittenauer LA. A fluorescent calmodulin that reports the binding of hydrophobic inhibitory ligands. Biochem J. 1983;211:473–479. doi: 10.1042/bj2110473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zimmer M, Hofmann F. Calmodulin antagonists inhibit activity of myosin light-chain kinase independent of calmodulin. Eur J Biochem. 1984;142:393–397. doi: 10.1111/j.1432-1033.1984.tb08300.x. [DOI] [PubMed] [Google Scholar]