

Abstract
The increase in multidrug resistant bacteria has sparked an interest in the development of novel antibiotics. Antimicrobial peptides (AMPs) that operate by crossing the cell membrane may also have the potential to deliver drugs to intracellular targets. Buforin 2 (BF2) is an AMP that shares sequence identity with a fragment of histone subunit H2A and whose bactericidal mechanism depends on membrane translocation and DNA binding. Previously, novel histone-derived antimicrobial peptides (HDAPs) were designed based on properties of BF2, and DesHDAP1 and DesHDAP3 showed significant antibacterial activity. In this study, their DNA binding, permeabilization, and translocation abilities were assessed independently and compared to antibacterial activity to determine whether they share a mechanism with BF2. To investigate the importance of proline in determining the peptides' mechanisms of action, proline to alanine mutants of the novel peptides were generated. DesHDAP1, which shows significant similarities to BF2 in terms of secondary structure, translocates effectively across lipid vesicle and bacterial membranes, while the DesHDAP1 proline mutant shows reduced translocation abilities and antimicrobial potency. In contrast, both DesHDAP3 and its proline mutant translocate poorly, though the DesHDAP3 proline mutant is more potent. Our findings suggest that a proline hinge can promote membrane translocation in some peptides, but that the extent of its effect on permeabilization depends on the peptide's amphipathic properties. Our results also highlight the different antimicrobial mechanisms exhibited by histone-derived peptides and suggest that histones may serve as a source of novel AMPs with varied properties.
Keywords: Antimicrobial peptide, histone-derived peptide, translocation, membrane permeabilization, proline hinge, amphipathic
1.0 Introduction
Growing concern over the rise of multidrug resistant bacterial strains has led to increased interest in the discovery of novel antibiotics. Antimicrobial peptides (AMPs) are a class of short amino acid sequences produced by animals, plants, and even bacteria themselves to ward off infection. Most naturally occurring AMPs contain 10–50 amino acid residues, multiple positive charges, and several hydrophobic residues. Many AMPs exhibit relatively non-specific bactericidal activity against both gram-positive and gram-negative species and selectively kill prokaryotic rather than eukaryotic cells [1].
AMPs are often truncated versions of larger proteins within the cell, many of which also show some antibacterial activity [2]. For example, although histone core particles are generally located within the cell nucleus as part of condensed chromatin, recent studies have isolated cytoplasmic and secreted histone proteins with antimicrobial activity from a wide range of organisms and tissues, such as mollusks [3], fish [4], amphibians [5], and the human gastrointestinal tract [6]. The antimicrobial role of histones has been discussed further in recent reviews [7, 8]. All four histone core subunits as well as H1 are capable of crossing cell membranes as well as sometimes mediating the crossing of other small molecules [9, 10]. Recent work has further investigated the antibacterial mechanism of histones H3 and H4 [11]. Additionally, several naturally occurring AMPs share sequence identity with portions of various histone subunits, implying that both whole and fragmented histone proteins may possess antibacterial properties [7, 8].
The best-studied histone-derived antimicrobial peptide (HDAP) is buforin II (BF2), which is identical to a part of histone subunit H2A. BF2 is noteworthy because it uses an uncommon mechanism wherein it kills bacteria without causing cell lysis and is able to cross bacterial membranes [12] and enter lipid vesicles [13]. AMPs that operate through this type of cell-penetrating mechanism are of particular interest to the medical and scientific communities for both their potential to assist in targeting drug molecules to the cytoplasm and for their bactericidal properties [14]. After entering cells, BF2 is believed to kill microbes through interactions with nucleic acids [12, 15].
Several studies have focused on the importance of BF2's sole proline residue on its function [13, 16, 17]. This proline provides a hinge in the helical structure of the peptide and plays a critical role in promoting peptide translocation. BF2 adopts a partially α-helical conformation in the presence of anionic lipid vesicles [13]. Proline cannot form the hydrogen bonds necessary to stabilize an α-helix, thus the presence of proline in the generally helical structure of BF2 introduces flexibility and distorts the N-terminal region of the helix, which in turn increases the helix's amphipathic nature [18]. The BF2 P11A mutant has a longer undistorted helical region but shows significantly decreased translocation across lipid vesicle membranes and reduced antibacterial potency [13]. Subsequent work has shown that the proline in BF2 is in an ideal position for translocation, as moving it one turn towards either the N- or C-terminus significantly reduces the translocation ability of the peptide [17].
Tsao et al previously described three novel designed histone-derived antimicrobial peptides (DesHDAPs 1–3) [19]. These peptides were designed to incorporate several characteristics of BF2. All three peptides share exact sequence identity to a fragment of a histone subunit predicted to bind to DNA; DesHDAP1 is derived from H2A, DesHDAP2 from H3, and DesHDAP3 from H4. The peptides also all possess multiple positive charges and one proline residue that disrupts a C-terminal helical region.
Although these peptides were designed based on properties of BF2, it is unclear whether these characteristics imbued all the designed peptides with a BF2-like mechanism. DesHDAPs 1 and 3 show significantly stronger antibacterial activity against both gram-negative and gram-positive bacteria than DesHDAP2, and are thus the primary focus of this study. However, the growth curves of bacteria exposed to DesHDAPs 1 and 3 show dissimilar shapes, implying possible mechanistic differences [19]. In order to characterize these potential differences, we have measured the translocation ability, membrane permeabilization, and DNA binding of these two peptides. The results clearly show that the two peptides utilize divergent mechanisms, with DesHDAP1 translocating much more readily across membranes than DesHDAP3. Moreover, proline mutations in the two peptides exhibit opposite effects on antimicrobial activity and lead to distinct changes in membrane translocation and permeabilization properties.
2.0 Materials and Methods
2.1 Peptides
Buforin II F10W, DesHDAP1, DesHDAP2, and DesHDAP3 (Table 1) were synthesized and purified by GenScript (Piscataway, NJ) to >95% purity. Proline to alanine mutant versions of each peptide were also purchased at >95% purity. Wild type and mutant peptides with an N-terminal biotin group were also obtained at >95% purity. All peptides had an unmodified C-terminus with a free carboxylic acid, and peptides that did not have an N-terminal biotin group had a free amine group at the N-terminus with no other modification. Peptides were dissolved in nanopure H2O and stored at −20°C. Peptide concentrations were determined from the absorbance signals of native tryptophan residues. Absorbance was measured with a Biorad SmartSpec Plus spectrophotometer (Philadelphia, PA) or a Thermo Scientific Nanodrop 2000 (Wilmington, DE).
Table 1.
Sequences of buforin II, DesHDAP1, DesHDAP2, DesHDAP3, with proline shown in bold.
| Peptide | Histone Subunit Source | Amino Acid Sequence | # of Amino Acids |
|---|---|---|---|
| Buforin II (BF2) | H2A | TRSSRAGLQWPVGRVHRLLRK | 21 |
| DesHDAP1 | H2A | ARDNKKTRIWPRHLQLAVRN | 20 |
| DesHDPA2 | H3 | HRYRPGTVALREIRRYQKST | 20 |
| DesHDAP3 | H4 | KVLRDNIQGWTKPAIRRLARRG | 22 |
| DesHDAP1 P11A mutant | H2A | ARDNKKTRIWARHLQLAVRN | 20 |
| DesHDAP2 P5A mutant | H3 | HRYRAGTVALREIRRYQKST | 20 |
| DesHDAP3 P13A mutant | H4 | KVLRDNIQGWTKAAIRRLARRG | 22 |
2.2 Circular Dichroism Measurements
Circular dichroism spectra were collected from 190–250 nm using an Olis Multiscan 1622 spectrometer (Bogart, GA) in Starna 1 mm path-length quartz cuvettes (Atascadero, CA). All peptides were at a concentration of 25 μM. Spectra were collected in a 1:1 trifluoroethanol (Arcos Organics, NJ):phosphate buffer (10 mM Na3PO4, 45 mM NaCl, 1 mM EDTA, pH 7.4) solution. Reported spectra are the average of 10 scans collected at 25 °C with an integration time of 2.0 s.
2.3 Radial Diffusion Assay for Antibacterial Potency
The radial diffusion assay was performed as described elsewhere [20] on two gram-negative bacterial strains (E. coli ATCC #25922, S. marcesens Carolina #155450A) and three gram-positive bacterial strains (E. faecalis ATCC #29212, S. aureus Carolina #155554A, B. subtilis ATCC #6051). Cells picked from frozen bacterial stocks were incubated overnight at 37 °C in trypticase soy broth (30% TSB w/v) (Sigma Aldrich) to log phase. The overnight culture was diluted 1:1000 in fresh TSB and grown for 2.5 hrs. Bacteria were pelleted via centrifugation at approximately 880 × g for 10 min at 4°C and washed with 10 mM phosphate buffer (10 mM Na3PO4, pH 7.4). 10 mL of molten agarose gel (10 mM Na3PO4, 1% TSB v/v, 1% agarose w/v, pH 7.4) was inoculated with 4×106 CFU of bacteria in phosphate buffer and allowed to solidify on a petri dish. 2.5 μL of 3×10−4 M peptide solution was added to 1-mm wells in the underlay gel and incubated at 37°C for 3 hours. 10 mL of overlay gel (30% w/v TSB, 1% w/v agarose) was poured over the underlay gel and incubated overnight at 37°C. The area of bacterial clearing was measured at 7× magnification.
2.4 Lipid Vesicle Translocation Assay
Lipid vesicles were prepared essentially as described in Torchilin and Weissig [21]. Phospholipids dissolved in chloroform were purchased from Avanti Polar Lipids (Alabaster, AL). Chloroform was evaporated from a 50:45:5 mixture of phosphatidylcholine (POPC), phosphatidylglycerol (POPG), and 5-dimethylaminonaphthalene-1-sulfonyl phosphatidylethanolamine (DNS-POPE) using a nitrogen gas stream. Following overnight desiccation, anhydrous lipid cakes were rehydrated in 10 mM HEPES buffer (10 mM HEPES, 45 mM NaCl, 1 mM EDTA, pH 7.4) containing either 0.2 mM porcine trypsin (Sigma-Aldrich) or 0.2 mM porcine trypsin and 2.0 mM Bowman-Birk trypsin inhibitor (Sigma Aldrich) and subjected to five freeze-thaw cycles. Vesicles were extruded 21 times through a nuclepore track etch membrane with 0.1 μm pores (Whatman, United Kingdom) in an Avanti Polar Lipids extruder to ensure uniform vesicle size. Vesicle concentration was measured in triplicate as a function of total phosphorus content in solution (http://www.avantilipids.com/index.php?Option=com_content&view=article&id=1686&Itemid=405).
Translocation ability was measured essentially as described by Kobayashi et al [13]. Trypsin located outside of experimental, trypsin-containing vesicles was inhibited by the addition of Bowman-Birk trypsin inhibitor such that the final inhibitor concentration was 10× that of the trypsin. Fluorescence emission at 525 nm was then monitored for 25 min following peptide exposure to vesicles with an excitation wavelength of 280 nm. For all samples, peptides were at a concentration of 3 μM and vesicles were at a concentration of 250 μM. Fluorescent emission was also monitored under the same conditions described above using vesicles containing 0.2 mM trypsin and 2 mM Bowman-Birk inhibitor to control for changes in fluorescent signal unrelated to translocation. The final average fluorescence value (Ffavg) for both control and experimental conditions was calculated by dividing the average of the fluorescence values collected during the last minute of the experiment by the fluorescence at 15 s after combining peptide and vesicle samples (Fo). The F0 was taken at 15 s to reduce artifacts in the fluorescence signal that can occur immediately after mixing in the sample. A quantitative translocation ratio (TR) was calculated for each peptide by dividing the Ffavg for the control vesicles by the Ffavg for the experimental vesicles.
2.5 Confocal Microscopy
Peptide translocation into E. coli cells (ATCC #25922) was visualized essentially as described by Park et al. [22]. Briefly, an overnight culture of Top10 E. coli (Invitrogen) was diluted 1:100 in a TSB media and allowed to grow to mid-logarithmic phase. Bacteria were pelleted via centrifugation at approximately 880 × g and resuspended in 10 mM phosphate buffer, pH 7.4. Bacterial cells at a concentration of 107 CFU/mL were incubated with 16 μg/mL biotinylated peptide for 30 minutes at 37°C. Cells were then placed on poly-L-lysine coated glass slides and exposed to Triton-X with a final concentration of 0.066% Triton-X/phosphate buffer for 1–2 minutes. Biotinylated peptides were visualized following the addition of a streptavidin-AlexaFluor488 conjugate (Invitrogen) to a final concentration of 5 μg/mL. Cells were observed using a Leica TCS-SP1 confocal microscope with excitation at 488 nm. All images represent the average of at least four scans.
2.6 DNA Binding
Peptide binding to nucleic acids was measured using a fluorescence intercalator displacement assay [23]. The fluorescence intensity of 0.55 μM Thiazole Orange in STE buffer (10 mM Tris, 50 mM NaCl, 1 mM EDTA, pH 8.0) was measured using a Varian Cary Eclipse fluorescence spectrophotometer (excitation wavelength: 509 nm; emission wavelength: 527 nm) and normalized to 0% relative fluorescence. dsDNA (IDT, Coralville, IA) used in previous studies of BF2 DNA binding [15] (AAATACACTTTTGGT) was prepared in STE buffer and added to the thiazole orange solution to a final concentration of 1.1 μM base pairs. The solution was allowed to equilibrate for several minutes before the fluorescence was measured. The solution was then titrated with a 78 μM peptide solution, allowing five minutes equilibration prior to measurement of fluorescence after each peptide addition. The peptide concentration at which fluorescence intensity was decreased to half was extrapolated, and usually fell within a concentration of 1–10 mM. Reported measurements are from at least three independent experiments for each peptide.
2.7 Membrane Permeabilization
Bacteria picked from a frozen stock were allowed to grow overnight at 37° in TSB media (30% w/v TSB) (Sigma). The overnight culture was diluted 1:1000 into fresh TSB and allowed to grow to an optical density of between 0.2 and 1.0. Bacteria were pelleted by at 880 × g, resuspended in sterile phosphate buffer (100 mM Na3PO4 in H2O, pH 7.4), pelleted again, and finally suspended in sterile phosphate buffer to an optical density of 0.5. Propidium iodide (PI) was added at a concentration of 20 μg/mL and the system allowed to equilibrate. PI complexed with intracellular DNA was excited at 535 nm and fluorescence measured at 617 nm on a Varian Cary Eclipse fluorescence spectrophotometer. After the untreated cells had equilibrated, peptide was added to a concentration of 2 μM and fluorescence was monitored. The increase in fluorescence due to presence of peptide was determined by comparing the fluorescence 5 minutes after peptide addition to the averaged value for the fluorescence in the minute prior to peptide addition.
3.0 Results and Discussion
3.1 Designed HDAPs Do Not Share a Mechanism of Action
BF2's antimicrobial potency is dependent on its ability to cross bacterial and lipid vesicle membranes [13, 22]. The absorbance profiles of bacteria exposed to DesHDAP1 resemble those of BF2, suggesting that it too might use a translocation-based antimicrobial mechanism [19]. In contrast, the absorbance profile of cells exposed to DesHDAP3 shows a dramatic drop signifying cell lysis, which raises questions about whether it shares a mechanism with BF2. The designed peptides' translocation abilities into bacteria were assessed qualitatively using confocal laser microscopy. In these experiments, E. coli were exposed to biotinylated DesHDAP1 and DesHDAP3 and visualized with a streptativdin-AlexaFluor488 conjugate. Like BF2, DesHDAP1 generally appeared to be internalized by bacteria that showed fluorescence (Fig. 1A). In contrast, DesHDAP3 showed little entry into cells and was primarily localized to bacterial cell membranes (Fig. 1B).
Figure 1.

Translocation of DesHDAP1 (A), DesHDAP3 (B), DesHDAP1 P11A (C–D), DesHDAP 3 P13A (E) into E. coli. 107 CFU/mL bacteria were incubated with biotinylated peptides (16 μg/mL). Peptides localization was assessed following treatment with a streptavidin-AlexaFluor488 conjugate and visualized on a Leica TCS-SP1 confocal microscope with excitation at 488 nm. A 2 μm scale bar is provided in the images.
The designed HDAPs' translocation abilities were determined more quantitatively using the vesicle based translocation assay previously used by Matsuzaki and co-workers to characterize the function of BF2 [16]. In this assay, the native tryptophan residues in each peptide act as FRET donors when associated with 50:45:5 POPC:POPG:DNS-POPE lipid vesicles, producing a fluorescent signal. As peptides cross the lipid bilayer, they are digested by trypsin trapped within the vesicle, leading to a loss of FRET signal. To control for loss of FRET signal due to incomplete inhibition of the trypsin outside vesicles or other factors unrelated to translocation ability, the FRET signal between the peptide and vesicles encapsulating both trypsin and trypsin inhibitor was also monitored. A significant decrease in FRET signal as compared to this control is therefore indicative of a peptide that readily translocates across membranes.
The drop in fluorescence observed for DesHDAP1 (Fig. 2B) is significantly greater than the drop observed in its control scenario. In our experience, the relative fluorescence drop observed for DesHDAP1 is even larger than that observed for BF2 in our vesicle samples (Fig. 2A).. Together with the observed translocation into E. coli cells, this suggests that DesHDAP1, like BF2, is a cell-penetrating AMP that spontaneously crosses lipid membranes.
Figure 2.

Translocation of BF2 (A), DesHDAP1 (B), DesHDAP3 (C), DesHDAP1 P11A (D), and DesHDAP3 P13A (E) across lipid vesicle membranes. Peptides (3 μM) were exposed to lipid vesicles (250 μM) containing trypsin. The FRET signal at 525 nm (black) is plotted as the fluorescent signal throughout the experiment relative to the initial fluorescent signal (F/Fo). In control traces (gray), peptides were exposed to vesicles containing both trypsin and trypsin inhibitor.
In order to quantify the translocation, we averaged the ratio of the control and experimental signals over three independent experiments. Our quantitative translocation ratio for BF2 is somewhat lower than the apparent value in data shown by Kobayashi et al.[13] This difference was likely due to the different sources of lipid (e.g. egg lipids versus chemically synthesized POPC and POPG used in this study) and different sources of trypsin and trypsin inhibitor. However, our quantitative translocation ratios for both BF2 and DesHDAP1 are indicative of membrane translocation. In contrast, DesHDAP3 showed a much smaller decrease in fluorescence when compared to controls (Fig. 2C), indicating that it did not exhibit appreciable translocation (Table 2).
Table 2.
Translocation of DesHDAP1, DesHDAP3 and their proline to alanine mutants into lipid vesicles. The reported translocation ratios represent the ratio of the retention of fluorescence after 25 minutes for the translocation control to that of the experimental conditions. Uncertainty is reported as a standard deviation.
| Peptide | Translocation ratio |
|---|---|
| DesHDAP1 | 1.13 ± 0.08 |
| DesHDAP1 P11A | 1.11 ± 0.01 |
| DesHDAP3 | 0.97 ± 0.04 |
| DesHDAP3 P13A | 0.982 ± 0.005 |
| Buforin II | 1.07 ± 0.06 |
After translocating across the cell membrane, cell-penetrating AMPs must be able to reach and interact with their intracellular target(s). In BF2, DNA binding is correlated with overall potency, indicating that DNA binding is indeed critical to the bactericidal mechanism [15]. The DNA binding strength of DesHDAPs 1 and 3 was assessed by measuring the peptide concentration required to displace thiazole orange from double stranded DNA (Fig 3, Table 3). In this assay, DesHDAP1 bound DNA somewhat more weakly than BF2, which may partially explain why BF2 is more potent against some bacterial strains than DesHDAP1 [19]. In contrast, DesHDAP3 bound to DNA with approximately the same affinity as BF2. However, since DesHDAP3 is unable to cross membranes effectively, its enhanced DNA binding is clearly less relevant for its antimicrobial activity. Moreover, the differences in DNA binding between these peptides are relatively small and therefore assumedly less important in determining functional differences than the differing translocation and permeabilization abilities of these peptides.
Figure 3.
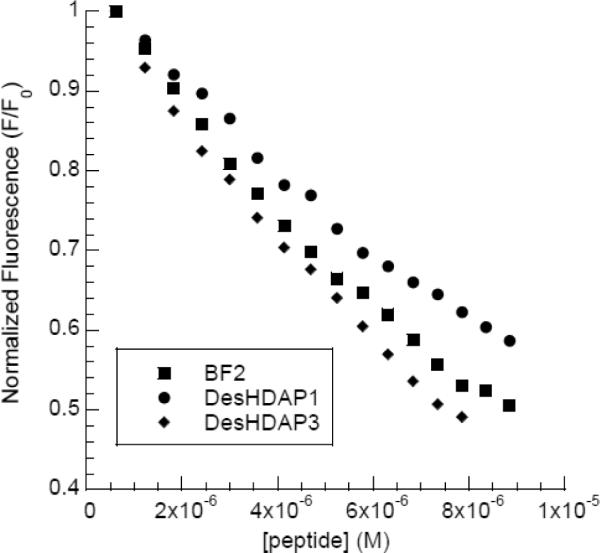
Representative titration data from fluorescent intercalator assays measuring the DNA binding of BF2, DesHDAP1 and DesHDAP3.
Table 3.
DNA binding of the novel HDAPs measured by fluorescent intercalator displacement. The concentration of peptide required to decrease the fluorescence of thiazole orange by 50% (C50) was calculated by monitoring the decrease in fluorescence. Relative binding is normalized compared to BF2. Uncertainty is reported as a standard deviation.
| Peptide | C50 (μM) | Relative C50 |
|---|---|---|
| BF2 | 8.0 ± 1 | 1 |
| DesHDAP1 | 10.2 ± 0.5 | 1.28 |
| DesHDAP3 | 8.7 ± 1 | 1.09 |
If DesHDAP3 does not function through effective translocation across lipid membranes, its antimicrobial activity may arise from an ability to permeabilize membranes. To test this possibility, we measured membrane permeabilization with a propidium iodide based assay. Although these peptides do have some affinity for DNA, appreciable competition between peptide and PI binding should not occur because of the high DNA binding constant of PI and the 750-fold higher concentration of PI compared to peptide in these experiments. Both designed peptides were relatively unable to permeabilize E. coli membranes (Fig. 4, Table 4), showing a lower fluorescence from PI entering the cell than observed for BF2. The observations that DesHDAP3 is less effective at membrane translocation and no better at causing membrane permeabilization than DesHDAP1 are consistent with the relatively lower antibacterial activity of the wild type DesHDAP3 peptide.
Figure 4.

Representative fluorescence traces from membrane permeabilization experiments using E. coli cells exposed to peptide (2 μM) in the presence of propidium iodide (20 μg/mL). Fluorescence was monitored at 617 nm with excitation at 535 nm.
Table 4.
The effect of proline-to-alanine mutations on the permeabilizing ability of DesHDAP1 and DesHDAP3. Propidium iodide, a membrane impermeable DNA intercalator, can enter bacterial cells only after membrane disruption. Data is presented as the ratio of propidium iodide fluorescence at 617 nm 5 minutes after addition of 2 μM peptide to the fluorescence before peptide addition. Uncertainty is reported as standard deviation.
| F5-/F0 | ||
|---|---|---|
| Wildtype | Proline mutant | |
| BF2 | 1.29 ± 0.1 | - |
| Magainin | 1.52 ± 0.2 | - |
| DesHDAP1 | 1.12 ± 0.06 | 1.06 ± 0.05 |
| DesHDAP3 | 1.13 ±0.08 | 1.57 ±0.4 |
3.2 The Proline Hinge Plays a Different Role in the Function of DesHDAP1 and DesHDAP3
DNA binding and translocation studies indicate that DesHDAP1 likely uses a mechanism similar to the one employed by BF2, while DesHDAP3 is significantly less able to translocate across membranes. Both BF2's translocation ability and ultimately its bactericidal activity are closely tied to the presence of a helix-breaking proline hinge in its structure [13, 22], as mutating this proline to alanine significantly decreases the antimicrobial potency and translocation ability of BF2 while increasing its membrane permeabilization [13, 16]. Both DesHDAP1 and DesHDAP3 were designed to have an analogous proline hinge, so proline to alanine mutants of DesHDAPs 1 and 3 were generated to assess the role that the proline hinge plays in determining the mechanism and biological activity of these novel HDAPs.
CD spectra of the peptides in a solution of 50% trifluoroethanol confirm the structural changes that result from the proline to alanine mutations (Fig. 5). As observed previously [19], the wild type DesHDAP1 peptide has a CD spectrum very similar to that of BF2, implying that it likely shares a partially helical structure with BF2, whereas wild type DesHDAP3 has significantly greater helical character. As expected, the proline mutation increases the α-helical structure of both peptides. However, the effect is more dramatic for the DesHDAP1 peptide, which was originally less helical.
Figure 5.

Circular dichroism spectra of all peptides were taken in 50% trifluoroethanol: 50% phosphate buffer solutions. A. CD spectra of DesHDAPs1 (black) and DesHDAP1 P11A (gray). B. CD spectra of DesHDAP3 (black) and DesHDAP3 P13A (gray).
Antibacterial potency of the DesHDAP1 and DesHDAP3 peptides and their proline mutants was measured using a radial diffusion assay (Fig. 6). As seen in previous studies, DesHDAP1 showed antibacterial activity similar to that of BF2, while DesHDAP3 had somewhat lower activity [19]. We note that the activity of the DesHDAP1 sample used in this work appeared somewhat higher against some strains than observed previously, but general trends in activity against differing strains were consistent [19]. Like BF2 P11A, DesHDAP1 P11A showed reduced activity against all bacterial species tested (Fig. 6A). In contrast, removing the proline hinge improved the bactericidal activity of DesHDAP3, as DesHDAP3 P13A showed greater activity against all bacterial strains than the parent peptide (Fig. 6C). In fact, the potency of DesHDAP3 P13A is comparable to that of DesHDAP1 and BF2.
Figure 6.

The antibacterial activity of the original DesHDAP1 (A), DesHDAP2 (B) and DesHDAP3 (C) peptides (solid bars) and their respective proline-to-alanine mutants (white bars) were assessed with a radial diffusion assay. Error bars represent standard deviations.
Because the proline to alanine mutation dramatically increased DesHDAP3's antibacterial potency, we decided to determine whether a similar mutation would improve its activity of the relatively weaker peptide DesHDAP2. In fact, DesHDAP2 P5A did show greater bactericidal activity than wild type DesHDAP2 (Fig. 6B), and may therefore share some mechanistic or structural similarities with DesHDAP3. However, DesHDAP2 P5A remained a significantly weaker antimicrobial agent than BF2, DesHDAP1, and DesHDAP3 P13A, and was therefore not characterized any further in this study.
In order to explain these divergent trends in activity for DesHDAPs1 and 3, we characterized how the proline mutations altered the membrane translocation and permeabilization of the two peptides. The vesicle-based translocation assay suggested a potential decrease in DesHDAP1 P11A's translocation abilities compared with wildtype peptide (Table 2, Fig. 2D), although this decrease was within the experimental error of our method. Decreased translocation also arose in microscopy data, where some bacteria exposed to DesHDAP1 P11A displayed peptide internalization while others within the same slide showed localization of the peptide to the membrane (Fig. 1C–D). Overall these observations are consistent with the proline mutation somewhat reducing, but not eliminating, DesHDAP1 translocation. This reduction in translocation efficiency may explain the decrease in DesHDAP1 bactericidal activity and suggests that the proline hinge plays a role in peptide translocation for both BF2 and DesHDAP1. Like the wild type DesHDAP3, DesHDAP3 P13A showed minimal translocation in both lipid vesicle and bacterial studies (Table 2, Fig. 1E, Fig. 2E). This suggests that the proline hinge aids in translocation for some HDAPs, but that the proline hinge alone does not necessarily confer translocation abilities.
To assess whether any of the peptides might be working through a lytic mechanism rather than a cell-penetrating mechanism, membrane permeabilization in the presence of peptide was tested using the PI assay (Fig. 3, Table 4). As shown above, both of the wild type DesHDAPs caused minimal membrane permeabilization. However, while DesHDAP1 P11A caused even less permeabilization than the wildtype designed peptide, DesHDAP3 P13A caused membrane permeabilization at a level comparable to magainin, a known lytic peptide [24]. Thus, it appear that the P13A mutation enhances the antibacterial activity of DesHDAP3 by converting the peptide into a much more effective membrane permeabilizing agent.
3.3 Relevance of DesHDAP Data on the Role of Proline in Antimicrobial Peptides
These results for DesHDAP1 and DesHDAP3 also provide more general insight into the role played by proline in the membrane permeabilization of antimicrobial peptides. Several previous studies had observed that adding a proline residue to an antimicrobial peptide decreased its ability to permeabilize membranes [25–27]. Since proline disrupts α-helical structures, it is tempting to interpret the decreased permeabilization in peptide variants containing proline as being due to their decreased α-helical structure. However, our data on designed HDAP proline mutants emphasize that increased permeabilization is not a direct effect of a longer α-helical structure, but instead arises because of changes in amphipathic structure that can occur when adding or removing a proline residue from a peptide sequence. For example, the α-helicity of both DesHDAP1 and DesHDAP3 increased in their respective proline to alanine mutations (P11A and P13A). However, this mutation only increased permeabilization in DesHDAP3, while it actually decreased permeabilization in DesHDAP1. A helical wheel representation highlights that DesHDAP3 is very amphipathic if allowed to form a fully α-helical structure, as seems to occur in DesHDAP3 P13A (Fig. 7). However, DesHDAP1 would not be particularly amphipathic in its fully α-helical structure (Fig. 7), as would be promoted by its P11A mutation. In fact, the DesHDAP1 P11A mutation likely forces some polar residues (e.g. Asp 3, Lys 6 and Thr 7) to lie on the more hydrophobic face of the α-helix, making the peptide potentially less amphipathic than in its original form that contains proline. These differences in amphipathic nature, and not the extent of α-helicity, therefore dictate the permeabilization of these peptides.
Figure 7.

HDAP helical wheel depictions generated using the GROMACS suite [35]. Hydrophobic residues are shown in grey, and the proline hinge is outlined.
A similar effect was seen in studies by Oh et al. of cecropin-magainin hybrid peptides [28]. In this study, a variant containing a central proline residue increased membrane permeabilization despite inducing a kink in the peptide that disrupted the helical structure in the middle portion of the peptide. However, this kink allowed the peptide to form two independent amphipathic helices rather than one longer α-helix that would have no clear hydrophobic and hydrophilic faces. Similarly, Xie et al. observed that BF2 variants containing either a proline at residue 7 (P11A/G7P BF2) or no proline (P11A BF2) have very similar membrane permeabilization abilities despite the increased α-helicity that results from removing the proline [17]. Similar to our observations for DesHDAP1, this likely arises because extending the α-helical structure of BF2 to the N-terminal side of residue 7 would not effectively increase the amphipathic nature of the α-helix as the N-terminal residues do not fit well into the amphipathic pattern in the rest of the peptide (Fig. 7). A molecular dynamics study of another α-helical AMP, ovispirin, also suggests that the ability to form an amphipathic structure may correlate with membrane lytic behavior [29].
Our data on DesHDAP1 and DesHDAP3 also provide additional insight into the role of proline in membrane translocation. In particular, a proline hinge may be more likely to induce translocation in peptides with less amphipathic character that do not readily form stable membrane-disrupting structures, such as DesHDAP1 and BF2, by increasing their ability to interact with lipids upon membrane entry. In fact, deformations around proline that increase membrane interactions have been observed in molecular dynamics simulations of BF2 interacting with lipid membranes [30]. The inclusion of a proline hinge may be less effective in inducing translocation in peptides that exhibit greater lytic potential, such as DesHDAP3, although additional examples of similar peptides with these properties would be helpful to support this assertion.
4.0 Conclusions
The present study evaluated the mechanism of two recently designed peptides, DesHDAP1 and DesHDAP3 [19]. In general, our results suggest that DesHDAP1 shares a mechanism of action with BF2 and are consistent with the hypothesis that both operate by crossing bacterial membranes and interacting with intracellular targets like DNA. In addition to sharing a similar secondary structure with BF2, DesHDAP1 readily translocates across cell membranes without causing significant membrane permeabilization. In contrast, DesHDAP3's relatively weak bactericidal activity compared to BF2 and DesHDAP1 appears to result from it having both relatively poor translocation and membrane permeabilization abilities. Interestingly, a DesHDAP3 proline to alanine mutant makes that peptide much more active against bacteria. Although that mutation does not enhance translocation, it does cause an increase in membrane permeabilization that appears to be primarily responsible for the peptide's bactericidal potency. While it is clear that membrane permeabilization plays a more central role in the antibacterial activity of DesHDAP3 than DesHDAP1, based on our data, we cannot determine whether the wild type DesHDAP3 peptide's mechanism of action relies solely on membrane permeabilization or on some combination of weak membrane permeablization and weak translocation.
Our results on DesHDAP1 and DesHDAP3 also provide insight into the roles of proline residues in antimicrobial peptides and the role of α-helicity as a predictor of antimicrobial peptide properties. In particular, our data emphasize the observation that the membrane permeabilization caused by antimicrobial peptides is not a direct effect of the extent of α-helical structure, but instead arises because of the amphipathic structure of a peptide.. Similarly, our data also suggest that the correlation of α-helicity with antimicrobial potency observed for truncated BF2 mutants is not universally true for all HDAPs [17, 22]. In spite of DesHDAP1's relatively low α-helical content compared to that of the other HDAPs, it shows the greatest antimicrobial potency. DesHDAP3 is a weaker antimicrobial agent and translocates poorly, despite its significant α-helical content. As with membrane permeabilization, although α-helical character may be correlated with antimicrobial activity in many cases, the overall activity of a peptide results from a combination of its different properties that directly impact microbial health, such as membrane permeabilization, membrane translocation or the ability to interact with intracellular components. These properties are all dictated by the overall physiochemical properties of the peptide. While these properties are certainly related to peptide structure, they are not necessarily dictated by the extent of helical secondary structure, especially for peptides that do not follow a straightforward amphipathic α-helical pattern
Overall, the effectiveness of our designed HDAPs suggests that histone derivatives could be a rich source of AMPs. However, our results highlight that different histone fragments with antimicrobial activity may function in different manners. Although many histone-related proteins with antimicrobial activity have been isolated from natural sources, the mechanism is only well characterized in a few cases. In particular, the mechanism of BF2, which involves membrane translocation and nucleic acid interactions, has been fairly well established [12, 13, 15, 31]. More recently, researchers determined that parasin kills bacteria through membrane lysis but does not readily translocate into bacterial cells [32]. Since both parasin and BF2 are derived from histone H2A, these studies have shown that antimicrobial fragments of the same histone will not necessarily utilize an identical mechanism of action. Strikingly, parasin was not derived from a DNA binding region of histone H2A, opening up the possibility that only histone fragments derived from DNA binding regions of the protein would function via a BF2-like mechanism. However, the data presented here show that at least some DNA-binding fragments from histones, such as DesHDAP3, do not operate in a manner similar to that of BF2. Ongoing work on other naturally occurring and synthetic HDAPs as well as on the properties of full histones will be necessary to obtain a systematic understanding of trends in HDAP activity. Multiple studies have shown the utility of histones and histone fragments not only as antimicrobial molecules but also as anti-cancer agents [33] and in cellular transfection applications [34]. Thus, an increasingly complete understanding of how histone derivatives function on the molecular level will provide a foundation for future work in which researchers attempt to rationally design and prepare histone-derivatives with desired properties.
Highlights
-
•
Not all histone-derived antimicrobial peptides (HDAPs) use the same mechanism.
-
•
DesHDAP1 shows proline-dependent translocation, analogous to buforin II.
-
•
In DesHDAP3, proline to alanine mutation increases membrane lysis and potency.
-
•
Proline promotes membrane translocation in some antimicrobial peptides.
-
•
Amphipathicity, not 〈-helicity, determines lytic abilities of proline mutants.
5.0 Acknowledgements
Research Funded by National Institute of Allergy and Infectious Diseases (NIH-NIAID) award R15AI079685, National Science Foundation award CHE-0922860, the Wellesley College Staley Fund and funding to Wellesley College through the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hancock RE, Patrzykat A. Clinical development of cationic antimicrobial peptides: from natural to novel antibiotics. Curr Drug Targets Infect Disord. 2002;2:79–83. doi: 10.2174/1568005024605855. [DOI] [PubMed] [Google Scholar]
- [2].Nguyen LT, Haney EF, Vogel HJ. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011;29:464–472. doi: 10.1016/j.tibtech.2011.05.001. [DOI] [PubMed] [Google Scholar]
- [3].Seo JK, Stephenson J, Noga EJ. Multiple antibacterial histone H2B proteins are expressed in tissues of American oyster. Comp Biochem Physiol B Biochem Mol Biol. 2011;158:223–229. doi: 10.1016/j.cbpb.2010.11.011. [DOI] [PubMed] [Google Scholar]
- [4].Noga EJ, Borron PJ, Hinshaw J, Gordon WC, Gordon LJ, Seo JK. Identification of histones as endogenous antibiotics in fish and quantification in rainbow trout (Oncorhynchus mykiss) skin and gill. Fish Physiol Biochem. 2011;37:135–152. doi: 10.1007/s10695-010-9422-7. [DOI] [PubMed] [Google Scholar]
- [5].Kawasaki H, Koyama T, Conlon JM, Yamakura F, Iwamuro S. Antimicrobial action of histone H2B in Escherichia coli: Evidence for membrane translocation and DNA-binding of a histone H2B fragment after proteolytic cleavage by outer membrane proteinase T. Biochimie. 2008;90:1693–1702. doi: 10.1016/j.biochi.2008.07.003. [DOI] [PubMed] [Google Scholar]
- [6].Rose FR, Bailey K, Keyte JW, Chan WC, Greenwood D, Mahida YR. Potential role of epithelial cell-derived histone H1 proteins in innate antimicrobial defense in the human gastrointestinal tract. Infect Immun. 1998;66:3255–3263. doi: 10.1128/iai.66.7.3255-3263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kawasaki H, Iwamuro S. Potential roles of histones in host defense as antimicrobial agents. Infect Disord Drug Targets. 2008;8:195–205. doi: 10.2174/1871526510808030195. [DOI] [PubMed] [Google Scholar]
- [8].Smith VJ, Desbois AP, Dyrynda EA. Conventional and unconventional antimicrobials from fish, marine invertebrates and micro-algae. Mar Drugs. 2010;8:1213–1262. doi: 10.3390/md8041213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hariton-Gazal E, Rosenbluh J, Graessmann A, Gilon C, Loyter A. Direct translocation of histone molecules across cell membranes. J Cell Sci. 2003;116:4577–4586. doi: 10.1242/jcs.00757. [DOI] [PubMed] [Google Scholar]
- [10].Rosenbluh J, Singh SK, Gafni Y, Graessmann A, Loyter A. Non-endocytic penetration of core histones into petunia protoplasts and cultured cells: a novel mechanism for the introduction of macromolecules into plant cells. Biochim Biophys Acta. 2004;1664:230–240. doi: 10.1016/j.bbamem.2004.06.003. [DOI] [PubMed] [Google Scholar]
- [11].Tagai C, Morita S, Shiraishi T, Miyaji K, Iwamuro S. Antimicrobial properties of arginine- and lysine-rich histones and involvement of bacterial outer membrane protease T in their differential mode of actions. Peptides. 2011;32:2003–2009. doi: 10.1016/j.peptides.2011.09.005. [DOI] [PubMed] [Google Scholar]
- [12].Park CB, Kim HS, Kim SC. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochemical and Biophysical Research Communications. 1998;244:253–257. doi: 10.1006/bbrc.1998.8159. [DOI] [PubMed] [Google Scholar]
- [13].Kobayashi S, Takeshima K, Park CB, Kim SC, Matsuzaki K. Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: Proline as a translocation promoting factor. Biochemistry. 2000;39:8648–8654. doi: 10.1021/bi0004549. [DOI] [PubMed] [Google Scholar]
- [14].Morris MC, Deshayes S, Heitz F, Divita G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell. 2008;100:201–217. doi: 10.1042/BC20070116. [DOI] [PubMed] [Google Scholar]
- [15].Uyterhoeven ET, Butler CH, Ko D, Elmore DE. Investigating the nucleic acid interactions and antimicrobial mechanism of buforin II. Febs Letters. 2008;582:1715–1718. doi: 10.1016/j.febslet.2008.04.036. [DOI] [PubMed] [Google Scholar]
- [16].Kobayashi S, Chikushi A, Tougu S, Imura Y, Nishida M, Yano Y, Matsuzaki K. Membrane translocation mechanism of the antimicrobial peptide buforin 2. Biochemistry. 2004;43:15610–15616. doi: 10.1021/bi048206q. [DOI] [PubMed] [Google Scholar]
- [17].Xie Y, Fleming E, Chen JL, Elmore DE. Effect of proline position on the antimicrobial mechanism of buforin II. Peptides. 2011;32:677–682. doi: 10.1016/j.peptides.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yi GS, Park CB, Kim SC, Cheong C. Solution structure of an antimicrobial peptide buforin II. Febs Letters. 1996;398:87–90. doi: 10.1016/s0014-5793(96)01193-3. [DOI] [PubMed] [Google Scholar]
- [19].Tsao HS, Spinella SA, Lee AT, Elmore DE. Design of novel histone-derived antimicrobial peptides. Peptides. 2009;30:2168–2173. doi: 10.1016/j.peptides.2009.09.011. [DOI] [PubMed] [Google Scholar]
- [20].Lehrer RI, Rosenman M, Harwig SS, Jackson R, Eisenhauer P. Ultrasensitive assays for endogenous antimicrobial polypeptides. J Immunol Methods. 1991;137:167–173. doi: 10.1016/0022-1759(91)90021-7. [DOI] [PubMed] [Google Scholar]
- [21].Torchilin VP, Weissig V. Liposomes. 2nd ed. Oxford University Press; New York: 2003. [Google Scholar]
- [22].Park CB, Yi KS, Matsuzaki K, Kim MS, Kim SC. Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: the proline hinge is responsible for the cell-penetrating ability of buforin II. Proc Natl Acad Sci U S A. 2000;97:8245–8250. doi: 10.1073/pnas.150518097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tse WC, Boger DL. A fluorescent intercalator displacement assay for establishing DNA binding selectivity and affinity. Acc Chem Res. 2004;37:61–69. doi: 10.1021/ar030113y. [DOI] [PubMed] [Google Scholar]
- [24].Matsuzaki K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim Biophys Acta. 1998;1376:391–400. doi: 10.1016/s0304-4157(98)00014-8. [DOI] [PubMed] [Google Scholar]
- [25].Lee SA, Kim YK, Lim SS, Zhu WL, Ko H, Shin SY, Hahm KS, Kim Y. Solution structure and cell selectivity of piscidin 1 and its analogues. Biochemistry. 2007;46:3653–3663. doi: 10.1021/bi062233u. [DOI] [PubMed] [Google Scholar]
- [26].Thennarasu S, Nagaraj R. Specific antimicrobial and hemolytic activities of 18-residue peptides derived from the amino terminal region of the toxin pardaxin. Protein Eng. 1996;9:1219–1224. doi: 10.1093/protein/9.12.1219. [DOI] [PubMed] [Google Scholar]
- [27].Zhang L, Benz R, Hancock RE. Influence of proline residues on the antibacterial and synergistic activities of alpha-helical peptides. Biochemistry. 1999;38:8102–8111. doi: 10.1021/bi9904104. [DOI] [PubMed] [Google Scholar]
- [28].Oh D, Shin SY, Lee S, Kang JH, Kim SD, Ryu PD, Hahm KS, Kim Y. Role of the hinge region and the tryptophan residue in the synthetic antimicrobial peptides, cecropin A(1–8)-magainin 2(1–12) and its analogues, on their antibiotic activities and structures. Biochemistry. 2000;39:11855–11864. doi: 10.1021/bi000453g. [DOI] [PubMed] [Google Scholar]
- [29].Khandelia H, Kaznessis YN. Molecular dynamics investigation of the influence of anionic and zwitterionic interfaces on antimicrobial peptides' structure: implications for peptide toxicity and activity. Peptides. 2006;27:1192–1200. doi: 10.1016/j.peptides.2005.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fleming E, Maharaj NP, Chen JL, Nelson RB, Elmore DE. Effect of lipid composition on buforin II structure and membrane entry. Proteins-Structure Function and Bioinformatics. 2008;73:480–491. doi: 10.1002/prot.22074. [DOI] [PubMed] [Google Scholar]
- [31].Cho JH, Sung BH, Kim SC. Buforins: Histone H2A-derived antimicrobial peptides from toad stomach. Biochimica Et Biophysica Acta-Biomembranes. 2009;1788:1564–1569. doi: 10.1016/j.bbamem.2008.10.025. [DOI] [PubMed] [Google Scholar]
- [32].Koo YS, Kim JM, Park IY, Yu BJ, Jang SA, Kim KS, Park CB, Cho JH, Kim SC. Structure-activity relations of parasin I, a histone H2A-derived antimicrobial peptide. Peptides. 2008;29:1102–1108. doi: 10.1016/j.peptides.2008.02.019. [DOI] [PubMed] [Google Scholar]
- [33].Lee HS, Park CB, Kim JM, Jang SA, Park IY, Kim MS, Cho JH, Kim SC. Mechanism of anticancer activity of buforin IIb, a histone H2A-derived peptide. Cancer Lett. 2008;271:47–55. doi: 10.1016/j.canlet.2008.05.041. [DOI] [PubMed] [Google Scholar]
- [34].Kaouass M, Beaulieu R, Balicki D. Histonefection: Novel and potent non-viral gene delivery. J Control Release. 2006;113:245–254. doi: 10.1016/j.jconrel.2006.04.013. [DOI] [PubMed] [Google Scholar]
- [35].Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. Journal of Molecular Modeling. 2001;7:306–317. [Google Scholar]