


Abstract
The FokI restriction endonuclease is a monomeric protein that recognizes an asymmetric sequence and cleaves both DNA strands at fixed loci downstream of the site. Its single active site is positioned initially near the recognition sequence, distant from its downstream target 13 nucleotides away. Moreover, to cut both strands, it has to recruit a second monomer to give an assembly with two active sites. Here, the individual steps in the FokI reaction pathway were examined by fluorescence resonance energy transfer (FRET). To monitor DNA binding and domain motion, a fluorescence donor was attached to the DNA, either downstream or upstream of the recognition site, and an acceptor placed on the catalytic domain of the protein. A FokI variant incapable of dimerization was also employed, to disentangle the signal due to domain motion from that due to protein association. Dimerization was monitored separately by using two samples of FokI labelled with donor and acceptor, respectively. The stopped-flow studies revealed a complete reaction pathway for FokI, both the sequence of events and the kinetics of each individual step.
INTRODUCTION
Type II restriction endonucleases recognize short DNA sequences, typically 4–8 bp long, and cleave both DNA strands at fixed positions relative to their target sequence (1). Many of these enzymes recognize palindromic sites, with the same 5′–3′ sequence in both strands, and cut both strands at symmetrically equivalent positions within the sequence. The enzymes that recognize such sites, such as EcoRI, EcoRV and BamHI, are often dimers of identical subunits that bind to DNA with matching symmetry, to position the active site from one subunit against the scissile bond in one strand and that from the second subunit against the equivalent bond in the opposite strand (2–4). The only cofactor that most Type II enzymes need for their DNA cleavage reactions is Mg2+. Though some other metal ions can support low levels of activity, Ca2+ usually gives no activity though it often enables specific binding (5).
Enzymes like EcoRV and BamHI are by no means representative of all Type II restriction enzymes. Perhaps the only feature common to all Type II nucleases is that they cleave DNA at specified positions relative to their recognition sites. Nevertheless, they achieve this goal in many different ways (6–12). They can thus be categorized into numerous subtypes by several different criteria: the nature of their recognition sequence, the loci at which they cleave the DNA; their mode of action; their genetic organisation; and their cofactor requirements (13).
Many Type II nucleases recognize asymmetric rather than palindromic sequences and cleave the DNA at a fixed distance away from the recognition sequence: in some cases on one side of the site; in others, on both sides (1,6,11,12). The enzymes that recognize asymmetric sites and cleave the DNA asymmetrically on one side of the site are known as Type IIS restriction endonucleases since they show a shifted pattern of DNA cleavage (6). In the recognition of a symmetrical site by a homodimeric protein, the contacts made by one protein subunit to one half of the target DNA are normally duplicated by the other subunit with the second half (3,4). An asymmetric sequence cannot be recognized in the same way, simply because the site cannot be divided into two equal halves. Instead, the recognition of an asymmetric site requires either a monomeric protein to contact the entire length of the sequence, or an oligomeric protein with individual subunits contacting separate segments of the DNA (12,14). Recognition by a monomer poses a further problem since Type II restriction enzymes generally possess only one catalytic centre for phosphodiester hydrolysis per protein subunit. Hence it might be expected that a monomer at an asymmetric site would cleave only one strand of the DNA. Yet nearly all of the Type IIS enzymes cut both strands, a process that usually requires two catalytic centres.
The Type IIS endonuclease studied most deeply to date is FokI. FokI recognizes a 5 bp asymmetric sequence and cleaves downstream of the site, 9 and 13 nt away in ‘top’ and ‘bottom’ strands, respectively:
5′-G-G-A-T-G-n-n-n-n-n-n-n-n-n↓n-n-n-n-n
3′-C-C-T-A-C-n-n-n-n-n-n-n-n-n-n-n-n-n↑n
where n indicates any nucleotide and arrows mark cleavage loci (15). FokI is a monomeric protein of 66 kDa, both in free solution and when bound to DNA in the absence of divalent metal ions (16,17). It contains two domains connected by a flexible linker: an N-terminal domain of 41 kDa that binds to but does not cleave the cognate sequence; and a C-terminal domain of 25 kDa that cleaves DNA non-specifically (18,19). The crystal structure of FokI bound to a 21 bp DNA carrying both recognition and cleavage sites (20), but without metals, confirmed both its monomeric structure and its two-domain organisation (Figure 1). In the crystal structure, the catalytic domain is packed against the recognition domain, far away from the cleavage sites. The interface between catalytic and recognition domains appears to be functionally significant: mutations that relax the specificity of FokI for its unmodified target sequence (21) are often at this interface (20). In footprinting studies, FokI protects the recognition but not the cleavage site, so the latter must initially be unoccupied (22,23). Hence, at some stage in the reaction pathway, the catalytic domain must move from its starting position by the recognition domain to a location where it can engage its target phosphodiester bond.
Figure 1.
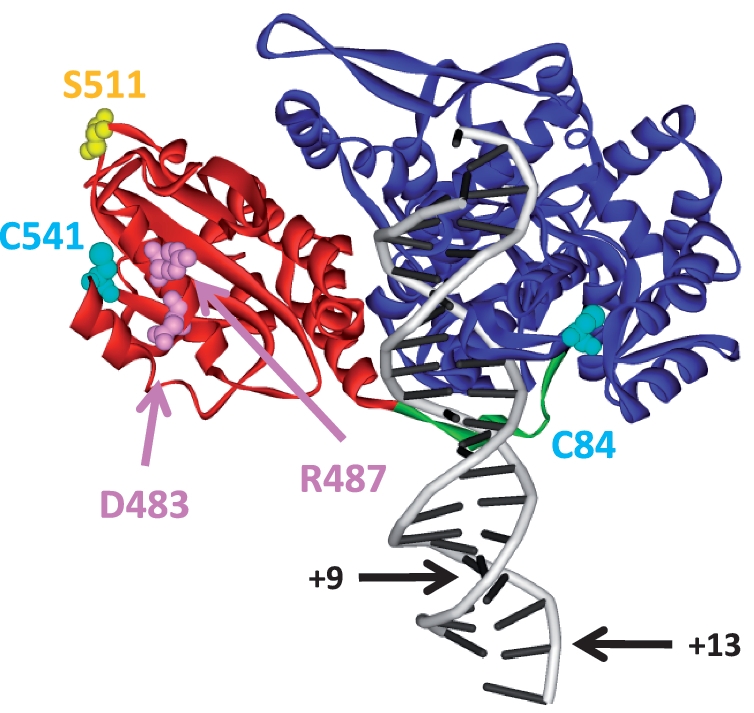
Crystal structure of FokI bound to specific DNA in the absence of divalent metal ions (20). The complex contains one monomer of the FokI endonuclease bound to a 21 bp DNA duplex (in grey) carrying the recognition sequence for FokI and the DNA cleavage loci 9 and 13 nt downstream of the recognition site (indicated by arrows marked +9 and +13, respectively). The main chain of the FokI protein is shown as a ribbon, with the N-terminal (DNA recognition) domain in blue, the C-terminal (catalytic) domain in red and the linker connecting the domains in green. Also marked, in space-filling representation, are the following amino acids: in cyan, C84 and C541, the natural cysteines in FokI; in pink, D483 and R487, the sites of the dimerization-defective mutations in FokI; in yellow, S511, the site for labelling the FokI protein used in this study (Figure drawn from co-ordinates at doi:10.2210/pdb1fok/pdb).
Mutational studies suggested that the FokI monomer has only one catalytic centre and that it uses this active site to cut both DNA strands (22). However, the use of a single active site to hydrolyse phosphodiester bonds in both 5′–3′ and 3′–5′ strands poses significant difficulties: that centre must undergo not only a 180° rotation parallel to the DNA axis to switch between strands, but also a 180° rotation perpendicular to the axis, to accommodate the anti-parallel nature of duplex DNA (24). While the structure of the FokI monomer bound to its recognition site without metal ions confirmed the presence of a single catalytic centre (20), a subsequent crystal structure of the free protein showed direct contact between two catalytic domains, to give a unit with two active sites that could potentially each cleave one strand of the DNA, much like BamHI (25). The FokI monomer at the recognition site is now known to have no activity until it associates with a second monomer to form a dimer (17,26–28). Disruption of the interface between the catalytic domains seen in the crystal structure of the free protein, by two amino acid substitutions (Figure 1), obliterates FokI activity, presumably by blocking dimerization (26). The monomeric protein–DNA complex formed in the absence of metal ions associates to a dimer in the presence of Ca2+ (17).
The monomer of FokI bound directly to the recognition site uses its active site on the bottom strand of the DNA, to cut the scissile bond 13 nt downstream of the site, while the second monomer, tethered to that site by protein–protein interactions, cuts the top strand 9 nt away (29). On a DNA molecule with one FokI site, the second monomer comes from free solution. However, the association is relatively weak: the interface between the catalytic domains has a small surface area (25). The KD (equilibrium dissociation constant) for the second monomer is ∼100 nM, so only a fraction of the DNA carries the dimeric form unless the protein concentration is >>100 nM (27). In contrast, on a DNA with two or more FokI sites, two monomers bound to separate sites in the same molecule are restrained close together and so interact with each other much more readily than with a protein in free solution or one on a different molecule of the DNA (28). Consequently, DNA substrates with one FokI site are cleaved slowly except at elevated protein concentrations (26,27), while plasmids with two FokI sites are cleaved rapidly even at low enzyme concentrations (28,30).
The reaction pathway for the FokI endonuclease contains many steps that are common among enzymes acting at specific DNA sites: DNA sequence recognition; structural rearrangements; domain motion; protein assembly on DNA; phosphodiester hydrolysis; and dissociation from DNA. The objective of this study is to establish the sequence of steps during DNA cleavage by FokI and the dynamics of the individual steps, and thus to elucidate the complete reaction pathway for FokI on a DNA with one recognition site.
MATERIALS AND METHODS
Proteins
Wild-type (WT) FokI endonuclease was purified from a transformant of Escherichia coli MM294[λAFB] (a strain carrying the FokI methyltransferase on a λ lysogen) with pAFP1, a plasmid expressing the FokI nuclease from a tac promoter (from W.E. Jack and J. Bitinaite, New England Biolabs). Site-directed mutants of FokI were created by the QuikChange method (Stratagene) using pAFP1 as the template. The mutated DNA was used to transform E. coli MM294[λAFB] and the plasmids isolated from several individual transformants sequenced across the FokI gene (University of Dundee Sequencing Service): in all cases noted here, the only change(s) to the WT sequence were those specified by the mutagenic primer(s). FokI variants were purified to >95% homogeneity by the same procedure as WT FokI (27) and kept at −80°C in Storage Buffer [5 mM K2HPO4, 5 mM KH2PO4, 300 mM NaCl, 1 mM EDTA and 10% (v/v) glycerol]. Protein concentrations were assessed from A280 measurements using the WT extinction coefficient and are given as molar concentrations of monomeric protein.
DNA
The supercoiled (SC) form of the plasmid pIF190 (27) was purified from transformants of E. coli HB101, grown in broth containing [methyl-3H]thymidine, by CsCl density gradient centrifugation as before (10,30).
Unless noted otherwise, oligodeoxyribonucleotides were obtained from Purimex (Grebenstein, Germany), dissolved in H2O to give stocks of 100 µM and stored at −20°C: those labelled with Alexa Fluor 488 (Ax488) at either 5′ or 3′ termini were initially synthesized with a 5′ or 3′ C6 amino-linker, purified by high pressure liquid chromatography (HPLC), reacted with the succinimidyl ester of Ax488 carboxylic acid and then purified again by HPLC. To anneal the duplexes (Figure 2), the ‘top-strand’ oligonucleotide was mixed with a 1.5-fold excess of the ‘bottom-strand’ oligonucleotide to give, following the addition of buffer components, a solution containing 30 µM duplex in 20 mM Tris–HCl (pH 8.0), 100 mM NaCl. [For 24 s-HEX, a 1.5-fold excess of top over bottom strand was used: the latter was from Sigma-Genosys]. The solutions were protected from UV light, heated to 95°C for 10 min and then cooled slowly to room temperature.
Figure 2.

Oligoduplexes. Synthetic oligonucleotides were annealed to give the duplexes shown. The species 24 s-Ax488 and 34 s-Ax488 carry the recognition sequence for FokI (underlined in bold), the downstream sites of DNA cleavage (arrows) and an Alexa Fluor 488 dye attached via a C6 amino-linker to the 3′-end of the top strand (noted as Ax488): i.e. downstream of the recognition site. Two further duplexes, 24 s and 24 s-HEX (not shown), had the same sequence as 24 s-Ax488 but instead of the 3′-Ax488 label on the top strand have: in the case of 24 s, no label; in the case of 24 s-HEX, a hexachlorofluorescein moiety at the 5′-end of the bottom strand, again downstream of the site. A further derivative, 24 ns, has the same sequence as 24 s apart from alterations at all 5 bp of the specific sequence, to give a non-specific sequence for FokI. The duplex Ax488-28 s carries the recognition and cutting sites for FokI and an Ax488 dye (all noted as above), but the latter is attached, again via a C6 amino-linker, to the 5′-end of the top strand: i.e. upstream of the site. The 13 s species has the recognition site but no sites for DNA cleavage.
FokI activity
Activities of WT and mutant forms of FokI were assessed from reactions containing the FokI protein (2 nM) and the SC form of 3H-labelled pIF190 (10 nM) in M-Buffer [20 mM Tris–acetate (pH 7.9), 50 mM potassium acetate, 10 mM magnesium acetate, 1 mM DTT] at 37°C. Samples were withdrawn from the reactions at varied times after adding the protein and analysed as before (10,30) to measure the residual concentration of the SC DNA at each time point. In all cases, the decline in the concentration of SC DNA with time followed an exponential progress curve (Supplementary Figure S1) that yielded, from the best fit to an exponential decline, a first-order rate constant. The first order rate constants were normalized against the enzyme concentration (31) to yield kcat/Km values (Supplementary Table S1).
Protein labelling
The C5 maleimide derivatives of Ax488 and Alexa Fluor594 (Ax594) (Invitrogen Ltd) were dissolved in dimethylsulphoxide to give 22 mM stock solutions. For protein labelling, a small volume of fluorophore solution was added with gentle mixing to the FokI mutant (typically 50 µM) in Storage Buffer, in a 5-fold molar excess of dye over protein. The mixture, protected from light, was incubated at room temperature for 15 min before stopping the reaction with a 2-fold excess of β-mercaptoethanol over dye. To separate the labelled protein from the free dye, the sample was loaded onto a 5 ml HiTrap desalting column (in Storage Buffer) attached to an AKTA Purifier system (GE Healthcare). The column was developed with Storage Buffer and fractions collected. The eluate was monitored at both 280 nm and at either 488 or 594 nm, depending on the fluorophore: the labelled protein eluted at ∼1 ml and the free label at ∼4 ml (data not shown). Extents of labelling were calculated from the ratio of absorbance readings at the maxima for the protein (280 nm) and for the dye (32).
Rapid reactions
A Hi-Tech SF61-DX2 stopped-flow fluorimeter (TgK Scientific Ltd, UK) was used to mix rapidly equal volumes (75 µl) of two solutions, both in either M- or C-buffer (as M-buffer but with 2 mM CaCl2 in place of the magnesium acetate), to give reactions at 20°C containing one reagent labelled with Ax488 and one labelled with Ax594. The fluorescence from the mixture in the observation cell was excited with monochromatic light at 492 nm (the optimum for excitation of Ax488, but distant from the excitation peak for Ax594) and emission observed through a Schott OG590 long-pass filter: this filter cuts off wavelengths <590 nm and so excludes most of the emission from Ax488 while permitting emission from Ax594. The signal from this set-up thus arises primarily from fluorescence resonance energy transfer (FRET) between the Ax488 and Ax594 moieties. Changes in emission were recorded over time: 2048 data points were collected over 0–6 s on a linear or a logarithmic scale. For each trace, records from ≥8 repeat reactions were averaged. To permit comparisons of the amplitudes of the fluorescence signal from different experiments, none of the settings on the instrument were altered between experiments.
Data fitting was done in BERKELEY MADONNA (http://www.berkeleymadonna.com/). This programme solves the differential rate equations for each species in any given reaction scheme by numerical integration, using specified values for each individual rate constant. It was then used to fit globally multiple data sets by varying iteratively the rate constants and the amplitude of the FRET signal from each species until it reached a minimum in the sum-of-squares deviations from the complete data set.
RESULTS AND DISCUSSION
Experimental design
The active complex of FokI on a DNA with one cognate site contains two monomers of the protein with their catalytic domains each positioned to cleave their target strand. There exist several possible pathways for its assembly (Figure 3). One is that two monomers associate in free solution to form the dimer, which then binds to the DNA via the recognition domain in one monomer before finally re-positioning its two catalytic domains against their target bonds downstream of the site (Figure 3, black route). In free solution, the equilibrium between monomeric and dimeric forms of FokI strongly favours the monomer to the exclusion of the dimer (17), but it may still permit a transient dimerization in solution immediately followed by DNA binding, which in turn fixes the enzyme as a dimer. Alternatively, a monomer of FokI binds to its recognition site and then, without moving its catalytic domain, uses that domain to capture a second monomer from solution: the catalytic domains from the DNA bound and the captured monomers then translocate together to their DNA cleavage loci, in bottom and top strands, respectively (Figure 3, yellow route). A further option is that FokI binds to its DNA site as above, as a monomer, but then moves its catalytic domain to its target bond in the bottom strand before associating with a second monomer (Figure 3, red route).
Figure 3.

Possible pathways to impact by FokI. The FokI restriction enzyme contains a recognition domain (in blue) and a catalytic domain (in red) connected by a flexible linker (black line). The oligoduplex carries the recognition site for FokI (yellow arrow, oriented 5′–3′ on the top strand) and the downstream sites for DNA cleavage 9 and 13 nt distant in top and bottom strands, respectively (black chevron). Initially, before binding to the DNA, the catalytic domain is sequestered by the recognition domain. One possible pathway (shown by the black connectors) is that two FokI monomers in free solution dimerize via their catalytic domains, then bind to DNA as a dimer before moving the two catalytic domains to the cleavage loci. Another possibility (yellow connectors) is that FokI binds to its recognition site as a monomer but without releasing its catalytic domain from its recognition domain, then recruits a second monomer from free solution and finally swivels the pair of catalytic domains to the cleavage sites. A third possibility (red connectors) is that the monomer of FokI at the recognition site undergoes the conformational change to position its catalytic domain at the cleavage site on the bottom strand after binding to the DNA and that the catalytic domain at the cleavage site then captures a second monomer to form the active protein–DNA complex.
These schemes may be differentiated by FRET. FRET exploits the ability of a pair of fluorophores to transfer energy between themselves, provided the emission spectrum of one (the donor) overlaps the excitation spectrum of the other (the acceptor), and that the two dyes are in close proximity (33,34). The excitation of the donor then results in emission from the acceptor. However, the efficiency of the transfer varies steeply with the distance between the fluorophores, especially over distances around the Förster radius (R0) for that particular pair of fluorophores (34). Consequently, FRET is a powerful method for detecting changes in the distance between two fluorescently labelled molecules, typically over 30–100 Å (33). Throughout this study, Ax488 was used as the donor and Ax594 as the acceptor: this pair has an R0 of 60 Å (32).
Two sorts of FRET experiments were carried out: firstly between DNA and protein and secondly between protein and protein. The former used oligoduplexes tagged at one end with the donor Ax488, either upstream or downstream of the recognition site (Figure 2), and a variant of the FokI protein labelled in the catalytic domain with the acceptor Ax594. This arrangement ought to yield FRET changes from both the initial binding of the protein to the DNA and the motion of the catalytic domain(s) to the DNA cleavage loci. Potentially, further FRET changes could come from the dimerization of the labelled protein on the DNA as this would position two acceptor fluorophores near the donor on the DNA: this possibility was examined by comparing signals from dimerization-competent (d-c) and dimerization-defective (d-d) variants of FokI. To monitor directly the protein–protein interaction by FRET, the reactions contained one sample of FokI that had been labelled with the donor fluorophore and a second of acceptor-labelled FokI. The FRET changes were monitored in a stopped-flow fluorimeter set to excite the Ax488 donor and to detect emission from the Ax594 acceptor.
Labelling FokI domains
For the above approaches, a fluorophore needs to be attached to a specific site in a given domain of the FokI protein. Fluorophores can be coupled to proteins by using maleimide chemistry to modify a cysteine residue (32,35). FokI might appear to be an ideal candidate for this strategy as it has only two cysteines: C84 in the DNA recognition domain and C541 in the catalytic domain (Figure 1). Hence, by mutating each Cys in turn, it should be possible to modify selectively the residual Cys and thus place a label specifically on the DNA recognition or the catalytic domain. Two mutants of FokI were initially constructed, each with a serine, a near-isosteric substitution, in place of a Cys, C84S and C541S. However, both mutants had <15% of the activity (kcat/Km) of WT FokI (Supplementary Table S1) and were much less stable than the native protein (data not shown). Several other amino acids were subsequently chosen as replacements, by computational design (R. Sessions, personal communication): for C84, Thr, Leu and Ala; for C541, Thr, Ile and Ala. All six proteins were purified and tested for catalytic activity: the best residues found for positions 84 and 541 were Ala and Thr, respectively.
The C84A and C541T proteins were treated with the maleimide conjugate of Ax594 but under all conditions and over all reaction times tested, <50% of the residual Cys in these proteins (C541 in the C84A variant, C84 in C541T) became modified with the dye (data not shown). The low levels of modification are probably due to the native cysteines being partially buried. A Cys-free (CF) variant of FokI was therefore made by removing both of its cysteines, before putting novel cysteines into the CF protein at surface-exposed positions. The CF form was constructed by introducing into the C84A protein the C541T substitution, to give the double mutant, C84A-C541T, with the optimal replacements at both positions.
The activity of the CF enzyme was similar (within a factor of 2) to WT FokI (Supplementary Table S1). The double mutant was then manipulated further at several solvent-accessible positions in the catalytic domain: S446, S511 and R570 were each mutated to Cys. The triple mutants were purified and labelled as before. Though all three mutants could be labelled with a maleimide derivative with near 100% yield, the CF-S446 and CF-R570 proteins had severely reduced activities, which fell still further upon labelling with Ax594. On the other hand, the triple mutant C84A-C541T-S511C (noted as CF-S511C) was almost as active—and as stable—as WT FokI and retained much of its activity after labelling. The latter was also shown to bind cognate DNA (Supplementary Figure S2). As in previous studies with other restriction enzymes (36,37), binding was monitored from the increase in the fluorescence anisotropy of a HEX-labelled duplex upon adding the protein. At saturation of the DNA, the anisotropy of the complex with the mutant protein was similar to that with WT FokI, indicating a similar rotational correlation time (34), which in turn indicates an assembly of comparable size.
DNA to protein FRET
To look for a FRET signal between appropriately labelled samples of the CF-S511C protein and a DNA duplex, the protein was tagged at its solitary Cys with the FRET acceptor Ax594 and then added to the duplex 24 s-Ax488 in a buffer containing Ca2+ (Figure 2). This 24 bp DNA carries the cognate site for FokI and the downstream loci for DNA cleavage. It also has the FRET donor Ax488 attached to the 3′-end of the top strand: the length of DNA between recognition site and label, 17 bp (≡58 Å), matches the R0 for this FRET pair. Following excitation of the donor, the emission spectrum from this system was scanned in a spectrofluorimeter (Supplementary Figure S3). In the absence of protein, the DNA showed the expected emission peak from Ax488 at 520 nm and no further peaks at higher wavelengths. When increasing amounts of CF-S511C-Ax594 were added to this DNA, the peak at 520 nm fell and a second peak arose at 620 nm, the emission maximum for Ax594. Increased acceptor emission with parallel falls in donor emission is the hallmark of FRET (34). The changes at both 520 and 620 nm developed progressively as the protein concentration was increased, up to a maximum when 2 mol protein had been added per mole DNA: further increases caused no further changes to the FRET signal (data not shown). In principle, the distance between donor and acceptor can be evaluated from the efficiency of FRET (33,34) but here the FRET seems to arise from one donor (on the DNA) to two acceptors (on separate protein molecules), which effectively excludes any attempt to evaluate distances between donor and acceptor(s). Nevertheless, it should still be possible to monitor the kinetics of FokI–DNA interactions from changes in the FRET signal with time.
The kinetics of FokI binding to DNA were studied by using a stopped-flow fluorimeter to mix rapidly the CF-S511C-Ax594 protein with oligoduplexes tagged at various positions with Ax488 (Figure 2). The buffer contained Ca2+ rather than Mg2+, to permit DNA binding without cleavage. Following excitation of the Ax488 at 492 nm, emission at wavelengths >590 nm (primarily from Ax594) was recorded over time (Figure 4). The resulting changes in emission occurred in at least two phases. These included an initial rapid increase in emission over the first ∼0.2 s (τ½ = 0.014 s) and then, with the 24 s-Ax488 substrate, a slower increase (τ½ = 0.37 s) over the next ∼2 s: the latter may itself contain two or more phases (Figure 4, black trace). [Reaction records were taken with the data points distributed uniformly over either linear or logarithmic time scales (‘Materials and Methods’ section). Data from a linear time-scale record is shown in Figure 4. The fast phase was analysed from logarithmic time-scale records (data not shown), with more data points at shorter time intervals].
Figure 4.

Binding of acceptor-labelled FokI to donor-labelled DNA. Equal volumes of the CF-S511C-Ax594 form of FokI and one of the following oligoduplexes were mixed by stopped-flow to give reactions in C-buffer at 20°C that contained 200 nM FokI protein and 100 nM duplex: either 24 s-Ax488 (black trace); 34 s-Ax488 (cyan trace); or Ax488-28 s (red trace). Excitation was at 492 nm and the change in emission at >590 nm recorded. The series of experiments shown were performed consecutively, with in all cases the same optical and amplification settings on the stopped-flow instrument. The cartoons illustrate the different positions of the donor fluorophore (green circle) on each oligoduplex relative to the recognition sequence (yellow arrow) and the cleavage loci (black chevron), respectively.
To examine whether the changes in fluorescence emission were due to alterations in the distance between the acceptor fluorophore(s) on the protein and the donor on the DNA, the 24 bp DNA was replaced with a 34 bp substrate, 34 s-Ax488 (Figure 2). The 24 and 34 bp duplexes are essentially identical apart from the latter having an additional 10 bp between cleavage loci and FRET donor: the length of DNA between label and recognition site, 92 Å, is now longer than the R0 for this FRET pair. When the 34 bp duplex tagged with Ax488 was mixed with the FokI variant carrying Ax594, the fluorescence record again showed multiple phases, with both fast and slow phases displaying an increase in emission. The rates of the fast (τ½ = 0.018 s) and the slow (τ½ = 0.63 s) phases with the 34 bp DNA were comparable to those with the 24 bp duplex but both phases featured much smaller amplitudes. The signal recorded in the stopped-flow thus denotes changes in the distance between the donor on the DNA and the acceptor(s) on the protein molecule(s). The increases in the amplitude of the signal show that the distance becomes progressively shorter in at least two stages during the binding of FokI to the downstream-labelled DNA.
The crystal structure of the FokI monomer bound to its specific site in the absence of metal ions (20) implies that at some stage during the reaction the catalytic domain must move from its initial position on the recognition domain to the downstream cleavage site. To see if the catalytic domain actually moves in the direction implied by the crystal structure, the 28 bp duplex Ax488-28 s was designed. This DNA carries recognition and cleavage sites as before but it has the Ax488 donor located upstream of the recognition site, rather than downstream. The catalytic domain and its attached acceptor should thus move away from the donor, not towards it. When the Ax488-28 s duplex was mixed with the acceptor-labelled CF-S511C mutant, the FRET signal again contained at least two phases: an initial rapid increase in acceptor emission but this time followed by a slower decline to ∼75% of the maximum (Figure 4, red trace). This second phase had similar kinetics (τ½ = 0.25 s) to the slow increase seen with DNA carrying downstream labels (τ½ = 0.37 s). The decrease in emission indicates that the acceptor on the catalytic domain moves away from the upstream donor after the protein binds the DNA. Presumably the catalytic domain is initially sequestered by the recognition domain, but then moves to its target locus, 13 nt downstream of the recognition site in the bottom strand.
Protein to protein FRET
The overall reaction of the FokI nuclease in the presence of metal ions involves not only the motion of the catalytic domain to its cleavage locus but also the formation of the protein dimer (17,26,27). It had yet to be determined at what stage during the reaction the dimer materializes. To monitor the protein–protein association, one batch of the CF-S511C variant of FokI was labelled with the acceptor fluorophore Ax594 and a separate batch with the donor Ax488. In the first test for dimer formation, the two labelled samples of FokI were mixed together in the absence of DNA: no change in emission was detected (Figure 5A, black trace). In contrast, when a solution containing both donor- and acceptor-labelled proteins was mixed by stopped-flow with an unlabelled DNA that possessed both recognition and cleavage sites (24 s, Figure 2), a relatively slow increase in acceptor emission was observed (Figure 5B, green trace) (This duplex has the same sequence as the 24 s-Ax488 duplex used above but just lacks the 3′ label). The protein–protein FRET in the presence of cognate DNA thus lacks the fast phase observed over the first 0.2 s of the DNA–protein FRET records (Figure 4) and instead occurs over the same time scale as the slow phases(s) seen with DNA–protein FRET.
Figure 5.

Association of acceptor-labelled and donor-labelled FokI. Reaction A: equal volumes of solutions of the CF-S511C-Ax488 and the CF-S511C-Ax594 forms of FokI were mixed together by stopped-flow to give a reaction containing both FokI derivatives at 100 nM, in C-buffer at 20°C with no other components (black trace). Reactions B–D: the stopped-flow device was used to mix equal volumes of one solution containing both the Ax488- and the Ax594 labelled forms of FokI with a second containing one of the following duplexes, to give reactions in C-buffer at 20°C that contained, after mixing, the duplex and both FokI derivatives all at 100 nM. The duplexes were: in (B), 24 s, green trace; in (C), 24 ns, cyan trace; in (D), 13 s, red trace. Excitation was at 492 nm and the change in emission at >590 nm recorded. The cartoons adjacent to the letters B–D, illustrate the characteristics of the relevant duplex: the recognition sequence (only in B and D) is noted as a yellow arrow and the cleavage loci (only in B) by a chevron (the duplex for C lacks the recognition sequence and is not a substrate).
Further tests employed either a non-specific DNA duplex that lacked the recognition sequence for FokI (24 ns, Figure 2), or a truncated duplex that had the recognition site but only 4 bp downstream of the sites and so lacked the scissile bonds 9 and 13 nt away (13 s). When added to the mixture of the donor- and acceptor-labelled varieties of FokI, neither the non-specific nor the truncated duplex caused any change in acceptor emission (Figures 5C and D, cyan and red traces, respectively). In control experiments, all three of the duplexes used here were able to displace a HEX-labelled specific duplex bound to the FokI protein, albeit with varying degrees of efficiency: 24 s was the most effective, followed by 24 ns and then 13 s: these experiments (data not shown) were carried out by adding to an equilibrium mixture of FokI with the 24 s-HEX duplex increasing amounts of each duplex and then measuring the decrease in the anisotropy of the HEX (as in Supplementary Figure S2). The latter two duplexes are thus able to bind the protein, yet they fail to induce the protein-protein FRET.
The lack of a protein–protein signal in the absence of DNA concurs with previous studies showing that FokI is a monomer in free solution (16,18). This approach thus failed to provide any support for schemes involving the dimerization of FokI in free solution before DNA binding, but it did reveal the assembly of the dimer on the DNA. The generation of a FRET signal from the donor- and acceptor-labelled proteins in the presence of a cleavable DNA substrate shows that the two proteins must be physically close to each other on the DNA, presumably by forming the dimer required for cutting both DNA strands. However, dimer assembly requires the recognition sequence, since no FRET was seen with a non-specific duplex even though FokI binds to non-specific DNA. But the recognition sequence is not in itself sufficient for dimer formation: it also requires the downstream cleavage loci, as no assembly occurred on the truncated DNA that lacked the scissile bonds. Dimer formation thus presumably occurs after the catalytic domain of the monomer at the recognition site has engaged its target phosphodiester bond 13 nt away: i.e. domain motion precedes the dimerization step.
Strategy for kinetic analysis
The FRET experiments noted above (Figures 4 and 5), together with previous data (26,27,29), establish that, out of the various possibilities shown in Figure 3, FokI assembles its active complex on a DNA with one recognition site by the pathway marked in red: first, the protein binds to the cognate site via its DNA binding domain, during which stage the catalytic domain remains pinioned against the binding domain; the catalytic domain of the monomer at the recognition site then moves to its target phosphodiester bond 13 nt away in the bottom strand; that catalytic domain then associates with the catalytic domain from a second monomer, to position the active site from the latter against the scissile bond 9 nt away in the top strand; only then can the complex cleave the DNA, and eventually disassemble. However, in this scheme, the formation of the catalytically active complex with two protein monomers at a single recognition site, the (ES)*E species, is defined by six rate constants (see below), while the complete reaction in the presence of Mg2+ requires yet more rate constants for the DNA cleavage and product release steps. Attempts to fit data from binding reactions in Ca2+ to all six rate constants resulted in highly degenerate and poorly determined sets of values: each set appeared to fit the data equally well but the values for the individual rate constant in one set often varied wildly from that for the same constant in the next set. Consequently, to obtain robust values for the rate constants for each step, the separate stages of the reaction needed to be examined in isolation of each other (Figure 6A).
Figure 6.

Reactions of donor-labelled DNA with acceptor-labelled variants of FokI. (A) Rate constants for the individual steps in the overall reaction of FokI at its recognition site (Equation 3) were obtained in stages as indicated by the brackets below the scheme: k1, k-1, k2 and k−2 from (B); k3 and k−3 from (C); and k4 from (D). The reactions in (B–D) were carried out at 20°C by mixing in the stopped-flow device the oligoduplex 24 s-Ax488 with either the d-d or the d-c form of the CF-S511C species of FokI labelled with Ax594: concentrations noted refer to final concentrations after mixing. (B) The reactions, in C-buffer, contained 30 nM duplex and d-d protein at either 40 nM (red trace) or 60 nM (cyan trace). (C) The reactions, in C-buffer, contained 30 nM duplex and d-c protein at either 60 nM (cyan trace) or 80 nM (green trace). (D) The reactions, in M-buffer, contained 100 nM duplex and 200 nM d-c protein (orange trace). For (B–D), the best fits to the integrated rate equations from the relevant reaction scheme, Equations 1–3, respectively, are shown in black.
Dimerization of FokI can be blocked by mutating two amino acids on the surface of the catalytic domain, D483 and R487 (Figure 1), to alanines (26). Preventing dimerization should stop the reaction of FokI on DNA after the translocation of the catalytic domain, so that the reaction is defined by just four rate constants and two fluorescence amplitude changes.
| (1) |
[(ES)* denotes the complex with the protein in an extended conformation engaging its phosphodiester bond.] It may thus be possible to evaluate all four of the rate constants in Equation 1 by fitting the data from stopped-flow experiments with a d-d variant of FokI to the relevant kinetic equations. The reactions with an active d-c version of FokI in the presence of Ca2+ extends this scheme to
| (2) |
Values for the forward and reverse rate constants for the dimerization step should then be attainable by keeping the above values for the first four rate constants and then fitting the data with the d-c variant by floating just k3 and k−3. Finally, in the presence of Mg2+, the active enzyme will proceed to cleave the DNA and release the products
| (3) |
The data from cleavage reactions in the stopped-flow can then yield the best fit to a value for k4 alone, given the previously determined values for all six preceding rate constants. The dissociation of the cleaved DNA fragment that carries the fluorescence donor will result in the decay of the FRET signal from the (ES)*E complex but the rate constant (k4) for this step also encompasses the two hydrolysis steps, in bottom and top strands.
Dimerization-defective FokI
The mutations known to block FokI dimerization, D483A and R487A (26), were introduced into the CF-S511C variant of FokI to give a species with five amino acid substitutions, C84A-C541T-S511C-D483A-R487A, named ddCF-S511C. This quintuple mutant was tested for catalytic activity and DNA binding. It had, as expected, no detectable activity (Supplementary Table S1): the d-d mutations inactivate FokI (26). Binding was monitored from the change in fluorescence anisotropy upon adding the protein to a HEX-labelled duplex with the specific sequence (Supplementary Figure S2). The d-d derivative bound the duplex like its parent but it produced a complex that had a lower anisotropy than those with either the CF-S511C or the WT proteins, indicating faster rotational motion (Supplementary Figure S2). In gel-filtration experiments, the ddCF-S511C protein bound to a DNA duplex eluted after the equivalent complex with its parental protein, CF-S511C (data not shown), again indicating a smaller molecular mass. Moreover, unlike the d-c variant (Figure 5B), no FRET was obtained upon adding the cognate DNA 24 s to a mix of d-d proteins labelled with either Ax488 or Ax594 (data not shown). Hence, under conditions where the specific complex with active FokI contains two protein molecules, the complex formed with the d-d mutant seems to involve only one.
The ddCF-S511C protein was labelled at its single Cys with the FRET acceptor Ax594. In parallel experiments, this d-d protein, or the equivalent d-c species (CF-S511C-Ax594), were mixed in the stopped-flow with the donor-labelled DNA, 24 s-Ax488, in the presence of Ca2+. The changes in FRET during the binding reactions were recorded at varied concentrations of each protein (Figures 6B and C). In all cases, the FRET signal increased throughout the reaction, in two or more distinct phases. Since the kinetics of the reactions with the d-d protein (Figure 6B) reflect the initial binding of the protein to the DNA (Equation 1) while those with the active protein (Figure 6C) also involve the dimerization step (Equation 2), and since the KD for the former is much smaller than the latter (27,28), most of the reactions with the d-d protein were at lower protein concentrations than those with the active enzyme. Nevertheless, when tested at the same protein concentration (cyan traces in both Figures 6B and C), the amplitudes of the FRET changes, particularly those for the slower phases, were smaller with the d-d protein than the d-c species. Hence, the FRET changes seen during the binding of the acceptor-labelled CF-S511C protein to donor-labelled DNA are indeed due to the proximity of the donor to two molecules of the acceptor.
To obtain rate constants for the association of FokI to its recognition site and for the subsequent conformational change but without the added complication of the dimerization step, the FRET data at varied concentrations of the d-d protein was correlated to the reaction scheme in Equation 1. The rate equations for each species were solved by numerical integration in an iterative procedure that altered systematically the four rate constants (k1, k−1, k2 and k−2) and the FRET amplitudes for each step, until a minimal sum-of-squares deviations from the entire data set across the range of protein concentrations was reached (‘Materials and Methods’ section). The best fit found by this procedure (Figure 6B, black traces) indicated that ∼60% of the total increase in FRET occurred upon the initial binding of the d-d protein to the DNA, with association (k1) and dissociation (k−1) rate constants of 1.3 × 109 M−1s−1 and 6.3 s−1, respectively. This association rate constant is typical for the binding of proteins to specific DNA sites at low ionic strength (38) while the ratio of k−1:k1 yields a KD for the initial binding at 4.8 nM. In previous studies (28), the rates at which WT FokI had cleaved a plasmid substrate were measured in single turnover reactions at varied enzyme concentrations to yield a KD of 4 nM for the binding of the protein to a cognate site. The previous studies had however been carried out at 37°C rather than the 20°C employed here.
The best fit of the data with the d-d mutant stipulated a further increase in the FRET signal after the initial binding, corresponding to ∼40% of the total change. The increase in FRET during this slow phase shows that the acceptor on the catalytic domain of the DNA-bound protein must move closer to the donor attached to the DNA downstream of the recognition site: i.e. towards the DNA cleavage loci. The forward and reverse rate constants for this reconfiguration (k2 = 1.4 s−1; k−2 = 0.09 s−1) demonstrate that the preferred conformation of FokI bound to DNA in the presence of metal ions is with its catalytic domain located downstream of the recognition site, and not the juxtaposed arrangement of catalytic and recognition domains seen in the crystal structure without metal ions (20).
To extend the above analysis to include the dimerization stage but not the DNA cleavage or subsequent steps, the FRET records of the binding of the active CF-S511C enzyme to DNA in Ca2+ were correlated to the scheme in Equation 2 by numerical integration, as above, except this time fixed values were used for the first four rate constants (k1, k−1, k2 and k−2). These constants were held at the values for the corresponding steps with the d-d mutant and the only parameters allowed to float while fitting the data from the d-c variant were the forward and reverse rate constants for the dimerization step itself (k3 and k−3 in Equation 2) and the relative FRET signals from the ES, (ES)* and (ES)*E states. The data from the d-c variant could accommodate readily the numbers for the first four rate constants from the d-d mutant, as these still allowed for close fits to the experimental data (Figure 6C). The best global fit of the data with the d-c enzyme gave values of 4 × 107 M−1s−1 for k3, the rate constant for the association of the DNA-bound monomer with a second monomer from free solution, and 3 s−1 for k−3, the dissociation of the dimer. The former falls in the middle of the range observed for protein–protein association rates (39), while the ratio of k−3:k3 yields a KD of 75 nM for the dimerization equilibrium. The latter value obtained at 20°C can be compared with the KD values of 100–166 nM, that had been got from single-turnover reactions of plasmid cleavage by WT FokI at 37°C (27).
The complete reaction
The Ca2+ that had been present in all of the above experiments, to allow DNA binding and dimerization but not cleavage (18), was replaced with Mg2+. FokI requires Mg2+ for optimal activity (15,16), so this ought to reveal the complete turnover of the enzyme reaction, including the hydrolytic steps and the subsequent release of the cleaved DNA. Upon adding the CF-SC511C form of FokI carrying the FRET acceptor Ax594 to the donor-labelled DNA 24 s-Ax488, a rapid increase in fluorescence emission was observed followed by a slow decline back to the level before mixing (Figure 6D). The increase occurred in multiple phases, each of which had essentially the same kinetics as the binding reaction in the presence of Ca2+. The slow decline appeared to be governed by a single exponential. The FRET donor is located on the top strand of the DNA, beyond both recognition and cleavage sites so, even if the enzyme remains bound to its site after cutting the DNA, the donor will likely be liberated from the rest of the complex on an 8 nt segment of non-specific DNA, with concomitant loss of the FRET signal.
The sequential increases and the subsequent decrease in the FRET signal were related to the scheme in Equation 3 by the numerical integration procedure noted above. In fitting the data from the reactions of the active enzyme in Mg2+, the only parameters allowed to vary in the search for an optimal fit were the rate constant for the hydrolysis/product release step, k4, and the amplitudes of the FRET change at each stage. The estimates for the forward and reverse rate constants for the initial binding (k1 and k−1) and the domain motion (k2 and k−2) were all held at the values found with the d-d mutant in Ca2+ (Figure 6B), while the constants for the dimerization step (k3 and k−3) were likewise kept to the best-fit solutions found with the d-c protein in Ca2+ (Figure 6C: this fit had already employed fixed values for k1, k−1, k2 and k−2). Despite these constraints, this scheme still gave a close match to the experimental data (Figure 6D, orange trace): the above values for the six preceding rate constants thus seem to be applicable to the active enzyme with Mg2+. The best fit yielded a value of 0.5 s−1 for k4.
Though the decline in the FRET signal during the reaction in Mg2+ is due to the release of the 8 nt product from the top strand, the rate of this process could be determined by either strand scission event [FokI cuts the two strands at the same rate, in no particular order: (29)] or by the actual dissociation of the product. The value of 0.5 s−1 for k4 obtained from the FRET data is, however, about 10 times faster than the turnover rate of FokI on a one-site plasmid, measured under conditions when all of the DNA-bound enzyme is in the dimeric state (27). For FokI (27,28), as for most Type II restriction enzymes (37,40,41), the rate-limiting step of the overall reaction on plasmid substrates is the final dissociation of the cleaved DNA product(s) rather than phosphodiester hydrolysis. But they often leave the DNA product(s) more slowly if the substrate is a large plasmid rather than a short duplex (40,42). After cleaving its site on a plasmid, the dissociation of the enzyme from that site is then normally followed by multiple re-association/dissociation events with non-specific sites elsewhere in the DNA polymer before eventually escaping into bulk solution (38,43). In contrast, after cutting a short duplex, the enzyme dissociates directly into bulk solution. Hence, the relative high value for k4 may reflect a relatively rapid release of the labelled DNA product.
CONCLUSIONS
By combining distance sensitivity with the necessary time resolution, FRET measurements in the stopped-flow proved to be a powerful approach to elucidating the mode of action of the FokI endonuclease. It revealed essentially the complete pathway for the reaction of FokI on a DNA with a single recognition site, in terms of the sequence of the molecular events and the kinetics of virtually every step (Figure 7).
Figure 7.

The reaction pathway for FokI. The cartoons illustrate the individual steps in the pathway for the reaction of the FokI restriction endonuclease on a DNA substrate with one recognition site. The pathway identified for FokI corresponds to the scheme in Figure 3 marked with red connectors. As in Figure 3, the recognition and catalytic domains of the FokI protein are denoted by blue and red ovals, respectively while in the DNA the recognition site is represented by a yellow arrow and the cleavage loci by a black chevron. The values for each rate constant determined here are also noted at each step.
The three main stages during the assembly of the active complex of FokI at its recognition site are the initial binding, the domain motion and the dimerization step. The order of these events, and thus the distinction between the various possible pathways shown in Figure 3, was elucidated by protein–protein FRET (Figure 4), which showed that dimerization happened after domain motion. The DNA–protein FRET (Figure 3) revealed the nature of the domain motion: after binding to DNA, the catalytic domain of the FokI protein moves from its initial position, presumably against the recognition domain (20), to a position downstream of the recognition site, probably the scissile bond 13 nt away in the bottom strand. In the presence of Mg2+, the active complex proceeds to cleave both DNA strands and release the products. The loss of FRET at this stage is due to the release of the segment of the DNA carrying the donor but whether this is rate-limited by the release itself or by the hydrolysis reactions is not yet known.
The overall scheme (Equation 3) is defined by seven rate constants. It is impossible to establish meaningful values for all seven from the FRET records of the complete reaction (Figure 6D). Instead, a stepwise procedure was used: first, forward and reverse rate constants for the DNA binding and domain motion steps came from the data with the inactive d-d form of FokI (Figure 6B); next, rate constants for dimer formation were established with the active d-c protein in Ca2+ (Figure 6C); finally, a single rate constant encompassing, both DNA cleavage and product dissociation was obtained from the reaction of the active enzyme in Mg2+ (Figure 6D). The values given here for all seven constants (Figure 7) are justified by the fact that those for the first two steps, from the d-d mutant, could be applied to both dimerization and DNA-cleavage by the d-c protein, in Ca2+ and Mg2+, respectively: likewise the values for dimerization in Ca2+ could be carried forward to DNA cleavage in Mg2+. In addition, the rate constants for the two bimolecular steps, the protein–DNA and protein–protein associations, both fell within the expected range for each sort of reaction (38,39). Furthermore, the KD values for both bimolecular equilibria, from the ratio of forward and reverse rate constants, matched previous estimates from the kinetics of plasmid DNA cleavage, albeit at a different temperature (27,28).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1; Supplementary Figures 1–3; References [27,30,31,34].
FUNDING
The Wellcome Trust (grant 078794) and the Marie Curie Research Training Network on ‘DNA Enzymes’. Funding for open access charge: The Wellcome Trust and the Marie Curie Research Training Network.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Richard Sessions, Bernard Connolly and Mark Szczelkun for help in the design of the project. We also thank Stuart Bellamy, Lucy Catto and Yana Kovacheva for experimental aid and advice, Rachel Smith and David Rusling for comments on the manuscript and New England Biolabs for the supply of materials.
REFERENCES
- 1.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2010;38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts RJ, Halford SE. Type II restriction endonucleases. In: Linn SM, Lloyd RS, Roberts RJ, editors. Nucleases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 35–88. [Google Scholar]
- 3.Aggarwal AK. Structure and function of restriction endonucleases. Curr. Opin. Struct. Biol. 1995;5:11–19. doi: 10.1016/0959-440x(95)80004-k. [DOI] [PubMed] [Google Scholar]
- 4.Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell. Mol. Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PubMed] [Google Scholar]
- 5.Vipond IB, Halford SE. Specific DNA recognition by EcoRV restriction endonuclease induced by calcium ions. Biochemistry. 1995;34:1113–1119. doi: 10.1021/bi00004a002. [DOI] [PubMed] [Google Scholar]
- 6.Szybalski W, Kim SC, Hasan N, Podhajska AJ. Class-IIS restriction enzymes - a review. Gene. 1991;100:13–26. doi: 10.1016/0378-1119(91)90345-c. [DOI] [PubMed] [Google Scholar]
- 7.Mucke M, Kruger DH, Reuter M. Diversity of Type II restriction endonucleases that require two DNA recognition sites. Nucleic Acids Res. 2003;31:6079–6084. doi: 10.1093/nar/gkg836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirsanova OV, Baskunov VB, Gromova ES. Type IIE and IIF restriction endonucleases interacting with two recognition sites on DNA. Mol. Biol. 2004;38:886–900. [PubMed] [Google Scholar]
- 9.Siksnys V, Grazulis S, Huber R. Structure and function of the tetrameric restriction enzymes. In: Pingoud A, editor. Nucleic Acids and Molecular Biology, Restriction Endonucleases. Vol. 14. Berlin: Springer-Verlag; 2004. pp. 237–259. [Google Scholar]
- 10.Gowers DM, Bellamy SRW, Halford SE. One recognition sequence, seven restriction enzymes, five reaction mechanisms. Nucleic Acids Res. 2004;32:3469–3479. doi: 10.1093/nar/gkh685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marshall JJT, Halford SE. The Type IIB restriction endonucleases. Biochem. Soc. Trans. 2010;38:410–416. doi: 10.1042/BST0380410. [DOI] [PubMed] [Google Scholar]
- 12.Chan SH, Stoddard BL, Xu SY. Natural and engineered nicking endonucleases-from cleavage mechanism to engineering of strand-specificity. Nucleic Acids Res. 2011;39:1–18. doi: 10.1093/nar/gkq742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden DTF, Dybvig K, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aggarwal AK, Wah DA. Novel site-specific DNA endonucleases. Curr. Opin. Struct. Biol. 1998;8:19–25. doi: 10.1016/s0959-440x(98)80005-5. [DOI] [PubMed] [Google Scholar]
- 15.Sugisaki H, Kanazawa S. New restriction endonucleases from Flavobacterium okeanokoites (FokI) and Micrococcus luteus (MluI) Gene. 1981;16:73–78. doi: 10.1016/0378-1119(81)90062-7. [DOI] [PubMed] [Google Scholar]
- 16.Kaczorowski T, Skowron P, Podhajska AJ. Purification and characterization of the FokI restriction endonuclease. Gene. 1989;80:209–216. doi: 10.1016/0378-1119(89)90285-0. [DOI] [PubMed] [Google Scholar]
- 17.Vanamee ES, Santagata S, Aggarwal AK. FokI requires two specific DNA sites for cleavage. J. Mol. Biol. 2001;309:69–78. doi: 10.1006/jmbi.2001.4635. [DOI] [PubMed] [Google Scholar]
- 18.Li L, Wu LP, Chandrasegaran S. Functional domains in Fok I restriction endonuclease. Proc. Natl Acad. Sci. USA. 1992;89:4275–4279. doi: 10.1073/pnas.89.10.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim YG, Li L, Chandrasegaran S. Insertion and deletion mutants of FokI restriction endonuclease. J. Biol. Chem. 1994;269:31978–31982. [PubMed] [Google Scholar]
- 20.Wah DA, Hirsch JA, Dorner LF, Schildkraut I, Aggarwal AK. Structure of the multimodular endonuclease FokI bound to DNA. Nature. 1997;388:97–100. doi: 10.1038/40446. [DOI] [PubMed] [Google Scholar]
- 21.Waugh DS, Sauer RT. A novel class of FokI restriction endonuclease mutants that cleave hemi-methylated substrates. J. Biol. Chem. 1994;269:12298–12303. [PubMed] [Google Scholar]
- 22.Waugh DS, Sauer RT. Single amino acid substitutions uncouple the DNA binding and strand scission activities of Fok I endonuclease. Proc. Natl Acad. Sci. USA. 1993;90:9596–9600. doi: 10.1073/pnas.90.20.9596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yonezawa A, Sugiura Y. DNA binding mode of class-IIS restriction endonuclease FokI revealed by DNA footprinting analysis. Biochim. Biophys. Acta. 1994;1219:369–379. doi: 10.1016/0167-4781(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 24.Sasnauskas G, Zakrys L, Zaremba M, Cosstick R, Gaynor JW, Halford SE, Siksnys V. A novel mechanism for the scission of double-stranded DNA: BfiI cuts both 3′-5′ and 5′-3′ strands by rotating a single active site. Nucleic Acids Res. 2010;38:2399–2410. doi: 10.1093/nar/gkp1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wah DA, Bitinaite J, Schildkraut I, Aggarwal AK. Structure of FokI has implications for DNA cleavage. Proc. Natl Acad. Sci. USA. 1998;95:10564–10569. doi: 10.1073/pnas.95.18.10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bitinaite J, Wah DA, Aggarwal AK, Schildkraut I. FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA. 1998;95:10570–10575. doi: 10.1073/pnas.95.18.10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Catto LE, Ganguly S, Milsom SE, Welsh AJ, Halford SE. Protein assembly and DNA looping by the FokI restriction endonuclease. Nucleic Acids Res. 2006;34:1711. doi: 10.1093/nar/gkl076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catto LE, Bellamy SRW, Retter SE, Halford SE. Dynamics and consequences of DNA looping by the FokI restriction endonuclease. Nucleic Acids Res. 2008;36:2073–2081. doi: 10.1093/nar/gkn051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanders KL, Catto LE, Bellamy SRW, Halford SE. Targeting individual subunits of the FokI restriction endonuclease to specific DNA strands. Nucleic Acids Res. 2009;37:2105–2115. doi: 10.1093/nar/gkp046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bath AJ, Milsom SE, Gormley NA, Halford SE. Many type IIs restriction endonucleases interact with two recognition sites before cleaving DNA. J. Biol. Chem. 2002;277:4024–4033. doi: 10.1074/jbc.M108441200. [DOI] [PubMed] [Google Scholar]
- 31.Halford SE, Johnson NP, Grinsted J. The EcoRI restriction endonuclease with bacteriophage lambda DNA. Kinetic studies. Biochem. J. 1980;191:581–592. doi: 10.1042/bj1910581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson I, Spence MTZ. Molecular Probes Handbook, a Guide to Fluorescent Probes and Labeling Technologies. 11th edn. California: Invitrogen, Carlsbad; 2010. [Google Scholar]
- 33.Stryer L. Fluorescence energy transfer as a spectroscopic ruler. Annu. Rev. Biochem. 1978;47:819–846. doi: 10.1146/annurev.bi.47.070178.004131. [DOI] [PubMed] [Google Scholar]
- 34.Lakowizc JR. Principles in Fluorescence Spectroscopy. 3rd edn. New York, NY: Kulwer Academic/Plenum Press; 2006. [Google Scholar]
- 35.Lundblad RL. Chemical Reagents for Protein Modification. 3rd edn. Boca Raton, Florida: CRC Press; 2005. [Google Scholar]
- 36.Reid SL, Parry D, Liu HH, Connolly BA. Binding and recognition of GATATC target sequences by the EcoRV restriction endonuclease: A study using fluorescent oligonucleotides and fluorescence polarization. Biochemistry. 2001;40:2484–2494. doi: 10.1021/bi001956p. [DOI] [PubMed] [Google Scholar]
- 37.Bellamy SR, Kovacheva YS, Zulkipli IH, Halford SE. Differences between Ca2+ and Mg2+ in DNA binding and release by the SfiI restriction endonuclease: implications for DNA looping. Nucleic Acids Res. 2009;37:5443–5453. doi: 10.1093/nar/gkp569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halford SE. An end to 40 years of mistakes in DNA-protein association kinetics? Biochem. Soc. Trans. 2010;37:343–348. doi: 10.1042/BST0370343. [DOI] [PubMed] [Google Scholar]
- 39.Schreiber G, Haran G, Zhou H-X. Fundamental aspects of protein-protein association kinetics. Chem. Rev. 2009;109:839–860. doi: 10.1021/cr800373w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erskine SG, Baldwin GS, Halford SE. Rapid-reaction analysis of plasmid DNA cleavage by the EcoRV restriction endonuclease. Biochemistry. 1997;36:7567–7576. doi: 10.1021/bi970155s. [DOI] [PubMed] [Google Scholar]
- 41.Wright DJ, Jack WE, Modrich P. The kinetic mechanism of EcoRI endonuclease. J. Biol. Chem. 1999;274:31896–31902. doi: 10.1074/jbc.274.45.31896. [DOI] [PubMed] [Google Scholar]
- 42.Baldwin GS, Vipond IB, Halford SE. Rapid reaction analysis of the catalytic cycle of the EcoRV restriction endonuclease. Biochemistry. 1995;34:705–714. doi: 10.1021/bi00002a038. [DOI] [PubMed] [Google Scholar]
- 43.Gowers DM, Halford SE. Protein motion from non-specific to specific DNA by three-dimensional routes aided by supercoiling. EMBO J. 2003;22:1410–1418. doi: 10.1093/emboj/cdg125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.