

Abstract
InCl3, InBr3, and In(OTf)3 were tested as promoters in the preparation of glycosides from trichloroacetimidate precursors. A range of protecting groups and of alcohol acceptors were used to determine the versatility of these promoters. Disaccharide formation was demonstrated. In most cases, the In(III) compounds were shown to promote glycosylation better than the widely used promoter BF3•OEt2.
Keywords: Glycosylation, Trichloroacetimidate, Indium(III) promotion, InBr3, InCl3, In(OTf)3
Glycosylation is one of the most widely used processes in carbohydrate chemistry. It is one of the most common post translational modifications1 and thus is an important reaction in the synthesis and study of biologically relevant molecules. Synthetic methods of glycosylation require activation of the carbohydrate donor.2 These reactions have been researched and reviewed extensively,3-8 and promotion of a trichloroacetimidate donor with a Lewis acid is common.2,8 Due to the broad scope of glycosylation reactions, there is a constant need for new promoters to fine tune the reaction for specific donors and acceptors. Several of these promoters are heavy metal based, which can be impractical due to the waste generated.9-11 BF3•OEt2 and TMSOTf are common alternatives as they avoid heavy metal waste,2,8 however these promoters are hygroscopic, and BF3•OEt2 requires distillation prior to use, which creates difficulty in utility.
This paper describes the use of indium(III) as an alternative to current promoters. In(III) is desirable when compared to other Lewis acids due to its stability in air and water12,13 and relatively low toxicity.14 Since it is a weaker Lewis acid than the other heavy metal promoters, In(III) should be compatible with a wider range of substrates.15 In carbohydrate synthesis, In(III) has been reported to catalyze peracetylation,16 acetolysis, formation and hydrolysis of acetals, and formation of thioglycosides.17,18 In addition, InCl3 has been reported as a glycosylation promoter using glycohalide donors,19,20 as well as peracetylated trichloroacetimidates.21 While these sources focused on InCl3, this paper broadens the scope to include other In(III) glycosylation promoters in the presence of a variety of protecting groups and alcohol acceptors.
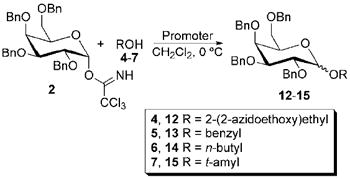
To investigate glycosylation reactions with In(III), InBr3, InCl3, and In(OTf)3 were used as promoters for the glycosylation of three trichloroacetimidate donors with four alcohol acceptors. The strategy for reaction development is shown in Scheme 1. Acetyl, benzyl, and acetonide protecting groups were chosen to provide a stability range that tested the limitations of the In(III) promoters. Primary, tertiary, and benzylic alcohols were chosen as glycosyl acceptors to demonstrate the steric range through which the varying In(III) compounds should be effective. The promoter BF3•OEt2 was used for comparison with In(III) reagents. Overall, this paper describes a simple reaction procedure for In(III) promoted glycosylation reactions.
Scheme 1.
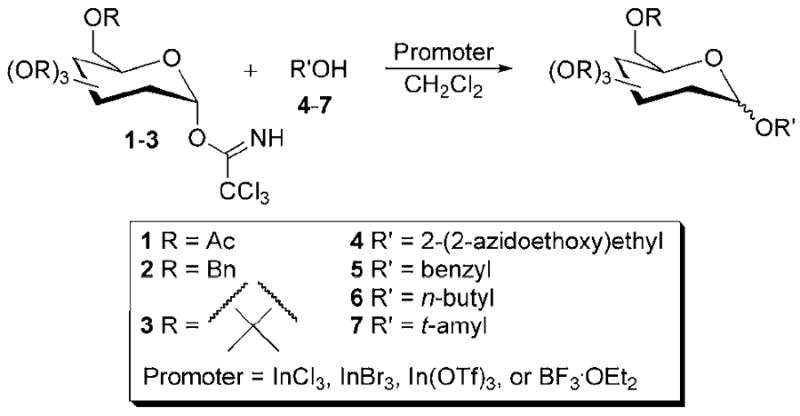
In(III)-promoted glycosylation.
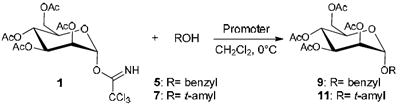
To identify optimal reaction conditions for each promoter, reactions were performed at a variety of different temperatures and with varying amounts of promoter. Reactions were monitored by 1H NMR for the presence of the trichloroacetimidate starting material to determine how long the reactions should be performed. For acetyl protected mannosyl donor 1, reactions at 0 °C were found to be optimal. As shown in Table 1, using alcohols 5 and 7 as the acceptor, 0.5 equivalents of InCl3 was necessary with this Lewis acid. Only 10 mol % of InBr3 was required (with decomposition becoming increasingly significant as larger amounts of InBr3 were added). A trace amount of In(OTf)3 was sufficient for glycosylation to occur in good yield. Thus, reactions using trichloroacetimidates 1-3 with alcohols 4-7 are reported using 0.1 equivalents of InBr3, 0.25-0.5 equivalents of InCl3, and 0.05 equivalents or a trace amount of In(OTf)3. Additional optimization reactions are summarized in Table S1 of the Supplementary Data.
Table 1.
Optimization of reaction conditions using 1 and 5 or 7.
 | |||
|---|---|---|---|
| Alcohol | Promoter | Time (min)a | Yield (%)b |
| 7 | InBr3 (0.9 equiv.) | 20 | 63 |
| 7 | InBr3 (0.2 equiv.) | 20 | 59 |
| 7 | InBr3 (0.1 equiv.) | 30 | 75 |
| 7 | InBr3 (0.06 equiv.) | 10 | 68 |
| 5 | InCl3 (0.5 equiv.) | 20 | 72 |
| 5 | InCl3 (0.25 equiv.) | 50 | 59 |
| 7 | In(OTf)3 (1.0 equiv.) | 10 | 37 |
| 7 | In(OTf)3 (0.2 equiv.) | 10 | 37 |
| 7 | In(OTf)3 (0.1 equiv.) | 10 | 52 |
| 7 | In(OTf)3 (0.06 equiv.) | 15 | 54 |
| 7 | In(OTf)3 (trace) | 20 | 54 |
| 7 | none | 60 | 0 |
Time to reaction completion.
Yields determined by 1H NMR spiked with a mesitylene standard.
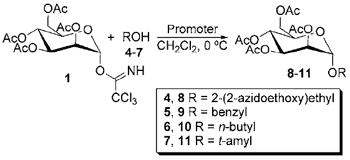
A series of experiments were performed to compare In(III) promoters against the standard method of BF3•OEt2 in glycosylation promotion, and these studies are summarized in Tables 2-4. Reactions using the peracetylated mannosyl trichloroacetimidate 1 are reported in Table 2. In general, InBr3 and InCl3 were shown to perform better than BF3•OEt2, with preference toward InBr3 or InCl3 being acceptor dependant. With alcohols 4-6, In(OTf)3 afforded lower yields of glycosylation products, but this trend is not observed with trichloroacetimidates 2 and 3 (vide infra). NMR yields with alcohol 4 were so low that no isolated yield was attempted. Only the alpha product was observed, presumably due to the neighboring group participation of the acetyl group at C2 on the mannoside and due to the anomeric effect.
Table 2.
Glycosylation results using peracetylated trichloroacetimidate 1.
 | |||
|---|---|---|---|
| Product | Promoter (equiv.) | Timea (min.) | Yield (%) |
| 8 | InBr3 (0.1) | 30 | 42 |
| 8 | InCl3 (0.5) | 20 | 45 |
| 8 | In(OTf)3 (0.05) | 15 | 6b |
| 8 | BF3•OEt2 (0.2) | 30 | 59 |
|
| |||
| 9 | InBr3 (0.1) | 30 | 53 |
| 9 | InCl3 (0.5) | 20 | 72 |
| 9 | In(OTf)3 (0.05) | 15 | 31 |
| 9 | BF3•OEt2 (0.2) | 30 | 59 |
|
| |||
| 10 | InBr3 (0.1) | 30 | 81 |
| 10 | InCl3 (0.5) | 20 | 42 |
| 10 | In(OTf)3 (0.05) | 15 | 33 |
| 10 | BF3•OEt2 (0.2) | 30 | 40 |
|
| |||
| 11 | InBr3 (0.1) | 30 | 60 |
| 11 | InCl3 (0.5) | 20 | 52 |
| 11 | In(OTf)3 (0.05) | 15 | 57 |
| 11 | BF3•OEt2 (0.2) | 30 | 43 |
Time to reaction completion.
Yields determined by 1H NMR spiked with a mesitylene standard.
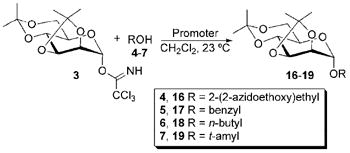
Table 4.
Glycosylation results using acetonide-protected trichloroacetimidate 3.
 | |||
|---|---|---|---|
| Product | Promoter (equiv.) | Timea (min.) | Yield (%) |
| 16 | InBr3 (0.1) | 25 | 29 |
| 16 | InCl3 (0.25) | 45 | 35 |
| 16 | In(OTf)3 (trace) | 30 | 92 |
| 16 | BF3•OEt2 (0.2) | 30 | 12 |
|
| |||
| 17 | InBr3 (0.1) | 25 | 37 |
| 17 | InCl3 (0.25) | 45 | 23 |
| 17 | In(OTf)3 (trace) | 25 | 90 |
| 17 | BF3•OEt2 (0.2) | 30 | 20 |
|
| |||
| 18 | InBr3 (0.1) | 25 | 12 |
| 18 | InCl3 (0.25) | 45 | 0 |
| 18 | In(OTf)3 (trace) | 25 | 78 |
| 18 | BF3•OEt2 (0.2) | 30 | 4 |
|
| |||
| 19 | InBr3 (0.1) | 25 | 3 |
| 19 | InCl3 (0.25) | 60 | 9 |
| 19 | In(OTf)3 (trace) | 25 | 83 |
| 19 | BF3•OEt2 (0.2) | 30 | 6 |
Time to reaction completion.
The yields for reactions using the perbenzylated galactosyl trichloroacetimidate 2 are summarized in Table 3. Used in these catalytic amounts, In(III) was shown to be an effective promoter in all cases tested, with In(OTf)3 giving yields of product formation consistently better than the BF3•OEt2 control. Yields for reactions with InBr3 were generally higher than those for reactions using InCl3, with the exception of the more hindered alcohol 7, which gave lower yields overall.
Table 3.
Glycosylation results using perbenzylated trichloroacetimidate 2.
 | ||||
|---|---|---|---|---|
| Product | Promoter (equiv.) | Timea (min.) | Yieldb (%) | α:β ratiob |
| 12 | InBr3 (0.1) | 30 | 57 | 1:10 |
| 12 | InCl3 (0.5) | 20 | 48 | 1:30 |
| 12 | In(OTf)3 (0.05) | 20 | 72 | 0:1 |
| 12 | BF3•OEt2 (0.2) | 30 | 65 | 0:1 |
|
| ||||
| 13 | InBr3 (0.1) | 30 | 61 | 0:1 |
| 13 | InCl3 (0.5) | 20 | 53 | 0:1 |
| 13 | In(OTf)3 (0.05) | 20 | 68 | 0:1 |
| 13 | BF3•OEt2 (0.2) | 30 | 60 | 0:1 |
|
| ||||
| 14 | InBr3 (0.1) | 30 | 57 | 0:1 |
| 14 | InCl3 (0.5) | 20 | 53 | 0:1 |
| 14 | In(OTf)3 (0.05) | 20 | 63 | 0:1 |
| 14 | BF3•OEt2 (0.2) | 30 | 61 | 0:1 |
|
| ||||
| 15 | InBr3 (0.1) | 30 | 30 | 1:3 |
| 15 | InCl3 (0.5) | 20 | 33 | 2:7 |
| 15 | In(OTf)3 (0.05) | 20 | 52 | 1:1 |
| 15 | BF3•OEt2 (0.2) | 30 | 24 | 2:7 |
Time to reaction completion.
Ratios determined by 1H NMR after column purification.
The experiments that are summarized in Table 3 using the perbenzylated galactosyl trichloroacetimidate 2 indicate that In(III) promoters can be catalytically used to effectively promote glycosylation reactions. However, since In(III) has been reported as a promoter of Friedel Crafts chemistry,22,23 formation of side products is likely to detrimentally affect reaction yields. Although the side products were not characterized, the increased complexity of the aromatic region in the 13C and 1H NMR spectra of crude product mixtures from 2 indicates their formation.
For reactions using benzylated donor 2, the β isomer is the major product in every case tested, and the most easily isolated, but α:β ratios vary depending on the acceptor used. Using In(OTf)3 afforded the best results, but the conditions were optimized based on overall yield rather than anomeric ratios. It should be noted that time and strength of the acid promoter (as well as reaction temperature) may have a significant effect on the anomeric ratio of products. While the α product gains some anomeric stability, electrostatic interactions in the oxocarbenium ion half-chair intermediates should have a strong effect on product formation.24 For 2, since some unfavorable interactions are likely in both half-chairs, mixtures of α and β products form. For 3, only one oxocarbenium ion can form, and only the α product is ovserved (see results below and Figure S1 in the Supplementary Data for half-chair diagrams).
Products 14 and 15 were independently resubjected to the reaction conditions using one equivalent of InBr3, InCl3, or BF3•OEt2, and interconversion of the α and β isomers was not observed. The α:β ratio did change over time for 14 (see Table S3 in the Supplementary Data), but the 1H NMR spectrum clearly indicated decomposition of 14, and the α:β ratio doesn’t reliably shift toward the ratio that was obtained in the reaction (in two trials, the α:β ratio shifts away from rather than toward the reaction ratio). These experiments indicate that the reaction occurs under kinetic control.
Glycosylation reactions using acetonide-protected mannoside 3 are summarized in Table 4. Optimization of these reactions requires a balance between the strength and quantity of the promoter that is present and the length of time that is required for the reaction to achieve completion. Reactions comparing temperatures and times were performed using alcohol 5, and details are provided in Table S1 in the Supplementary Data. Above 20 mol% for InBr3 and above trace amounts for In(OTf)3, significant product degradation was observed. The length of reaction time required when using InCl3 lead to significant product degradation. Removal of acetonide protecting groups using InCl3 has been reported,25 and deprotection is apparently a competitive reaction as the length of reaction and the quantity of promoter are increased, as seen by loss of protecting group peaks in the NMR spectra. Trace amounts of In(OTf)3 proved to be the most effective catalyst for glycosylation of 3 and required no column chromatography for purification. No beta product was observed for reactions using acetonide-protected mannoside 3, presumably due to the anomeric effect in conjunction with steric hindrance at the C2 position (see Figure S1 in the Supplementary Information for diagrams).
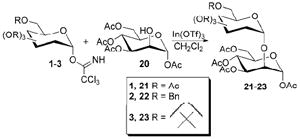
Overall, glycosylation of peracetylated mannosyl trichloroacetimidate 1 with simple alcohols afforded the highest yields using InBr3 or InCl3. In(OTf)3 was the best promoter for reactions with perbenzylated galactosyl trichloroacetimidate 2 and acetonide protected mannosyl trichloroacetimidate 3. Using these results, disaccharide-forming reactions were explored using 1-3 and acceptor 20. Initial tests with InBr3 afforded significant amounts of unreacted 1 even when 20 mol % was used (rather than the 10 mol % that was used with 4-7) and the reaction time was extended six-fold. In(OTf)3 proved to be the preferred promoter for all three donors in disaccharide formation with 20, and the results are shown in Table 5. Acetyl and acetonide protected mannosyl trichloroacetimidates gave high yields of disaccharides (78% and 94%, respectively) with only α-linked products observed and no column chromatography required in the case of acetonide protected mannosyl 3. Benzylated galactosyl trichloroacetimidate 2 afforded a lower yield of α-linked product (27% yield) due to β product also being formed.
Table 5.
In(OTf)3 promoted disaccharide formations.
 | ||||
|---|---|---|---|---|
| Product | Promoter (equiv.) | Temp (°C) | Timea (min.) | Yield (%) |
| 21 | In(OTf)3 (0.05) | 0 | 20 | 78 |
| 22 | In(OTf)3 (0.05) | 0 | 30 | 27b |
| 23 | In(OTf)3 (trace) | 23 | 25 | 94 |
Time to reaction completion.
Yield is for α-linked product only.
In conclusion, InBr3, InCl3 and In(OTf)3 were effective promoters of glycosylation reactions when used with the variety of protecting groups tested. With respect to acetylated mannosyl trichloroacetimidate 1, InBr3 or InCl3 afforded the highest yields. All three of the In(III) promoters that were tested worked well with benzylated galactosyl trichloroacetimidate 2, with In(OTf)3 giving slightly higher yields. When using acetonide protected mannosyl trichloroacetimidate 3, trace amounts of In(OTf)3 gave the highest yields and required no column chromatography for purification, while InBr3 and InCl3 gave lower yields equivalent to BF3•OEt2. A glycoside acceptor was also successfully added to the trichloroacetimidates using In(OTf)3 as the promoter. Overall, In(III) afforded good to excellent yields of products without the requirement of distillation or an inert reaction atmosphere. The ability to vary ligands on In(III) also provides a broad range of promotion strength, enabling optimization of the conditions for desired reactions.
1. Experimental
1.1. General Methods
To obtain pure products, crude products were purified by column chromatography on 60 Å silica gel for acetyl protected mannosyls and benzyl protected galactosyls with hexane: ethyl acetate mobile phase in the indicated ratio. Acetonide protected manosyls were purified by column chromatography on 32-63μm neutral alumina with hexane: ethyl acetate mobile phase. An ice bath was used for reactions run at 0 °C. 13C and 1H NMR were recorded for purified compounds on a Bruker DRX 500 MHz Spectrometer using TMS as an internal standard. Mass spectra were obtained using a high-resolution Bruker micro-TOF system with electrospray ionization. Yields are either isolated yields. or were calculated from crude 1H NMR spectra using an internal mesitylene standard as specified. Ratios of alpha to beta diastereomers were determined using integrations of resonances in the 1H NMR spectra or (for equivalent carbons) in the 13C NMR spectra.26 Trichloroacetimidate starting materials27 and acceptor 2028 were synthesized using previously described methods. Trace additions of In(OTf)3 were performed by the addition of a visible amount of In(OTf)3 that weighed less than 0.1mg.
1.2. General procedure for optimization of reaction conditions
Duplicate reactions were performed in parallel. A dry 4 mL scintillation vial was charged with the trichloroacetimidate starting material with a slight excess of alcohol and dissolved in enough dry CH2Cl2 to obtain a 50 mM solution of the trichloroacetimidate. The mixture was cooled if specified, and the promoter was added. BF3•OEt2 reactions were run under an argon atmosphere. Aliquots were taken from vial 1 and evaluated by 1H NMR for reaction completion. When no starting material was observed, vial 2 was quenched with approximately 0.2 g dry NaHCO3 for 20 min, diluted with 2 mL CH2Cl2 and filtered. The solvent was removed under reduced pressure. Yields of vial 2 were obtained from 1H NMR spectra using an internal mesitylene standard. Reaction conditions for vial 2 reactions are provided in Table S1.
1.3. General procedure for the preparation of mannopyranoside and galactopyranoside derivatives
A dry 4 mL scintillation vial was charged with the trichloroacetimidate starting material with a slight excess of alcohol and dissolved in enough dry CH2Cl2 to obtain a 50 mM solution of the trichloroacetimidate. The mixture was cooled if specified, and the promoter was added. BF3•OEt2 reactions were run under an argon atmosphere. The reaction stirred for the specified time, quenched with approximately 0.2 g dry NaHCO3 for 20 min, diluted with 2 mL CH2Cl2 and filtered. The solvent was removed under reduced pressure. Chromatographic purification was performed (galactopyranoside products 8:2 hexane : EtOAc; acetyl protected mannopyranoside products either 7:3 hexane : EtOAc or 1:1 hexane : EtOAc; acetonide protected mannopyranoside products 7:3 hexane : EtOAc + 0.5% DCM) to obtain pure samples. Exact reaction conditions are provided in Table S2 of the Supplementary Data.
1.4. General procedure for evaluation of the reversibility of the reaction
A dry 2 mL scintillation vial was charged with the glycoside and a slight excess of alcohol and dissolved in enough dry CH2Cl2 to obtain a 50 mM solution of the trichloroacetimidate. The mixture was cooled to 0 °C, and the promoter was added. Reactions run with BF3•OEt2 were additionally purged with Ar. The reaction then stirred for 2h, then was diluted with 2 mL CH2Cl2 and quenched with 4 mL saturated NaHCO3, washed with 4 mL brine, then dried over MgSO4 and filtered. The solvent was removed under reduced pressure. The α:β ratios of crude products were obtained from 1H NMR spectra. These reactions are summarized in Table S3 of the Supplementary Data.
1.5. General procedure for the preparation of acetonide protected mannopyranoside derivatives using In(OTf)3
A dry 4 mL scintillation vial was charged with the trichloroacetimidate 3 with a slight excess of alcohol and dissolved in enough dry CH2Cl2 to obtain a 50 mM solution of the trichloroacetimidate. Less than 0.1 mg In(OTf)3 was added, and reaction was stirred for 25 min. The reaction was quenched with approximately 0.2 g dry NaHCO3 for 20 min. The solution was then filtered over a medium-sized plug of alumina using CH2Cl2 as the eluent. The solvent was removed under reduced pressure affording clean product.
1.6. General procedure for preparation of disaccharides
A dry 4 mL scintillation vial was charged with the trichloroacetimidate starting material with a slight excess of 20 and dissolved in enough dry CH2Cl2 to obtain a 50 mM solution of the trichloroacetimidate. The mixture was cooled if specified, and the promoter was added. The reaction then stirred for the specified time, was quenched with 2 mL of saturated NaHCO3 for 20 min, diluted with 2 mL CH2Cl2 and filtered. The solvent was removed under reduced pressure. Chromatographic purification was performed (galactopyranoside product 6:4 hexane : EtOAc; acetyl protected mannopyranoside product 4:6 hexane : EtOAc). Acetonide protected mannopyranoside 23 was prepared via procedure 1.5 above. Exact reaction conditions are provided in Table S4 of the Supplementary Data.
Supplementary Material
Research Highlights.
In(OTf)3 and InBr3 are convenient catalytic promoters of glycosylation.
InCl3 is a convenient non-catalytic promoter of glycosylation.
In(III) was used effectively with trichloroacetimidate donors bearing acetyl, benzyl, and acetonide protecting groups.
Disaccharide formation using In(III) promoters was demonstrated.
Acknowledgments
This work was supported by NIH R01 GM62444.
Footnotes
Supplementary data associated with this article can be found, in the online version, at
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhao YY, Takahashi M, Gu JG, Miyoshi E, Matsumoto A, Kitazume S, Taniguchi N. Cancer Sci. 2008;99:1304–1310. doi: 10.1111/j.1349-7006.2008.00839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schmidt RR, Michel J. Angew Chem Int Ed Eng. 1980;19:731–732. [Google Scholar]
- 3.Fugedi P. Glycosylation Methods. In: Levy DE, Fugedi P, editors. The Organic Chemistry of Sugars. CRC Press; Boca Raton: 2006. pp. 89–179. [Google Scholar]
- 4.Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497–510. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 5.Smoot JT, Demchenko AV. Oligosaccharide Synthesis: from Conventional Methods to Modern Expeditious Strategies. In: Horton D, editor. Advances in Carbohydrate Chemistry and Biochemistry. Vol. 62. Academic Press; USA: 2009. pp. 161–250. [DOI] [PubMed] [Google Scholar]
- 6.Lindhorst TK. Essentials of carbohydrate chemistry and biochemistry. 3. Wiley-VCH; Weinheim: 2007. [Google Scholar]
- 7.Hanessian S, editor. Preparative carbohydrate chemistry. Marcel Dekker; New York: 1997. [Google Scholar]
- 8.Zhu XM, Schmidt RR. Angew Chem Int Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 9.Douglas SP, Whitfield DM, Krepinsky JJ. J Carbohydr Chem. 1993;12:131–136. [Google Scholar]
- 10.Adinolfi M, Barone G, Guariniello L, Iadonisi A. Tetrahedron Lett. 2000;41:9005–9008. [Google Scholar]
- 11.Adinolfi M, Barone G, Iadonisi A, Mangoni L, Schiattarella M. Tetrahedron Lett. 2001;42:5967–5969. [Google Scholar]
- 12.Loh TP, Chua GL. Chem Commun. 2006:2739–2749. doi: 10.1039/b600568n. [DOI] [PubMed] [Google Scholar]
- 13.Frost CG, Hartley JP. Mini-Rev Org Chem. 2004;1:1–7. [Google Scholar]
- 14.Asakura K, Satoh H, Chiba M, Okamoto M, Serizawa K, Nakano M, Omae K. J Occupational Health. 2009;51:498–512. doi: 10.1539/joh.l9080. [DOI] [PubMed] [Google Scholar]
- 15.Bandini M, Cozzi PG, Giacomini M, Melchiorre P, Selva S, Umani-Ronchi A. J Org Chem. 2002;67:3700–3704. doi: 10.1021/jo0163243. [DOI] [PubMed] [Google Scholar]
- 16.Bizier NP, Atkins SR, Helland LC, Colvin SF, Twitchell JR, Cloninger MJ. Carbohydr Res. 2008;343:1814–1818. doi: 10.1016/j.carres.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Das SK, Roy J, Reddy KA, Abbineni C. Carbohydr Res. 2003;338:2237–2240. doi: 10.1016/s0008-6215(03)00355-0. [DOI] [PubMed] [Google Scholar]
- 18.Giri SK, Verma M, Kartha KPR. J Carbohydr Chem. 2008;27:464–478. [Google Scholar]
- 19.Mukherjee D, Ray PK, Chowdhury US. Tetrahedron. 2001;57:7701–7704. [Google Scholar]
- 20.Ghosh RC, Chakraborty A, Maiti DK, Maiti SB. Ind J Chem. 2002;41B:583–585. [Google Scholar]
- 21.Ghosh RC, Chakraborty A, Maiti DK. Ind J Chemistry. 2003;42B:602–604. [Google Scholar]
- 22.Kaneko M, Hayashi R, Cook GR. Tetrahedron Lett. 2007;48:7085–7087. [Google Scholar]
- 23.Thirupathi P, Kim SS. J Org Chem. 2009;74:7755–7761. doi: 10.1021/jo9014613. [DOI] [PubMed] [Google Scholar]
- 24.Yang MT, Woerpel KA. J Org Chem. 2009;74:545–553. doi: 10.1021/jo8017846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pfrengle F, Dekaris V, Schefzig L, Zimmer R, Reissig HU. Synlett. 2008:2965–2968. [Google Scholar]
- 26.Decoster E, Lacombe JM, Strebler JL, Ferrari B, Pavia AA. J Carbohydr Chem. 1983;2:329–341. [Google Scholar]
- 27.Ren T, Liu DX. Tetrahedron Lett. 1999;40(43):7621–7625. [Google Scholar]
- 28.Gros EG, D JO, Mastronardi IO. Carbohydr Res. 1967;4:432–434. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.