

Abstract
Background
Hepatocellular carcinoma is difficult to diagnose early, and most patients are already in the late stages of the disease when they are admitted to hospital. The total 5-year survival rate is less than 5%. Recent studies have showed that brucine has a good anti-tumor effect, but high toxicity, poor water solubility, short half-life, narrow therapeutic window, and a toxic dose that is close to the therapeutic dose, which all limit its clinical application. This study evaluated the effects of brucine immuno-nanoparticles (BIN) on hepatocellular carcinoma.
Materials and methods
Anionic polymerization, chemical modification technology, and phacoemulsification technology were used to prepare a carboxylated polyethylene glycol-polylactic acid copolymer carrier material. Chemical coupling technology was utilized to develop antihuman AFP McAb-polyethylene glycol-polylactic acid copolymer BIN. The size, shape, zeta potential, drug loading, encapsulation efficiency, and release of these immune-nanoparticles were studied in vitro. The targeting, and growth, invasion, and metastasis inhibitory effects of this treatment on liver cancer SMMC-7721 cells were tested.
Results
BIN were of uniform size with an average particle size of 249 ± 77 nm and zeta potential of –18.7 ± 4.19 mV. The encapsulation efficiency was 76.0% ± 2.3% and the drug load was 5.6% ± 0.2%. Complete uptake and even distribution around the liver cancer cell membrane were observed.
Conclusion
BIN had even size distribution, was stable, and had a slow-releasing effect. BIN targeted the cell membrane of the liver cancer cell SMMC-7721 and significantly inhibited the growth, adhesion, invasion, and metastasis of SMMC-7721 cells. As a novel drug carrier system, BIN are a potentially promising targeting treatment for liver cancer.
Keywords: cancer targeting, hepatocellular carcinoma, nanoparticles, targeted drug delivery, anti-tumor effect
Introduction
Hepatocellular carcinoma (HCC) is the fifth most common malignant tumor, and there is only one cancer that causes more deaths worldwide. The overall 5-year survival rate is less than 5%. Each year some 564,000 new cases are diagnosed, and 549,000 people die. The poor prognosis is mainly due to common portal vein tumor invasion, which can cause high rates of intrahepatic metastasis and postoperative recurrence. Even in the early stages after radical resection, the metastasis and recurrence rate is 61.5%.1–3 Currently, the primary treatment for primary liver cancer is surgery resection.
Primary liver cancer is often accompanied by varying degrees of cirrhosis, which is reported in 70%–90% of all cases. The risk of cirrhosis is enormously increased with respect to liver cirrhosis due to the high sensitivity of the hardened liver to ischemia and blood loss. Long-term hepatic inflow occlusion and massive blood loss can easily lead to postoperative acute liver failure. The overall surgical resection rate is 15%–20%.4,5 Ziser et al found that patients with cirrhosis undergoing surgeries had a perioperative complication rate of 30.1% and mortality rate of 11.6%.6 Liver transplantation for the treatment of liver cancer can completely resect the tumor and also radically improve the liver function of the patient. Especially for unresectable liver cancer or patients who cannot undergo liver resection, liver transplantation is the only radical surgical treatment. Nevertheless, tumor recurrence and metastasis significantly restrict the feasibility of liver transplantation. Calne et al reported that 37.5% of patients receiving liver transplants died of tumor recurrence from 2 months to 5 years after the surgery. The 5-year survival rate was 18.6%.7
For patients who cannot tolerate surgery or bear a high risk of recurrence and metastasis after the surgery and transplantation, the alternative treatments include systemic and local chemotherapy. These can control tumor growth to some extent, but some side effects such as poor targeting, low sensitivity, short effecting time, and high toxicity all significantly restrict their clinical application.8–10 Therefore, the development of a new drug delivery system characterized by effective targeting of the cancer cells, strong anti-tumor effects, local drug accumulation, slow release, and lower systemic toxicity is the focus for liver cancer treatment and the prevention of recurrence and metastasis.
Brucine is a weak basic indole alkaloid. Its formula is C23H26N2O4 and its molecular weight is 394.47 Da. It is a white crystal that is highly toxic and odorless, with a very bitter taste. It is slightly soluble in water, is levorotatory, and is soluble in ether, chloroform, ethanol, methanol and other organic solvents. Its high toxicity, poor water solubility, short half-life, and low toxic dose for intravenous use limit its clinical application in cancer treatment.
Qin et al treated in vitro cultured human hepatoma cells SMMC-7721 with brucine and found that the inhibition rate grew as the amount of brucine increased. At a dosage of 320 μg/mL, the inhibition rate was close to 100%, which showed that brucine had a significant inhibitory effect on liver cancer cells. Further studies showed that brucine could induce cell apoptosis by increasing Fas expression.11 Deng et al found that brucine and its liposome complex showed significant growth inhibition on transplanted liver cancer in Heps tumor-bearing mice, and it stimulated the hematopoietic and immune systems. Liposomal brucine has targeting and slow-release effects, and showed more powerful anti-tumor effects than brucine monomer.12–14
Combining the drug-loaded nanoparticles with monoclonal antibodies (McAb) against human hepatocellular carcinoma, we produced a drug-nanoparticle-monoclonal antibody immune complex. With cellular-targeting capabilities, McAb could carry the drug-loaded nanoparticle to specific sites and enhance the interaction between the drug and liver cancer cells, thus elevating local drug concentration and increasing drug efficacy. This study employed anionic polymerization and chemical modification technology to prepare a carboxylated polyethylene glycol-polylactic acid block copolymer material, used phacoemulsification technology to prepare carboxylated polyethylene glycol-polylactic acid block copolymer brucine nanoparticles, and utilized chemical coupling technology to develop anti-human AFP McAb-polyethylene glycol-poly lactic acid copolymers with brucine immuno-nanoparticles. By culturing human liver cancer cells SMMC-7721 in vitro, as well as in matrix adhesion and transwell chamber experiments, we observed the effects of brucine immuno-nanoparticles on liver cancer cell growth, cell matrix adhesion, invasion, and migration ability.
Materials and methods
Materials
Brucine monomer (Chengdu Man Si Te Biotechnology Co, Ltd, batch number: 110706-200 505, purity >99%); 5-fluorouracil(5-Fu) (Shanghai Xudong Haipu Pharmaceutical Co, Ltd, batch number: 090315); carboxylated polyethylene glycol-poly lactic acid block copolymer (PLA-PEG-COOH, molecular weight, 40 kDa) (Jiangsu Paige Bio Co, Ltd, PA20100302); mouse anti-human AFP monoclonal antibody (McAb, 70 kDa, Hangzhou Hua Biotechnology Co, Ltd); 8% polyvinyl alcohol (PVA) aqueous solution, acetonitrile, methylene chloride, methanol, acetone, octanol, carbodiimide hydrochloride (EDAC) (Shanghai Xinhua Chemical Co, Ltd); goat anti-mouse IgG FITC (Shanghai Unitech Biotechnology Co, Ltd); human hepatocellular carcinoma SMMC-7721 cell line (provided by Shanghai Biological Sciences Institute affiliated to the Chinese Academy of Sciences); Tetrazolium salt (MTT), dimethyl sulfoxide (DMSO) (Sigma, Munich, Germany); Matrigel gel (BD Biosciences, Franklin Lakes, NJ); transwell (8 μm pore size; Corning, Inc, Corning, NY); fibronectin (Roche, Basel, Switzerland).
Methods
Preparation of the carboxylated polyethylene glycol-polylactic acid copolymers brucine nanoparticles
Anionic polymerization and chemical modification technology were used to prepare carboxylated polyethylene glycol-polylactide block copolymer materials. The following were mixed into 120 mL of brucine nano-emulsion: 0.8 g of oil phase carboxylated polyethylene glycol-polylactic acid block copolymer (PLA-PEG-COOH), 40 mg of brucine, 16 mL of dichloromethane (CH2Cl2), 8% aqueous polyvinyl alcohol (PVA), 40 mL of aqueous solution, and 80 mL of pure water. The mixture was dispersed by a high shear cutting homogenization machine, then dispersed under 12,000 rpm speed shear, and emulsified five times (for 30 seconds each time) under 300 W ultrasound. The emulsion was then added to 80 mL of purified water and magnetically stirred at 700 rpm at room temperature for 6 hours to volatilize the CH2Cl2. The dried emulsion was centrifuged at 5000 rpm for 10 minutes to remove any aggregates. The brucine nano-emulsion was then harvested by collecting the supernatant. The brucine nano-emulsion was put into an ultrafiltration tube (Millipore Amicon 100, 000; Millipore, Billerica, MA) and centrifuged at 3000 g for 30 minutes to concentrate the solution and remove free brucine. The solution was then washed with purified water and centrifuged with ultrafiltration again to obtain the brucine concentrate, which was stored at 4°C in the refrigerator.
Preparation of the anti-human AFP McAb-polyethylene glycol-polylactic acid copolymer BINs
Five milligrams of EDAC (carbodiimide hydrochloride) was added to 3 mL of brucine nanoparticle solution. The mixture was shaken for 15 minutes at 200 rpm, and antihuman AFP McAb (1 mg/mL) was added, oscillated at 200 rpm for 3 hours at 4°C, and centrifuged at 10,000 rpm for 5 minutes. The product of this was washed three times in 0.01 M PBS (pH 7.4). Nanoparticles were isolated by ultrafiltration centrifugation, and the BINs were stored at 4°C in the refrigerator.
Analysis of particle size and zeta potential
A PSS ZWL380 nano particle size analyzer (Particle Sizing Systems, Inc, Santa Barbara, CA) was used to measure BIN particle size, size distribution, and zeta potential by the dynamic light scattering method. The angle of observation was 90°, and the temperature of the measurement was 25°C.
Morphology
The prepared BIN suspension sample was put on aluminum foil, dried, and sprayed with platinum. A S4800 scanning electron microscope (Hitachi, Tokyo, Japan) was used to observe the morphology with an accelerating voltage of 1 kV.
Drug loading
Brucine acetonitrile solution (0.01 mg/mL) was prepared as the control solution. 0.1 mL of BIN concentrate was added to 5 mL of acetonitrile, and the concentrate was ultrasounded for 5 minutes to fully extract the brucine, then acetonitrile was added to make up the volume to 10 mL. After being shaken and centrifuged at 12,000 rpm for 20 minutes, the supernatant was collected to measure the UV absorption at 263 nm wavelength, and the absorbance A2 was recorded. The UV absorption of the control solution was measured at a wavelength of 263 nm, and the absorbance (As) was recorded. The brucine content in the concentrate was calculated as (A2/As) in mg/mL.
In vitro drug release
The dialysis method was employed to determine the drug release rate of BIN in vitro.
To calculate the stability of the brucine in the release medium, 20 mg of brucine was diluted to 0.01 mg/mL with 0.5% polyoxyethylene dehydrated sorbitol monooleate PBS solution. The solution was put into a water bath constant temperature oscillator (37°C ± 0.5°C) in the dark and 1.0 mL was sampled at 0, 0.5, 1, 2, 4, 8, 12, 24, 36, 48, and 60 hours, with 20 μL of this sample used to measure the brucine content. The content at 0 hours was set as 100% and changes in the brucine content were measured.
To calculate the drug release of BIN in vitro, dialysis bags were placed in distilled water for 24 hours. Eight milliliters of BIN suspension was put into the dialysis bags, the ends of the dialysis bags were clipped, and the bags were placed into the release medium with magnetic stirring (200 rpm). Of the solution, 1.0 mL was sampled at 0, 0.5, 1, 2, 4, 8, 12, 24, 36, 48, and 60 hours, with 20 μL of this sample used to measure the brucine content. The data were corrected in accordance with the degradation curve in the release medium. Concentrations of the brucine in the sample were measured at different time points. The cumulative drug release percentage (Q) was calculated and the cumulative release curve was plotted.
Determination of monoclonal antibody on brucine immuno-particle surface
The bicinchoninic acid (BCA) method was used to measure the antibody content of samples. After the BSA standard protein solution (10 μg/mL–750 μg/mL) and BCA working solution were allowed to react, the solution’s absorbance at 562 nm was measured and the concentration-absorbance curve and the curve equation were obtained. Forty microliters of BIN suspension reacted with the BCA working solution and its absorbance at 562 nm was determined. The concentration of the antibody on the brucine nanoparticles was then obtained using the curve equation.
Cell targeting and positioning
Human hepatoma cells (SMMC-7721) were diluted with 10% fetal calf serum medium (RPMI-1640) to a cell concentration of 1 × 105/mL. One milliliter of human hepatoma cells (SMMC-7721) was cultured in an incubator in 5% CO2 at 37°C for 24 hours. The culture fluid was discarded and the cells were washed twice with 0.01 M PBS. Then 100 μL of BIN solution was added and the cells were incubated in 5% CO2 at 37°C for 24 hours, after which the BIN solution was discarded and the cells were washed three times with 0.01 M PBS. One hundred microliters of FITC-labeled goat anti-mouse antibody (20 μL stock solution + 80 μL 0.01 M PBS) was added. After incubation for 2 hours, the cells were washed three times with 0.01 M PBS. A Zeiss LSM 510 META confocal microscope (Zeiss, Oberkochen, Germany) was used to observe the cells with 488 nm laser excitation.
Grouping
Based on the experimental requirements, eight groups were set up: a RPMI-1640 group, a PLA-PEG-COOH copolymer group, an anti-human AFP McAb group, an anti-human AFP McAb- PEG-PLA block copolymer group, a 5-FU group, a brucine group, a brucine nanoparticles group, and a BIN group.
Morphological changes of liver cancer cells
Human hepatoma cells SMMC-7721 were diluted with 10% fetal calf serum medium RPMI-1640 to a cell concentration of 4 × 104/mL. The cell suspension was seeded on a 96-well plate with 100 μL per well under conditions of saturated humidity, 5% CO2, and 37°C for 12 hours. After the medium was removed, 100 μL of 10% fetal calf serum RPMI-1640 culture medium containing different drug concentrations was added to each well. The different drug concentrations were 0.5, 1.0, 1.25, 2.5, 5.0, 7.5, 10, 15, 20, 25, 30, 40, 80, 160, 240, 320, 400, 480 μg/mL. Three wells were set up for each dose group.
The blank control group used 10% fetal calf serum medium RPMI-1640 and the control group used human hepatoma cells cultured with 10% fetal calf serum RPMI- 1640 culture medium. The content of PLA-PEG-COOH copolymer, anti-human AFP-McAb, and anti-human AFP McAb-PLA-PEG block copolymer nanoparticles corresponded to the amount of anti-human AFP -McAb and PEG-PLA block copolymer in the BIN. After placing the HCC cells under conditions of saturated humidity, 5% CO2, and 37°C for 72 hours, morphological changes as well as the growing state of cells were observed by inverted phase contrast microscope.
Cancer cell growth inhibition
After 72 hours of culture, 20 μL of MTT solution (5 mg/mL) was added to each well and cells were cultured for 4 hours under 37°C in the dark. Then the MTT-containing medium was removed and 150 μL DMSO was added to the culture dish to react for 10 minutes. The absorbance value A of the blank control group was detected at 570 nm and adjusted to zero. The inhibition rate was calculated by the following formula:
Effects of BIN on cell adhesion of liver cancer cells
Serum-free 1640 was used to dilute Matrigel (100 μg/mL), and 25 μL of the solution was then added to each of the 96 wells in the plate and left to dry in a biological safety cabinet at room temperature. Brucine, 5-FU, brucine nanoparticles, and BIN were applied to SMMC-7721 hepatoma cells for 72 hours, with each applied at five different concentrations: 20, 40, 80, 160, and 240 μg/mL. A cell suspension of 4.0 × 105 cells/mL was prepared and 100 μL was added to each well of a 96-well plate adhesion system containing Matrigel. Each of the concentration cell suspensions was added to three wells, and each plate included three empty holes that were left empty as the control (only 100 μL serum-free 1640 was added in the adhesion system). The 96-well plates containing hepatoma cells were placed in an incubator at 37°C, with 5% CO2 for 2 hours. The plates were then gently washed three times with 0.01 M PBS to remove non-adhesive cells. Twenty microliters of MTT solution (5 mg/mL) was added to each well, and the plates were then placed in the incubator at 37°C, with 5% CO2 for 4 hours. PBS (0.01 M) was used to wash the plates three times, and 100 μL of DMSO solution was added to each well and mixed for 10 minutes. The optical density of each well was measured under 570 nm to indirectly determine the cell-matrix adhesion. The average adhesion rate and the standard deviation were obtained by calculating the adhesion rate of each experimental concentration and that of the three blank holes.
Experimental adhesion rate of each well = (OD value of each hole – average OD value of blank wells)/(average OD value of control wells – average OD value of blank wells) × 100%
Effects of BIN on invasion of liver cancer cells
Serum-free 1640 was used to dilute Matrigel (100 μg/mL), which was added to the upper transwell chamber (50 μL/chamber) to dry in a biological safety cabinet at room temperature. Serum-free 1640 was added, and after 90 minutes the liquid and uncombined Martigel were removed. Brucine, 5-FU, brucine nanoparticles, and BIN at concentrations of 10, 20, 40, 80, 160, and 240 μg/mL were applied to SMMC-7721 hepatoma cells for 72 hours. A cell suspension of 4.0 × 105 cells/mL was prepared and added to the upper transwell chamber (150 μL/chamber). Six hundred microliters of RPMI-1640 containing 10 μg/mL fibronectin and 10% BSA serum was added to the lower transwell chamber. The transwell plates were then removed and placed in a 37°C incubator with 5% CO2 for 24 hours. The filter side of the upper chamber was then cleaned with a cotton swab and the filter was stabilized with ethanol and stained with H&E. The filter was carefully cut from the chamber and the cells that had migrated through the filter pores from the underside of the filter were counted in four high-power fields per insert, and average values were calculated based on five vision fields (the upper, lower, left, right, and central). For each migration condition, three replicates were performed.
Effects of BIN on the cell movement of liver cancer cells
Serum-free 1640 was added to the upper transwell chamber (100 μL/chamber). Brucine, 5-FU, brucine nanoparticles, and BIN were used on SMMC-7721 hepatoma cells for 72 hours at concentrations of 10, 20, 40, 80, 160, and 240 μg/mL. A cell suspension of 4.0 × 105 cells/mL was prepared and added to the upper transwell chamber (150 μL/chamber). Six hundred microliters of RPMI-1640 containing 10 μg/mL fibronectin and 10% BSA serum was added to the lower transwell chamber, and the transwell plates were placed in a 37°C incubator with 5% CO2 for 24 hours. The filter side of the upper chamber was then cleaned with a cotton swab and the filter was stabilized with ethanol and stained with H&E. The filter was carefully cut from the chamber and the cells that had migrated through the filter pores from the underside of the filter were counted in four high-power fields per insert, and average values were based on five vision fields (the upper, lower, left, right, and central). For each migration condition, three replicates were performed.
Statistical analysis
SPSS statistical software (v 13.0; SPSS, Inc, Chicago, IL) was used to analyze the data. Analysis of variance of the randomized design was employed within the group, and analysis of the data covariance of the randomized block design was used to compare the difference between groups. P < 0.05 was considered statistically significant.
Results and discussion
Preparation and properties of BIN
In this study, anionic polymerization, chemical modification technology, and phacoemulsification technology were used to prepare carboxylated polyethylene glycol-polylactic acid copolymer carrier material. Chemical coupling technology was utilized to develop anti-human AFP McAb-polyethylene glycol-polylactic acid copolymer BIN. BIN were successfully prepared and showed uniform size with an average particle size of 249 ± 77 nm and zeta potential of –18.7 ± 4.19 mV. The drug load was 5.6% ± 0.2% (Figures 1 and 2). Brucine was completely released within 2 hours. BIN were very stable in the medium with an accumulative release rate of over 80% in 24 hours and 100% in 48 hours (Figure 3).
Figure 1.
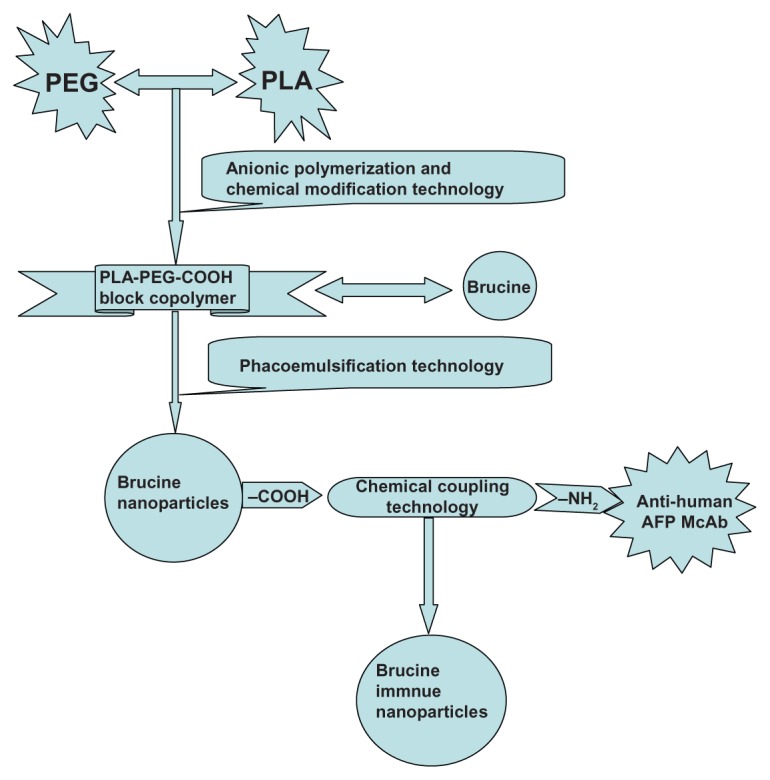
Synthesis scheme of brucine immuno-nanoparticles.
Abbreviation: PEG-PLA-COOH, carboxylated polyethylene glycol-polylactic acid block copolymer.
Figure 2.

Scanning electron microscope image of brucine immuno-nanoparticles (100,000× magnification).
Figure 3.

Release curve of brucine immuno-nanoparticles in vitro. Brucine was completely released within 2 hours. Brucine immuno-nanoparticles were very stable in the medium with an accumulative release rate of over 80% in 24 hours and 100% in 48 hours.
Determination of monoclonal antibodies on BIN surface
BCA was used to determine the concentration of AFP monoclonal antibodies on BIN, and the concentration was 15 μg antibodies/mg nanoparticles.
Brucine intake by cancer cells and its positioning
BIN were evenly distributed around the liver cancer cell membrane, showing consistent ring shapes and good target positioning (Figure 4).
Figure 4.

Cell targeting and positioning of the brucine immuno-nanoparticles. Complete uptake and even distribution of the brucine immuno-nanoparticles around the liver cancer cell membrane after incubation for 4 hours (A) ring green fluorescence; (B) without laser excitation; 400× magnification.
Liver cancer cell growth inhibition by BIN
Liver cancer cells in the blank control group showed adherent growth, clear cell outline, uniform cell arrangement, and vigorous growth under each experimental concentration. As the dose of BIN increased, the number of liver cancer cells dropped. Liver cancer cells that were arranged in sparse, round pseudopodia disappeared, normal cell structures were lost, and cytoplasm “bubble” phenomena could be seen. Other effects such as cell shrinkage and cell peripheral refraction changes decreased adhesion capacity, and more cell debris could be found (Figure 5).
Figure 5.

Growth effect of brucine immuno-nanoparticles on liver cancer cells. Liver cancer cells shrank and pseudopodia disappeared at a brucine concentration of 1.0 μg/mL in brucine immuno-nanoparticles for 72 hours in vitro (A). The number of liver cancer cells dropped, and liver cancer cells were sparse and round at a brucine concentration of 40 μg/mL in brucine immuno-nanoparticles for 72 hours in vitro (B). Liver cancer cells showed cytoplasm “bubble” phenomena and low adhesion capacity, and more cell debris could be found at a brucine concentration of 160 μg/mL in brucine immuno-nanoparticles for 72 hours in vitro (C) (200× magnification).
Negative control groups showed no significant growth inhibition on liver cancer SMMC-7721 cells. The difference between groups was not statistically significant (P > 0.05) after 72 hours. BIN had a significant inhibitory effect on the growth of hepatoma cells SMMC-7721, which was correlated with the drug concentration and showed a time and dose-dependent manner for 72 hours. The difference between the groups was statistically significant (F = 5.719, P < 0.01) (Figure 6). Compared with brucine and brucine nanoparticles, BIN had the strongest inhibitory effects on hepatoma cells SMMC-7721 and the IC50 was 28.2 μg/mL, close to that of 5-FU (IC50, 16.7 μg/mL) (Figure 7).
Figure 6.

The growth inhibition curve of the brucine immuno-nanoparticles on liver cancer cells. Brucine immuno-nanoparticles have more significant inhibitory effects on the hepatoma cells SMMC-7721 than brucine or brucine nanoparticles for 72 hours. Time- and dose-dependent effects were observed.
Figure 7.

Half maximal inhibitory concentration (IC50) of the brucine immuno-nanoparticles on liver cancer cells. The half maximal inhibitory concentration of the brucine immuno-nanoparticles was lower than that of brucine or brucine nanoparticles. The IC50 of the brucine immuno-nanoparticles (28.2 μg/mL) was close to that of 5-FU (16.7 μg/mL).
Effects of BIN on cell adhesion of liver cancer cells
In the negative control group, increased drug concentration had no significant effect on human hepatoma SMMC-7721 cell matrix adhesion. No significant difference was found between groups (F = 0.001, P > 0.05). 5-FU, brucine, brucine nanoparticles, and BIN all had significant inhibitory effects on human hepatoma SMMC-7721 cell matrix adhesion after 72 hours. As drug concentration increased, the inhibition effects were enhanced. The difference between groups was statistically significant (F = 125.194, P < 0.01). Compared with brucine and brucine nanoparticles, BIN had the strongest inhibition effects on liver cancer cell matrix adhesion (Figure 8).
Figure 8.

The matrix adhesive rate of the brucine immuno-nanoparticles on liver cancer cells. Compared with brucine and brucine nanoparticles, brucine immuno-nanoparticles had the strongest inhibition effects on liver cancer cell matrix adhesion for 72 hours.
Effects of BIN on invasion of liver cancer cells
In the negative control group, increased drug concentration had no significant effect on human hepatoma SMMC-7721 cell invasion. No significant difference was found between groups (F = 0.380, P > 0.05). 5-FU, brucine, brucine nanoparticles, and BIN all had significant inhibitory effects on human hepatoma SMMC-7721 cell invasion after 72 hours. As the drug concentration increased, the inhibition effects were enhanced. The difference between groups was statistically significant (F = 57.238, P < 0.01). Compared with brucine and brucine nanoparticles, BIN had the strongest inhibitory effect on liver cancer cell invasion (Figures 9 and 10).
Figure 9.
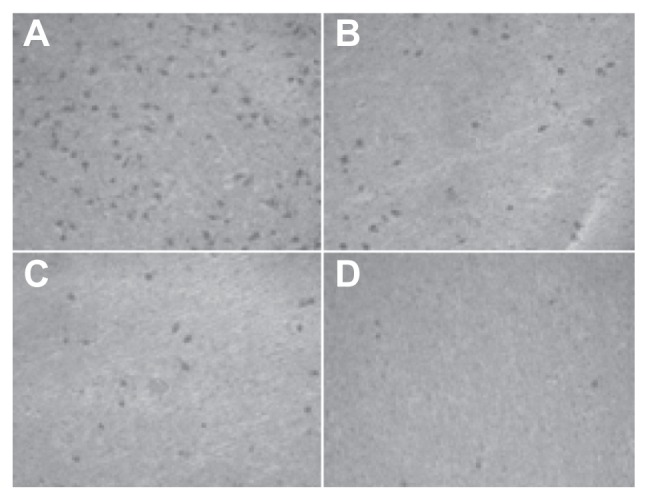
The number of cells permeating through the filter pores from the underside of the filter after brucine immuno-nanoparticles were applied at various concentrations to liver cancer cells for 72 hours (200× magnification). As the drug concentration increased, the number of cell invasions decreased. (A) 10 μg/mL; (B) 20 μg/mL; (C) 40 μg/mL; (D) 80 μg/mL.
Figure 10.

The invasion rate of the brucine immuno-nanoparticles on liver cancer cells. Compared with brucine and brucine nanoparticles, brucine immuno-nanoparticles had the strongest inhibition effects on liver cancer cell invasion after 72 hours.
Effects of BIN on cell movement of liver cancer cells
In the negative control group, increased drug concentration had no significant effect on human hepatoma SMMC-7721 cell migration. No significant difference was found between groups (F = 1.183, P > 0.05). 5-FU, brucine, brucine nanoparticles, and BIN all had significant inhibitory effects on human hepatoma SMMC-7721 cell migration after 72 hours. As the drug concentration increased, the inhibitory effects were enhanced. The difference between the groups was statistically significant (F = 51.237, P < 0.01). Compared with brucine and brucine nanoparticles, BIN had the strongest inhibitory effect on liver cancer cell migration. (Figures 11 and 12).
Figure 11.

The number of cells migrating through the filter pores from the underside of the filter after brucine immuno-nanoparticles were applied at various concentrations to liver cancer cells for 72 hours (200× magnification). As the drug concentration increased, the cell migration decreased. (A) 10 μg/mL; (B) 20 μg/mL; (C) 40 μg/mL; (D) 80 μg/mL; (E) 160 μg/mL; (F) 240 μg/mL.
Figure 12.

The migratory rate of the brucine immuno-nanoparticles on liver cancer cells. Compared with brucine and brucine nanoparticles, brucine immuno-nanoparticles had the strongest inhibitory effect on liver cancer cell migration.
Many anti-cancer drugs that are clinically employed at present have many problems and side effects such as poor water solubility, short half-life, poor targeting of cancer cells, high toxicity, and the suppression of bone marrow activity. In recent years, nanotechnology has been incorporated into the development of new nano-anti-cancer drugs which promise ideal targeting and sustained release. The concentration of the drug in the cancer-affected organ can be significantly increased, accompanied by significantly enhanced efficacy and reduced side effects. Thus, these drugs show promise in the treatment of liver cancer.15 Wang et al used tanshinone IIA and tanshinone IIA nanoparticles in the treatment of tumor-bearing mice with transplanted liver cancer H22 cells. Compared with the control group, tanshinone IIA and tanshinone IIA nanoparticles significantly inhibited tumor growth, and at the same level of drug concentration, tanshinone IIA nanoparticles inhibited tumor growth significantly better than tanshinone IIA, as demonstrated by significantly increased tumor necrosis and apoptosis, and lower cyclin E expression. This indicated that tanshinone IIA nanoparticles were superior to tanshinone IIA.16 The uptake of nanoparticles was affected by the size, shape, surface characteristics, medium concentration, incubation time, temperature, and many other factors.17–22
Combining drug-loaded nanoparticles with monoclonal antibodies against human hepatocellular carcinoma produces a drug-nanoparticle-monoclonal antibody immune complex. McAb can carry drug-loaded nanoparticles to specific target sites, thus enhancing specific cancer cell-drug combinations, increasing the drug concentration, and improving efficacy. Wu et al combined the highly specific anti-human liver acid ferritin monoclonal antibody with doxorubicin-poly butyl cyanoacrylate nanoparticles and prepared liver-specific doxorubicin immuno-nanoparticles. Experiments showed that the nanoparticles had a significantly longer half-life and high targeting in nude mice tumor inhibition experiments in vivo and cytotoxicity experiments in vitro. The drug accumulated in the liver tumor, greatly increasing the drug’s concentration, extending the time during which it is effective, enhancing the efficacy, and reducing toxicity to other organs.23 Liu et al connected anti-tumor monoclonal antibody HAb18 with mitoxantrone-bovine serum albumin nanoparticles and prepared liver-specific immuno-nanoparticles, which could effectively combine with human liver cancer cells. In vitro studies indicated that the nano-drug could significantly enhance the inhibition of human hepatoma SMMC-7721 cell growth.24 Chen et al wrapped doxorubicin in a phospholipid bilayer and prepared liver-specific immuno-phospholipid nanoparticles, which showed greatly increased inhibition of human liver cancer cell growth compared with doxorubicin or an ordinary plasmid. Compared with liposomes, lipid nanoparticles significantly improved the inhibition rate in mice bearing nude human liver cancer. Nanoparticles significantly increased the targeting of tumors, elevated drug concentration, extended the time during which it was effective, and reduced doxorubicin toxicity to other organs.25
Alpha-fetoprotein (AFP) is a major plasma protein produced by the yolk sac and the liver during fetal development. Normal adult liver cells lose the ability to produce AFP, and serum concentration is less than 20 μg/L. Cancerous liver cells can synthesize a large quantity of AFP, so AFP may be used as a biomarker for primary liver cancers. Liver cancer cells expressing AFP show a tendency towards early vascular invasion and intrahepatic metastasis. For primary liver cancer detection, the sensitivity for AFP is 79% and the specificity is 78%. The presence of AFP mRNA in peripheral blood reflects the level of free circulating peripheral liver cancer cells and allows early diagnosis, differential diagnosis, as well as signaling cancer recurrence or metastasis.26–28 Anti- tumor drugs connected with anti-AFP monoclonal antibodies can destroy tumor cells while causing only minor injury to normal cells, so are playing an increasingly important role in HCC treatment.29–31
This study exploited the hydrophilic and biocompatible characteristics of polyethylene glycol-polylactic acid block copolymers, and developed polyethylene glycol-polylactic acid block copolymer brucine nanoparticles. These have a number of advantages including good sustained release and strong targeting, and overcomes the disadvantages of brucine such as high toxicity, wide distribution, and short half-life. For the first time, we used phacoemulsification technology to combine brucine and carboxylated polyethylene glycol-polylactic acid block copolymers to produce brucine nanoparticles, and we coupled the C-terminal polyethylene glycol with anti-human AFP McAb to improve the link ability. The BIN was successfully prepared, and results showed that the BIN had a uniform size distribution. In experiments, brucine was completely released into the medium within 2 hours, while brucine in immuno-nanoparticles was completely released within 48 hours. In vitro experiments showed that BIN specifically targeted the liver cell membrane and had stronger liver cancer cell growth inhibition properties than brucine in a time- and dose-dependent manner. Regarding liver cancer cell inhibition, the IC50 of BIN was lower than that of brucine and brucine nanoparticles, and close to that of 5-FU. After 72 hours, an increased dose caused the number of liver cancer cells to reduce, pseudopodia disappeared, “bubble” phenomena appeared in the cytoplasm, cells shrank, cell peripheral refraction weakened, and adhesion capacity decreased.
Primary liver cancer invasion and metastasis is a multi-step and complex process involving a series of important changes including cell adhesion, matrix degradation, cell migration, proliferation, and angiogenesis, and leads to recurrence and metastasis which severely impair the effects and prognosis of treatment.4 Recurrence and metastasis are important biological behaviors of malignant tumors. Malignant cell invasion and metastasis begin with intercellular adhesion loss after early local infiltration. Laminin, fibronectin, collagen, and other integrins located on the basement membrane surface combine with laminin receptors, f ibronectin receptors, collagen receptors, and integrin receptors on the surface of the tumor cell, which allows a tumor cell to adhere to the basement membrane. A decrease is seen in the homogenous cell adhesion between tumor cells mediated by adhesion molecules, which promotes the shedding of tumor cells. The heterogenous adhesion between tumor cells and the matrix increases.32–35 Extracellular matrix degradation and basement membrane damage is one of the key steps in the process of tumor invasion and metastasis. After a number of hours of close contact between tumor cells and the basement membrane, tumor cells will directly induce host cells to secrete endothelial growth factors and epidermal growth factors, and activate endothelial cells to produce proteases which can digest laminin, fibronectin, proteoglycans, collagen, and other extracellular matrix components. The basement membrane is degraded with local defects, allowing tumor cells to move deeper into the tissue.36,37 Induced by growth factors and chemokines, tumor cells continue to move deeper, and after dissolving interstitial connective tissues, tumor cells can eventually reach blood vessels. Tumor cells can pass through the wall of a blood vessel in the same way and so enter the circulatory system, which can lead to recurrence and metastasis.38 The mechanisms of tumor recurrence and metastasis are still unclear, and we have not yet identified specific molecular markers or developed preventive measures to be used before these occur.
In vitro studies found that BIN could effectively inhibit cell matrix adhesion, invasion, and migration of SMMC-7721 human hepatocellular carcinoma cells in a dose-dependent manner. Brucine was superior to brucine nanoparticles in inhibiting cell matrix adhesion, invasion, and movement of SMMC-7721 cells, probably related to the nanoparticles’ slow drug release. Compared with brucine and brucine nanoparticles, BIN had the strongest inhibitory effects on cell matrix adhesion, invasion, and migration of human liver cancer SMMC-7721 cells. The results showed that brucine nanoparticles combined with anti-human AFP McAb had immunospecificity and increased drug-targeting effects, which promoted the specific interaction between the drug and the targeted cells. The drug concentration was significantly elevated around the tumor cells. The BIN with the application of polyethylene glycol-polylactic acid copolymers as drug carriers were macrocyclic and had good biocompatibility, large drug loading, wide drug range, good stability, and excellent bioavailability characteristics. Combining the monoclonal antibody with nano-anti-cancer drugs, we produced brucine immuno-targeting nanoparticles with greatly improved positioning and anti-tumor effects. Compared with brucine and brucine nanoparticles, BIN displayed more specific tumor cell targeting, increased local drug concentration, and effectively inhibited cancer cell growth, matrix adhesion, invasion, and metastasis. Therefore, BIN is a promising anti-cancer- targeting drug for inhibiting growth, recurrence, and metastasis of hepatocellular carcinoma.
Conclusion
A targeted drug delivery system was successfully prepared and its in vitro effects examined. This research shows that the BIN can specifically combine with liver cancer cells, target the liver cancer cell membrane, and exert anti-tumor effects. BIN inhibit the growth of human hepatoma SMMC- 7721 cells in a time- and dose-dependent manner. BIN have significant sustained release, high local drug concentration, longer duration of action, and improved efficacy. Compared with brucine and brucine nanoparticles, BIN displayed more specific tumor cell targeting, increased local drug concentration, and effectively inhibited cancer cell growth, matrix adhesion, invasion, and metastasis. BIN are a potentially promising anti-cancer targeting drug for inhibiting the growth, recurrence, and metastasis of hepatocellular carcinoma.
Acknowledgments
The authors thank Professor Yan Zhang for the correction and translation of the manuscript. The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (No 30873341), the Shanghai Education Commission (No 07CZ017), and the Shanghai Science and Technology Commission (No 1052nm060000).
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55(2):74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Tang ZY, Ye SL, Liu YK, et al. A decade’s studies on metastasis of hepatocellular carcinoma. J Cancer Res Clin Oncol. 2004;130(4):187–196. doi: 10.1007/s00432-003-0511-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mitsunobu M, Toyosaka A, Oriyama T, Okamoto E, Nakao N. Intrahepatic metastases in hepatocellular carcinoma: the role of the portal vein as an efferent vessel. Clin Exp Metastasis. 1996;14(6):520–529. doi: 10.1007/BF00115112. [DOI] [PubMed] [Google Scholar]
- 4.Wu MC, Chen H, Zhang XH, Yao XP, Yang JM. Surgical treatment of primary liver cancer: 1102 cases. Acad J Sec Mil Med Univ. 1993;14(3):201–204. [Google Scholar]
- 5.Belghiti J, Hiramatsu K, Benoist S, Massault P, Sauvanet A, Farges O. Seven hundred forty-seven hepatectomies in the 1990s: an update to evaluate the actual risk of liver resection. J Am Coll Surg. 2000;191(1):38–46. doi: 10.1016/s1072-7515(00)00261-1. [DOI] [PubMed] [Google Scholar]
- 6.Ziser A, Plevak DJ, Weisner RH, Rakela J, Offord KP, Brown DL. Morbidity and mortality in cirrhotic patients undergoing anesthesia and surgery. Anesthesiology. 1999;90(1):42–53. doi: 10.1097/00000542-199901000-00008. [DOI] [PubMed] [Google Scholar]
- 7.Calne R, Yamanoi A, Oura S, Kawamura M. Liver transplantation for hepatocarcinoma. Surg Today. 1993;23(1):1–3. doi: 10.1007/BF00308991. [DOI] [PubMed] [Google Scholar]
- 8.Tang ZY. The meaning and approach to carry out liver metastasis and recurrence research. Chin J Gen Surg. 2006;21(11):761. [Google Scholar]
- 9.Thomas MB, Zhu AX. Hepatocellular carcinoma: The need for progress. J Clin Oncol. 2005;23(13):2892–2899. doi: 10.1200/JCO.2005.03.196. [DOI] [PubMed] [Google Scholar]
- 10.Wu MC. Surgical treatment of primary liver cancer. Chin J Gen Surg. 2009;3(4):1–3. [Google Scholar]
- 11.Qin JM, Xu XJ, Sheng X, et al. The effect of brucine on hepatocellular carcinoma cell lines in vitro. Chin J Gen Surg. 2011;26(3):219–221. [Google Scholar]
- 12.Deng XK, Cai BC, Lv XY, et al. Anti-tumor effects comparision of the brucine and its liposomes in transplanted tumor-bearing mice. J Herb Med. 2006;37(3):389–393. [Google Scholar]
- 13.Deng XK, Cai BC, Yin W, Zhang XC, Li WD, Sun Q. Brucine on mouse tumor inhibition. Chin J Nat Med. 2005;3(6):392–396. [Google Scholar]
- 14.Deng XK, Cai BC, Yin W, Liu TS, Sun Q, Li WD. Research of the anti-tumor effect and toxicity of brucine in Heps tumor-bearing mice. Chin Pharmacol Bulletin. 2006;22(1):35–39. [Google Scholar]
- 15.Qin JM, Zhang YD, Wang HY, Wu MC. Nanotechnologies in the diagnosis and treatment of liver disease. Chin J Hepatobiliary Surg. 2004;10(9):646–648. [Google Scholar]
- 16.Wang Y, Li Q, Feng NP, et al. Tanshinone IIA nanoparticles in the treatment of mouse liver cancer and its effects on Cyclin E expression. Shanghai Traditional Chin Med. 2007;4(1):74–77. [Google Scholar]
- 17.Torchilin VP. Drug targeting. Eur J Pharm Sci. 2000;11(Suppl 2):S81–91. doi: 10.1016/s0928-0987(00)00166-4. [DOI] [PubMed] [Google Scholar]
- 18.Davda J, Labhasetwar V. Characterization of nanoparticle uptake by endothelial cells. Int J Pharm. 2002;233(1–2):51–59. doi: 10.1016/s0378-5173(01)00923-1. [DOI] [PubMed] [Google Scholar]
- 19.Sahoo SK, Panyam J, Prabha S, Labhasetwar V. Residual polyvinylalcohol associated with poly (D,L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J Control Release. 2002;82(1):105–114. doi: 10.1016/s0168-3659(02)00127-x. [DOI] [PubMed] [Google Scholar]
- 20.Panyam J, Labhasetwar V. Dynamics of endocytosis and exocytosis of poly (D,L-lactide-co-glycolide) nanoparticles in vascular smooth muscle cells. Pharm Res. 2003;20(2):212–220. doi: 10.1023/a:1022219003551. [DOI] [PubMed] [Google Scholar]
- 21.Lynch I. Are there generic mechanisms governing interactions between nanoparticles and cells? Epitope mapping the other layer of the protein– material interface. Physica A. 2007;373(3):511–520. [Google Scholar]
- 22.Rowland RES, Taylor PW, Florence AT. Attachment, uptake and transport of nanoparticles coated with an internal in A fragment in Caco-2 cell monolayer. J Drug Deliv Sci Technol. 2005;15(3):313–317. [Google Scholar]
- 23.Wu J, Nantz MH, Zern MA. Targeting hepatocytes for drug and gene delivery: emerging novel approaches and applications. Frontiers in Bioscience. 2002;7(1):717–725. doi: 10.2741/A806. [DOI] [PubMed] [Google Scholar]
- 24.Liu XB, Cai MY. Preparation and immunological identification of nanoparticles against human hepatocellular carcinoma in vitro. Chin J Immunology. 2000;16(5):262–265. [Google Scholar]
- 25.Chen Y, Tang R, Huang W. Study of hepatoma-specific immunal phospholipids doxorubicin nanoparticles. Chin Pharmacist. 2002;5(12):712–714. [Google Scholar]
- 26.Hu KQ, Kyulo NL, Lim N, Elhazin B, Hillebrand DJ, Bock T. Clinical significance of elevated alpha fetoprotein (AFP) in patients with chronic hepatitis C, but not hepatocellular carcinoma. Am J Gastroenterol. 2004;99(5):860–865. doi: 10.1111/j.1572-0241.2004.04152.x. [DOI] [PubMed] [Google Scholar]
- 27.Montaser LM, Abbas OM, Saltah AM, Waked IA. Circulating AFP mRNA as a possible indicator of hematogenous spread of HCC cells: a possible association with HBV infection. J Egypt Natl Canc Inst. 2007;19(1):48–60. [PubMed] [Google Scholar]
- 28.Kamiyama T, Takahashi M, Nakagawa T, et al. AFP mRNA detected in bone marrow by real-time quantitative RT-PCR analysis predicts survival and recurrence after curative hepatectomy for hepatocellular carcinoma. Ann Surg. 2006;244(3):451–463. doi: 10.1097/01.sla.0000234840.74526.2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Heiden PL, Jedema I, Willemze R, Barge RM. Efficacy and toxicity of gemtuzumab ozogamicin in patients with acute myeloid leukemia. Eur J Haematol. 2006;76(5):409–413. doi: 10.1111/j.1600-0609.2005.00623.x. [DOI] [PubMed] [Google Scholar]
- 30.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23(9):1147–1157. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 31.Wu AM, Senter PD. Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol. 2005;23(9):1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- 32.Jha RK, Ma Q, Chen S, Sha H, Ding S. Relationship of fibronectin and CD44v6 expression with invasive growth and metastasis of liver cancer. Cancer Invest. 2009;27(3):324–328. doi: 10.1080/07357900802375753. [DOI] [PubMed] [Google Scholar]
- 33.Sun H, Liu GT. Inhibitory effect of anti-hepatitis drug bicyclol on invasion of human hepatocellular carcinoma MHCC97-H cells with high metastasis potential and its relative mechanisms. J Asian Nat Prod Res. 2009;11(6):576–583. doi: 10.1080/10286020902942368. [DOI] [PubMed] [Google Scholar]
- 34.Garmy-Sudini B, Avraamides CJ, Schmid MC, et al. Integrin alpha4beta1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer Res. 2010;70(8):3042–3051. doi: 10.1158/0008-5472.CAN-09-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janik ME, Litynska A, Vereecken P. Cell migration – the role of integrin glycosylation D. Biochim Biophys Acta. 2010;1800(6):545–555. doi: 10.1016/j.bbagen.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 36.Kruegel J, Miosge N. Basement membrane components are key players in specialized extracellular matrices. Cell Mol Life Sci. 2010;67(17):2879–2895. doi: 10.1007/s00018-010-0367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiradjaja F, DiTommaso T, Smyth I. Basement membranes in development and disease. Birth Defects Res C Embryo Today. 2010;90(1):8–31. doi: 10.1002/bdrc.20172. [DOI] [PubMed] [Google Scholar]
- 38.Escaff S, Fernández JM, González LO, et al. Study of matrix metalloproteinases and their inhibitors in prostate cancer. Br J Cancer. 2010;102(5):922–929. doi: 10.1038/sj.bjc.6605569. [DOI] [PMC free article] [PubMed] [Google Scholar]