

Crystal structures of human tankyrase I in complex with small-molecule inhibitors PJ34 and XAV939 have been determined, both to 2.0 Å. The TNKS1-PJ34 system allows for displacement soaking and, therefore, is an ideal system for structure-based drug design.
Keywords: tankyrase, PARP domains, inhibitor complexes, displacement soaking
Abstract
The crystal structures of tankyrase 1 (TNKS1) in complex with two small-molecule inhibitors, PJ34 and XAV939, both at 2.0 Å resolution, are reported. The structure of TNKS1 in complex with PJ34 reveals two molecules of PJ34 bound in the NAD+ donor pocket. One molecule is in the nicotinamide portion of the pocket, as previously observed in other PARP structures, while the second molecule is bound in the adenosine portion of the pocket. Additionally, unlike the unliganded crystallization system, the TNKS1–PJ34 crystallization system has the NAD+ donor site accessible to bulk solvent in the crystal, which allows displacement soaking. The TNKS1–PJ34 crystallization system was used to determine the structure of TNKS1 in complex with XAV939. These structures provide a basis for the start of a structure-based drug-design campaign for TNKS1.
1. Introduction
Tankyrases (TRF1-interacting ankyrin-related ADP-ribose polymerases; TNKSs) are members of the poly-ADP-ribose polymerase (PARP) family of proteins, which play a role in cellular signaling through the ADP-ribosylation of target proteins (Schreiber et al., 2006 ▶). The two isoforms of TNKS, TNKS1 (PARP5a) and TNKS2 (PARP5b), are involved in telomere elongation (Smith & de Lange, 2000 ▶) as well as regulation of the Wnt/β-catenin signal transduction pathway (Huang et al., 2009 ▶). It has been shown that TNKS1 and TNKS2 work to stabilize axin, thereby upregulating the degradation of β-catenin and resulting in the inhibition of transcription of Wnt pathway-responsive genes. Because deregulation of this pathway has been identified in many cancers, TNKS1 and TNKS2 have become attractive drug targets. To this end, XAV939 (Fig. 1 ▶) has been identified as a small-molecule inhibitor of the Wnt/β-catenin pathway and, more specifically, a potent inhibitor of TNKS1 and TNKS2, with reported IC50 values of 0.011 and 0.004 µM, respectively (Huang et al., 2009 ▶). Additionally, PJ34 (Fig. 1 ▶), a well characterized general PARP inhibitor (Abdelkarim et al., 2001 ▶; Peukert & Schwahn, 2004 ▶), has also been shown to inhibit TNKS1 with an IC50 of 1 µM (internal data obtained using the assay described in Huang et al., 2009 ▶). Crystal structures have previously been determined for PJ34 in complex with PARP-3 (Lehtiö et al., 2009 ▶; PDB entry 3ce0) and PARP-15 (Karlberg et al., 2010 ▶; PDB entry 3gey).
Figure 1.

Molecular structures of (a) PJ34 (IC50 = 1.0 µM) and (b) XAV939 (IC50 = 0.011 µM).
Crystal structures of unliganded TNKS1 (Lehtiö et al., 2008 ▶) and TNKS2 (Karlberg et al., 2010 ▶) as well as of TNKS2 in the presence of small-molecule inhibitors, including XAV939 (Karlberg et al., 2010 ▶), have been determined. These inhibitors are bound in the donor site of the proteins in a position occupied by the nicotinamide portion of the substrate NAD+. In the unliganded structure of TNKS1, this portion of the donor site is occupied by residues Tyr1203 and Ile1204 from the D-loop (Phe1197–Gly1211). This structural feature, in addition to crystal contacts, reduces the possibility of achieving complex structures through soaking of the inhibitors into unliganded crystals. Here, we report the first structures of TNKS1 in complex with the small-molecule inhibitors PJ34 (PDB entry 3uh2) and XAV939 (PDB entry 3uh4) at 2.0 Å resolution. Additionally, we report the establishment of a displacement-soakable TNKS1 crystal system.
2. Materials and methods
2.1. Protein expression and purification
The gene encoding the PARP domain of TNKS1 (residues Gly1105–Thr1327) was inserted into a pET28a vector. A coding sequence for a six-histidine tag followed by a TEV protease consensus sequence (ATGGGCAGCAGCCATCATCATCATCATCACGAAAACCTGTATTTTCAGGGC) was added 5′ to the TNKS1 gene sequence. The construct was transformed into Escherichia coli BL21 Star (DE3) cells and grown at 310 K in Terrific Broth containing 100 µg ml−1 kanamycin. At an OD600 of 2.3, TNKS1 expression was induced using 1 mM IPTG. Cells were harvested following overnight growth at 291 K.
Cell pellets were resuspended in buffer A (50 mM Tris–HCl pH 8.0 and 5% glycerol) plus 500 mM NaCl and lysed using a microfluidizer, followed by ultracentrifugation. The supernatant was loaded onto a HisTrap HP chelating column in the same buffer and protein was eluted by the addition of 300 mM imidazole. The N-terminal histidine tag was removed by overnight incubation with TEV protease at 277 K. The protein was subsequently diluted eightfold with buffer A and then flowed through a HiTrap Q FastFlow column equilibrated with buffer A plus 50 mM NaCl. The flowthrough was concentrated and loaded onto a HiLoad Superdex 75 PG 16/100 column, exchanging the protein into the crystallization buffer: 25 mM Tris–HCl pH 7.5, 200 mM NaCl, 1 mM TCEP. The protein was concentrated to 9 mg ml−1 for use in crystallization.
2.2. Crystallization
For crystallization of the protein with PJ34, 1 mM PJ34 (Tocris Bioscience) was added to 9 mg ml−1 TNKS1. The sitting-drop vapor-diffusion method was used for crystallization, with the crystallization well containing 100 mM bis-tris pH 5.8, 16% PEG 3350 and 300 mM ammonium sulfate and the drop consisting of a 1:1 volume ratio of protein and crystallization solutions. Crystals formed within 3 d and were subsequently cryoprotected using the crystallization solution with the addition of 20% glycerol, followed by flash-cooling directly in liquid nitrogen.
To obtain crystals containing XAV939, TNKS1–PJ34 complex crystals were transferred into a soaking solution consisting of 100 mM bis-tris pH 5.8, 18% PEG 3350, 320 mM ammonium sulfate and 200 µM XAV939 (Huang et al., 2009 ▶). Transfer of the crystals into fresh solution occurred once per hour over a 4 h period. Crystals were cryoprotected using the same soaking solution with the addition of 20% glycerol, followed by flash-cooling directly in liquid nitrogen.
2.3. Data collection and structure determination
Diffraction data for the TNKS1–PJ34 complex were collected using a Quantum 315R CCD detector on ALS beamline 5.0.3 at a wavelength of 1 Å. The data were measured from a single crystal maintained at 100 K and the reflections were indexed, integrated and scaled using XDS (Kabsch, 2010 ▶). The space group of the complex was C2, with two molecules in the asymmetric unit. The structure was determined by molecular-replacement methods with the program Phaser (McCoy et al., 2007 ▶) using the unliganded TNKS1 structure (PDB code 2rf5; Lehtiö et al., 2008 ▶) with active-site waters removed as the starting model.
Diffraction data for the TNKS1–XAV939 complex were collected using a Dectris Pilatus 6M detector on Swiss Light Source beamline X06SA at a wavelength of 1 Å. The data were measured from a single crystal and the reflections were indexed, integrated and scaled using XDS (Kabsch, 2010 ▶). The resolution was limited to 2.0 Å owing to a rapid drop-off in data quality. When the data were processed to 1.9 Å resolution a significant degradation of data-quality indicators was observed. The space group of the complex was C2, with two molecules in the asymmetric unit. The structure was determined with Fourier methods using the TNKS1–PJ34 structure (PDB entry 3uh2) with active-site waters and inhibitors removed as the starting model.
Structure determination for both structures was achieved through iterative rounds of positional and simulated-annealing refinement using PHENIX (Adams et al., 2010 ▶) and/or BUSTER (Bricogne et al., 2009 ▶) with model building using Coot (Emsley et al., 2010 ▶). Individual B factors were refined using an overall anisotropic B-factor refinement along with bulk-solvent correction. The solvent, sulfate ions and inhibitor were built into the density in later rounds of refinement. Data-collection and refinement statistics are shown in Table 1 ▶.
Table 1. Crystallographic data and refinement information.
Values in parentheses are for the highest resolution shell.
| TNKS1PJ34 complex | TNKS1XAV939 complex | |
|---|---|---|
| Space group | C2 | C2 |
| Unit-cell parameters (, ) | a = 125.3, b = 43.3, c = 88.5, = 91.4 | a = 123.9, b = 45.2, c = 88.1, = 90.4 |
| Resolution range () | 62.612.00 (2.102.00) | 44.072.00 (2.052.00) |
| Total observations | 114726 | 108366 |
| Unique reflections | 31795 | 32886 |
| Completeness (%) | 97.7 (96.5) | 98.5 (98.1) |
| Multiplicity | 3.6 (3.5) | 3.3 (3.3) |
| I/(I) | 11.1 (2.6) | 13.6 (4.0) |
| R merge † | 0.070 (0.415) | 0.059 (0.285) |
| R cryst/R free ‡ | 0.175/0.217 | 0.180/0.234 |
| Protein atoms | 3292 | 3374 |
| Heterogen atoms | 115 | 97 |
| Solvent molecules | 176 | 316 |
| Average B factor (2) | 27.4 | 28.4 |
| R.m.s.d. bond lengths () | 0.01 | 0.01 |
| R.m.s.d. bond angles () | 1.05 | 1.04 |
| Ramachandran plot§ (%) | ||
| Most favored | 92.8 | 92.8 |
| Additionally allowed | 7.2 | 7.2 |
| Generously allowed | 0 | 0 |
| Disallowed | 0 | 0 |
R
merge = 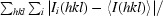
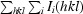 , where I(hkl) is the intensity of reflection hkl.
, where I(hkl) is the intensity of reflection hkl.
R
cryst and R
free = 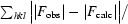
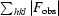 , where F
obs and F
calc are observed and calculated amplitudes, respectively. R
free was calculated using 5% of data excluded from the refinement.
, where F
obs and F
calc are observed and calculated amplitudes, respectively. R
free was calculated using 5% of data excluded from the refinement.
Laskowski et al. (1993 ▶).
3. Results and discussion
3.1. Structure of TNKS1–PJ34
The structure of the catalytic domain of human TNKS1 in complex with PJ34 was determined at 2.0 Å resolution. The final model was refined to an R factor of 0.175 and included residues Gly1105–Ala1202, Gly1206–Arg1281 and Ala1290–Pro1313 in molecule A, residues Gly1105–Tyr1203 and Gly1206–Glu1314 in molecule B, one zinc ion and two molecules of inhibitor PJ34 per protein molecule, five sulfate ions and 176 solvent molecules. Residues Tyr1203–Gly1205, Pro1282–Tyr1289 and the C-terminal residues Glu1314–Thr1327 in molecule A, as well as residues Ile1204–Gly1205 and the C-terminal residues Ala1315–Thr1327 in molecule B, were omitted from the final model because of poor electron density.
The overall structure of the PARP domain of TNKS1 has been described previously (Lehtiö et al., 2008 ▶). In Fig. 2 ▶(a), two PJ34 molecules are shown bound to the NAD+ donor pocket of TNKS1. Complex structures with PJ34 have previously been determined for the catalytic domains of both PARP-3 (Lehtiö et al., 2009 ▶; PDB entry 3ce0) and PARP-15 (Structural Genomics Consortium, unpublished work; PDB entry 3gey). In both cases only one molecule is bound to the nicotinamide portion of the NAD+ donor site. The key interactions between this PJ34 molecule and the PARP catalytic domains are conserved. The interactions in TNKS1 consist of three hydrogen bonds, the first two between the phenanthridinone O atom of PJ34 and both the side-chain hydroxyl of Ser1221 as well as the backbone amide of Gly1185 and the third between the phenanthridinone N atom of PJ34 and the carbonyl O atom of Gly1185. Additionally, the phenanthridinone of PJ34 makes π-stacking interactions with Tyr1224 and van der Waals interactions with Tyr1213. The most striking difference between the structures is the presence of a second PJ34 molecule in the adenosine-binding region of the NAD+ donor site within the TNKS1 structure. This second PJ34 molecule binds between Phe1188 and His1201, residues that are not conserved in PARP-3 or PARP-15. Additional interactions within this pocket include a hydrogen bond between the phenanthridinone O atom of PJ34 and the backbone amide of Asp1198, as well as a water-mediated hydrogen bond between the phenanthridinone N atom of PJ34 and the carbonyl O atom of Gly1196. This phenanthridinone O atom of PJ34 sits in the same proximity as an O atom from a glycerol molecule in the unliganded TNKS1 structure (Lehtiö et al., 2008 ▶).
Figure 2.

(a) Structure of TNKS1 in complex with PJ34, revealing two molecules bound in the NAD+ donor-binding site, with the bottom molecule in the nicotinamide-binding site and the top molecule in the adenosine-binding site. (b) Structure of unliganded TNKS1 (gray) superimposed on the TNKS1–PJ34 complex structure (yellow). The D-loop residues Phe1197–Gly1211 (cyan) of the unliganded TNKS1 structure, specifically Tyr1203 and Ile1204, occupy the same space in the NAD+ donor pocket as the PJ34 molecule.
In the previously reported structure of unliganded TNKS1 (Lehtiö et al., 2008 ▶) the donor site is unoccupied, allowing a closed conformation of the D-loop. This closed conformation is anchored through a hydrogen bond between the hydroxyl group of Tyr1203 and the O atom of the backbone carbonyl of Tyr1224, which also places Ile1204 in the donor binding-site groove (Fig. 2 ▶ b). This closed conformation, along with crystal contacts, hinders the soaking of NAD+-competitive inhibitors in this unliganded crystal form. The crystal contacts include two salt bridges between Glu1199 and Arg1296 (of a symmetry molecule) as well as Arg1200 and Glu1172 (of a symmetry molecule), in addition to hydrogen bonds involving residues Arg1200 and Gly1209 of a symmetry molecule. In the TNKS1–PJ34 complex structure the D-loop adopts an open conformation (Fig. 2 ▶ b) comparable to that seen in the PARP-3–PJ34 structure (Tyr1203–Gly1205 of chain A and Ile1204–Gly1205 of chain B of the D-loop are disordered in the TNKS1–PJ34 structure). The changes in the D-loop also affect the conformation of the loop containing Gln1262–His1270 between the β6 and β7 strands. This rearrangement is necessary to accommodate the new position of the D-loop in which Met1207 and Phe1208 are directed towards the protein core instead of bulk solvent. In this conformation, the NAD+ donor site can be occupied by PJ34, accessible to bulk solvent and not blocked by crystal contacts. Hence, the PJ34 cocrystallization system is an ideal candidate for displacement soaking of novel inhibitors into the NAD+ donor site.
3.2. Structure of TNKS1–XAV939
To show that the PJ34 cocrystal system was suitable for displacement soaking, we determined the crystal structure of XAV939 at 2.0 Å resolution by displacement soaking XAV939 into the TNKS1–PJ34 crystals. The final model was refined to an R factor of 0.180 and included residues Gly1105–His1201 and Gly1205–Pro1316 in molecule A, residues Gly1105–Ala1315 in molecule B, one zinc ion and one molecule of inhibitor XAV939 per protein molecule, eight sulfate ions, three glycerol molecules and 316 solvent molecules. The loop from Ala1202 to Ile1204 as well as the C-terminal residues Ser1317–Thr1327 in molecule A and the C-terminal residues Pro1316–Thr1327 of molecule B were omitted from the final model because of poor electron density.
The electron density clearly revealed that both PJ34 molecules were displaced from the NAD+ donor-binding site and were replaced by one XAV939 molecule. The interaction between TNKS1 and XAV939 closely mimics that in the published structure of the TNKS2–XAV939 complex (Karlberg et al., 2010 ▶), as expected since the XAV939-interacting residues are conserved between the two isoforms. Here, we see the same stacking interactions of the pyrimidine ring with Tyr1224 and hydrogen bonds between the hydroxyl of Ser1221 and Gly1185 with the pyrimidine hydroxyl and pyrimidine N atom, respectively (Fig. 3 ▶). Tyr1203 of the D-loop in the TNKS1–XAV939 structure can be seen in one molecule of the asymmetric unit, where it makes van der Waals interactions with the phenyl ring of XAV939. This same conformation of the D-loop was reported in the TNKS2–XAV939 complex structure. Additionally, interactions seen between the trifluoromethyl group of XAV939 and TNKS2 are retained in the TNKS1–XAV939 structure, in which nonpolar interactions occur with Pro1187, Phe1188 and Ile1228.
Figure 3.

Structure of TNKS1 in complex with XAV939. The inhibitor molecule occupies the nicotinamide-binding site of the NAD+-binding groove.
Using the described protocol, we have demonstrated that the PJ34 inhibitor can be displaced from this crystal form. The use of this displacement soaking system enables the rapid determination of new inhibitor complexes. Compounds that interact with the NAD+ donor pocket should be amenable to structure determination, thus creating an ideal system for structure-based drug design for the tankyrase 1 PARP domain. Additionally, the structure of PJ34 with a second molecule in the adenosine portion of the NAD+ donor pocket opens new directions for tankyrase drug design.
Supplementary Material
PDB reference: TNKS1–PJ34, 3uh2
PDB reference: TNKS1–XAV939, 3uh4
Acknowledgments
We would like to acknowledge the work of James Groarke and Guoping Xiao in protein purification of TNKS1. The TNKS1–PJ34 data collection was performed on beamline 5.0.3 at the Advanced Light Source at Berkeley Laboratory. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231. The TNKS1–XAV939 data collection was performed on the X06SA beamline at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland.
References
- Abdelkarim, G. E., Gertz, K., Harms, C., Katchanov, J., Dirnagl, U., Szabó, C. & Endres, M. (2001). Int. J. Mol. Med. 7, 255–260. [PubMed]
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Bricogne, G., Blanc, E., Brandl, M., Flensburg, C., Keller, P., Paciorek, W., Roversi, P., Smart, O. S., Vonrhein, C. & Womack, T. O. (2009). BUSTER v.2.8.0. Cambridge: Global Phasing Ltd.
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Huang, S.-M. A. et al. (2009). Nature (London), 461, 614–620.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Karlberg, T., Markova, N., Johansson, I., Hammarström, M., Schütz, P., Weigelt, J. & Schüler, H. (2010). J. Med. Chem. 53, 5352–5355. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Lehtiö, L., Collins, R., van den Berg, S., Johansson, A., Dahlgren, L. G., Hammarström, M., Helleday, T., Holmberg-Schiavone, L., Karlberg, T. & Weigelt, J. (2008). J. Mol. Biol. 379, 136–145. [DOI] [PubMed]
- Lehtiö, L., Jemth, A. S., Collins, R., Loseva, O., Johansson, A., Markova, N., Hammarström, M., Flores, A., Holmberg-Schiavone, L., Weigelt, J., Helleday, T., Schüler, H. & Karlberg, T. (2009). J. Med. Chem. 52, 3108–3111. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Peukert, S. & Schwahn, U. (2004). Expert Opin. Ther. Pat. 14, 1531–1551.
- Schreiber, V., Dantzer, F., Ame, J. C. & de Murcia, G. (2006). Nature Rev. Mol. Cell Biol. 7, 517–528. [DOI] [PubMed]
- Smith, S. & de Lange, T. (2000). Curr. Biol. 10, 1299–1302. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: TNKS1–PJ34, 3uh2
PDB reference: TNKS1–XAV939, 3uh4