

The interleukin-2 tyrosine kinase Src homology 2 domain was crystallized and its structure was solved to 2.35 Å resolution. The structure reveals a domain-swapped dimer that is related to other dimeric SH2 domains solved previously. The cis–trans-prolyl isomerization that is evident from solution studies of Itk SH2 cannot be observed in the crystal structure.
Keywords: IL-2-inducible T-cell kinase, Src homology 2 domain, proline isomerization, domain swapping
Abstract
The crystal structure of the interleukin-2 tyrosine kinase Src homology domain (Itk SH2) is described and it is found that unlike in studies of this domain using NMR spectroscopy, cis–trans-prolyl isomerization is not readily detected in the crystal structure. Based on similarities between the Itk SH2 crystal form and the cis form of the Itk SH2 NMR structure, it is concluded that it is likely that the prolyl imide bond at least in part adopts the cis conformation in the crystal form. However, the lack of high-resolution data and the dynamic nature of the proline-containing loop mean that the precise imide-bond conformation cannot be determined and prolyl cis–trans isomerization in the crystal cannot be ruled out. Given the preponderance of structures that have been solved by X-ray crystallography in the Protein Data Bank, this result supports the notion that prolyl isomerization in folded proteins has been underestimated among known structures. Interestingly, while the precise status of the proline residue is ambiguous, Itk SH2 crystallizes as a domain-swapped dimer. The domain-swapped structure of Itk SH2 is similar to the domain-swapped SH2 domains of Grb2 and Nck, with domain swapping occurring at the β-meander region of all three SH2 domains. Thus, for Itk SH2 structural analysis by NMR spectroscopy and X-ray crystallography revealed very different structural features: proline isomerization versus domain-swapped dimerization, respectively.
1. Introduction
Cis–trans-prolyl isomerization at specific imide-bond locations within folded proteins modulates protein structure and function (Sarkar et al., 2011 ▶; Andreotti, 2003 ▶; Brazin et al., 2002 ▶; Breheny et al., 2003 ▶; Eckert et al., 2005 ▶; Fischer & Aumüller, 2003 ▶; Lu, 2000 ▶; Lummis et al., 2005 ▶; Nelson et al., 2006 ▶; OuYang et al., 2008 ▶; Pastorino et al., 2006 ▶). In spite of the emerging functional importance of prolyl isomerization, isomerization about the prolyl imide bond is elusive since it is not readily detected using standard biochemical tools. The discovery of proline switches across a number of unrelated proteins has to date been serendipitous, often occurring for protein systems that are tractable by NMR spectroscopy (Sarkar et al., 2007 ▶; Koo et al., 2011 ▶; Mallis et al., 2002 ▶; OuYang et al., 2008 ▶; Pastorino et al., 2006 ▶). NMR permits the direct detection of prolyl isomerization because cis–trans isomerization is slow on the NMR timescale, giving rise to doubled resonances (Koo et al., 2011 ▶; Sarkar et al., 2007 ▶; Mallis et al., 2002 ▶).
Solution NMR studies of the Src homology 2 domain (SH2) from the immunological Tec-family tyrosine kinase Itk revealed a single prolyl imide bond within the folded domain that exchanges between the cis and trans conformations (Fig. 1 ▶ a; Mallis et al., 2002 ▶). The major SH2 species in solution (∼65%) adopts a trans Asn286–Pro287 imide bond, while ∼35% adopts the cis conformation about the same bond. Ongoing functional studies are defining the role of this proline-driven conformational switch in controlling Itk function and ligand recognition during immune cell signaling (Brazin et al., 2000 ▶; Breheny et al., 2003 ▶; Colgan et al., 2004 ▶; Min et al., 2010 ▶; Pletneva et al., 2006 ▶; Severin et al., 2009 ▶). These current lines of investigation have been pursued based on the initial observation of proline isomerization in Itk SH2 using solution NMR spectroscopy.
Figure 1.

(a) Itk SH2 structures determined by NMR spectroscopy. The canonical fold of an SH2 domain consists of a central antiparallel β-sheet that is sandwiched between an N-terminal α-helix A and a C-terminal α-helix B. Pro287 (circled) interconverts between the trans and cis imide-bond conformations in solution. The trans and cis X–Pro imide bonds are shown next to each structure, with the imide bond itself indicated in red. The β-meander structure in the SH2 monomer containing the short E and F strands is circled in both structures and labeled in the cis structure. All structural figures in this report were made using PyMOL (DeLano, 2002 ▶). (b) Itk SH2 with residues targeted for mutation to methionine shown as gray sticks and labeled (Pro287 is shown in red). (c) 1H–15N HSQC spectrum of wild-type Itk SH2 (blue) overlaid with the corresponding spectrum of the Itk SH2 V275M mutant (red). Right, expanded HSQC spectra for three specific SH2 resonances, Gly260, Lys258 and Phe278, showing the cis and trans peaks for wild-type and mutant protein. The cis:trans ratio is unchanged by the mutation of Val275 to methionine.
Given that the vast majority of protein structures available in the Protein Data Bank (PDB) have been solved using X-ray crystallographic techniques and not NMR, it is likely that many functionally relevant dynamic prolines have been overlooked. Indeed, it is generally presumed that X-ray crystallography does not reliably reveal dynamic prolyl imide bonds or even the presence of a stable cis-proline owing to resolution limits and the nature of refinement protocols (Weiss et al., 1998 ▶). Itk SH2 and its well characterized cis–trans-prolyl isomerization is a useful test case in order to examine, for at least one protein, how crystallization might affect our ability to detect prolyl isomerization in a folded domain. Several possible outcomes are envisioned: (i) both prolyl imide-bond conformations will be detected in the Itk SH2 crystal structure, (ii) crystallization of Itk SH2 will stabilize a single prolyl imide-bond conformation (either cis or trans), obscuring the dynamic nature of the prolyl imide bond preceding Pro287, or (iii) the nature of the diffraction data (insufficient resolution, high B parameters) might prohibit the detection of the prolyl imide-bond conformation(s).
2. Experimental procedures
2.1. Constructs
The construct for Itk SH2 (ITK; gene ID 16428) has been described previously (Brazin et al., 2000 ▶). Briefly, mouse Itk SH2 (residues 231–338) was cloned into the pGEX-2T vector (GE Healthcare) with an N-terminal (vector-derived) GST tag and a thrombin cleavage site between the GST tag and Itk SH2. Post-thrombin cleavage, Itk SH2 has two vector-derived N-terminal residues (Gly and Ser) and eight vector-derived C-terminal residues (GSPGIHRD). The Itk L253M, V264M, R265M, V275M, S276M, P287A, I315M, L318M, I319M, Q320M and L333M mutations were introduced by using the site-directed mutagenesis (SDM) kit (Stratagene). All constructs were verified by sequencing at the Iowa State DNA Sequencing and Synthesis Facility.
2.2. Protein expression and purification
Itk SH2 (wild type or mutant) was expressed in Escherichia coli strain BL21 (DE3) in LB medium and purified in HEPES buffer [50 mM HEPES pH 7.4, 150 mM NaCl, 2 mM DTT, 0.02%(w/v) NaN3] as described previously (Joseph et al., 2011 ▶). To produce selenomethionyl-labeled Itk SH2 or for the preparation of uniformly 15N-labeled protein, the cells were grown in modified M9 medium containing l-selenomethionine (SeMet; Calbiochem) or modified M9 minimal medium containing 15N-enriched ammonium chloride (1 g l−1; Cambridge Isotope Laboratories) as described previously (Brazin et al., 2000 ▶). Incorporation of SeMet was confirmed by MALDI–TOF mass-spectrometric analysis of the tryptic digests of the labeled protein at the Iowa State Protein Facility. The purified Itk SH2 was concentrated to 13.5 mg ml−1 and filter-sterilized prior to setting up crystal trays.
2.3. NMR spectra
All two-dimensional 1H–15N HSQC spectra were acquired using a Bruker DRX500 spectrometer operating at a 1H frequency of 499.867 MHz at 298 K. 15N-labeled protein (300 µM) samples were prepared in 50 mM sodium phosphate pH 7.2, 75 mM NaCl, 2 mM DTT, 0.02%(w/v) NaN3. 1H and 15N chemical shifts were externally referenced to 2,2-dimethyl-2-silapentane-5-sulfonate sodium salt (DSS) in identical buffer.
2.4. Size-exclusion chromatography
Proteins were separated on a Superdex 75 10/300 GL column (GE Healthcare) using the Bio-Rad BioLogic DuoFlow FPLC system. The column was equilibrated with three column volumes of HEPES buffer [50 mM HEPES pH 7.4, 150 mM NaCl, 2 mM DTT, 0.02%(w/v) NaN3] at a flow rate of 0.4 ml min−1 prior to each run. The column was calibrated using 100 µl of a mixture of 0.5 mg ml−1 each (1:1:1:1) of low-molecular-weight standards (albumin, chymotrypsinogen A, ovalbulin and ribonuclease A) from the gel-filtration calibration kit (GE Healthcare). 100 µl of a 1.35 mg ml−1 solution of Itk SH2 was separated using the same buffer conditions as described above.
2.5. Analytical ultracentrifugation
Sedimentation equilibrium was performed on a Beckman Coulter ProteomeLab XL-A analytical ultracentrifuge using a six-channel (1.2 cm path) Epon cell and an An-60 Ti rotor at 277 K. Itk SH2 was dialyzed overnight into HEPES buffer [50 mM HEPES pH 7.4, 150 mM NaCl, 2 mM DTT, 0.02%(w/v) NaN3] at 277 K and filtered (0.2 µm filter) prior to use. Three Itk SH2 samples were prepared at 16, 27 and 38 µM. The sample volumes were 120 µl and the reference cells contained 130 µl filtered dialysis buffer. The sample was spun at 39 600, 46 400 and 56 900 rev min−1 until equilibrium was achieved. For each speed, three scans at 1 h intervals were recorded at 280 nm using 0.001 cm spacing and averaging 20 readings at each point. Scan analysis was performed using UltraScan II (Demeler, 2005 ▶). The molecular mass of Itk SH2 was derived from the slope of the line obtained by plotting ln(absorbance) against radius2,
where M is the molecular mass (g mol−1), 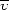 is the protein partial specific volume (0.7326 cm3 g−1), ρ is the solvent density (1.00835 g cm−3), ω is the angular velocity in radians s−1, R is the molar gas constant (8.314 × 104 kg cm2 s−2 mol−1 K−1) and T is the absolute temperature (277 K). The protein partial specific volume and the solvent density were calculated using the program SEDNTERP (Laue et al., 1992 ▶).
is the protein partial specific volume (0.7326 cm3 g−1), ρ is the solvent density (1.00835 g cm−3), ω is the angular velocity in radians s−1, R is the molar gas constant (8.314 × 104 kg cm2 s−2 mol−1 K−1) and T is the absolute temperature (277 K). The protein partial specific volume and the solvent density were calculated using the program SEDNTERP (Laue et al., 1992 ▶).
2.6. Crystallization of Itk SH2
The crystallization condition for Itk SH2 has been described previously (Joseph et al., 2011 ▶). Briefly, the crystals were grown by mixing 3.5 µl of a 1:1 ratio of 1.35 mg ml−1 Itk SH2 and reservoir solution [0.1 M sodium citrate pH 5.3, 10% isopropanol, 20%(w/v) PEG 4000 and 2 mM DTT] with 0.5 µl (12.5 mM final concentration) glycyl-glycyl-glycine (Sigma) as an additive in a final drop volume of 4 µl. All drops were equilibrated against 500 µl reservoir solution. The plate-like crystals grew to approximately 50–100 µm in two weeks and were flash-cooled directly (without additional cryoprotectants) in liquid nitrogen.
2.7. Data collection
Crystals were screened at Iowa State University on a Rigaku R-AXIS IV++ rotating-anode/image-plate system using Cu Kα radiation from an Osmic confocal optics system. Diffraction data for native wild-type Itk SH2 and Itk SH2 V275M SeMet crystals were collected from single crystals at a crystal-to-detector distance of 140 or 180 mm, respectively, on beamline 4.2.2 of the Advanced Light Source, Lawrence Berkeley National Laboratory. The crystal was rotated through 180° with a 1° oscillation range per frame. For Itk SH2 V275M SeMet crystals complete anomalous data sets were obtained at wavelengths corresponding to the peak absorbance (0.9790 Å), the inflection point (0.9793 Å) and a remote wavelength (0.9640 Å) from the absorption edge of selenium. The program d*TREK (Pflugrath, 1999 ▶) was used to index, integrate, scale and merge intensities, which were then converted to structure factors using the CCP4 program TRUNCATE (Winn et al., 2011 ▶; French & Wilson, 1978 ▶). The calculated Matthews coefficient (V M) of 2.07 Å3 Da−1, with a solvent content of 40.56%, suggests the presence of one molecule per asymmetric unit. Data-collection statistics are given in Table 1 ▶.
Table 1. Crystal parameters and data-collection statistics.
Values in parentheses are for the outermost resolution shell.
| Itk SH2 V275M, SeMet | ||||
|---|---|---|---|---|
| Itk SH2 WT, native | Peak | Inflection | Remote | |
| No. of crystals | 1 | 1 | ||
| Beamline | 4.2.2, Advanced Light Source, Lawrence Berkeley Laboratory | |||
| Wavelength (Å) | 1.2398 | 0.9790 | 0.9793 | 0.9640 |
| Detector | ‘NOIR-1’ MBC system | |||
| Crystal-to-detector distance (mm) | 140 | 180 | ||
| Rotation range per image (°) | 1 | |||
| Total rotation range (°) | 180 | |||
| Exposure time per image (s) | 12 | |||
| Resolution (Å) | 47.22–2.35 (2.43–2.35) | 43.39–2.50 (2.59–2.50) | 43.39–2.60 (2.69–2.60) | 43.40–2.60 (2.69–2.60) |
| Space group | I222 | C2221 | ||
| Unit-cell parameters (Å) | a = 53.6, b = 57.3, c = 83.1 | a = 53.9, b = 73.2, c = 57.1 | ||
| Mosaicity (°) | 2.00 | 0.94 | 0.92 | 0.93 |
| Total reflections | 29274 | 28895 | 25774 | 25847 |
| Unique reflections | 5487 | 4137 | 3687 | 3687 |
| Average multiplicity | 5.34 (4.42) | 6.98 (7.26) | 6.99 (7.18) | 7.01 (7.10) |
| Average I/σ(I) | 10.3 (4.1) | 9.6 (2.5) | 11.2 (3.6) | 10.9 (3.8) |
| Unaveraged I/σ(I) | 4.4 (1.9) | 3.9 (1.1) | 4.5 (1.6) | 4.3 (1.6) |
| Completeness (%) | 97.8 (93.1) | 100 (100) | 100 (100) | 100 (100) |
| Rmerge† (%) | 9.4 (34.7) | 9.3 (50.4) | 8.2 (38.4) | 8.2 (38.0) |
| Overall B factor from Wilson plot (Å2) | 51.80 | 63.00 | ||
R
merge = 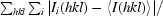
 , where Ii(hkl) is the scaled intensity of the ith observation of reflection hkl and I(hkl) is the mean intensity for this set of observations.
, where Ii(hkl) is the scaled intensity of the ith observation of reflection hkl and I(hkl) is the mean intensity for this set of observations.
2.8. Structure determination and refinement
The structure of the Itk SH2 V275M mutant was determined by the multiwavelength anomalous dispersion (MAD) phasing method using SOLVE (Terwilliger & Berendzen, 1999 ▶) and RESOLVE (Terwilliger, 2000 ▶). Two selenomethionine sites were located with a mean figure of merit of 0.52 and a Z score of 9.66. Itk SH2 was then built using the Src SH2 domain (PDB entry 1spr) as a model as follows. The nonconserved amino-acid residues within the Src SH2 domain were trimmed to the last common atom using the CCP4 program CHAINSAW (Stein, 2008 ▶). The trimmed Src model was then superimposed (rotated and translated) onto the model generated from RESOLVE using the CCP4 program LSQKAB (Kabsch, 1976 ▶). Improvement of the superimposed and trimmed Src model was carried out by manual building within the programs O (Jones et al., 1991 ▶) and XtalView (McRee, 1999 ▶) followed by refinement using REFMAC5 (Murshudov et al., 2011 ▶) and PHENIX (Adams et al., 2010 ▶). Both the Itk SH2 V275M SeMet and the native Itk SH2 wild-type data sets exhibited severe anisotropy. The Itk SH2 V275M SeMet data set had diffraction limits of 2.2 Å along the a* and c* directions but only 3.1 Å along the c* direction, while the native wild-type Itk SH2 data set had diffraction limits of 2.4 Å along the a* and b* directions but only 2.7 Å along the c* direction. Ellipsoidal truncation and anisotropic scaling was therefore carried out on both data sets to improve the electron-density maps (Strong et al., 2006 ▶). The initial model underwent simulated annealing from 1000 to 100 K in steps of 25 K, followed by 100 cycles of energy minimization and thermal parameter refinement with hydrogen-bond restraints using CNS (Brünger et al., 1998 ▶). Ligand and water molecules were fitted to OMIT electron density until no improvement in R free was evident. The native wild-type Itk SH2 structure was solved by molecular replacement using the Itk SH2 V275M structure as the search model in Phaser (McCoy et al., 2005 ▶; Read, 2001 ▶; Storoni et al., 2004 ▶). Water molecules with thermal parameters above 60 Å2 or that were more than 3.2 Å from the nearest hydrogen-bonding partner were removed from the final model. Refinement statistics are given in Table 2 ▶.
Table 2. Refinement and model statistics.
Values in parentheses are for the outermost resolution shell.
| Resolution (Å) | 47.22–2.35 (2.69–2.35) |
| Rcryst† | 23.7 (35.3) |
| Rfree‡ | 28.2 (42.9) |
| R.m.s.d. bond lengths (Å) | 0.009 |
| R.m.s.d. bond angles (°) | 1.263 |
| Average B parameter (Å2) | 50.4 |
| No. of atoms in model | 960 |
| Ligand | Citrate |
| No. of solvent molecules | 17 |
| MolProbity Ramachandran analysis | |
| Favored region (%) | 87.5 |
| Allowed region (%) | 9.82 |
| Outliers (%) | 2.68 |
| PDB code | 3s9k |
R
cryst = 100 × 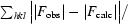
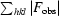 , where F
obs and F
calc are the observed and calculated structure factors, respectively.
, where F
obs and F
calc are the observed and calculated structure factors, respectively.
R free was determined using 9.5% of the data.
3. Results and discussion
To determine whether or not proline isomerization could be detected in a crystal form of Itk SH2, we set out to solve the structure of Itk SH2 by X-ray crystallography.
3.1. Identification of an Itk SH2 methionine mutant
Expression, purification and crystallization screens for Itk SH2 have been described in detail elsewhere (Joseph et al., 2011 ▶). After successful crystallization and data collection for the SeMet-derivatized wild-type Itk SH2 (Joseph et al., 2011 ▶), we found that the multiwavelength anomalous dispersion (MAD) data set was insufficient to provide adequate phase information, as the 118-residue Itk SH2 contains only a single methionine residue. Further attempts to solve the Itk SH2 structure by molecular replacement using either the NMR structures of Itk SH2 or the crystal structure of the Src SH2 domain (which has 34% sequence identity to Itk SH2) as a model were not successful.
We next mutated Itk SH2 to introduce an additional methionine residue to facilitate MAD phasing. Potential sites for the incorporation of a methionine residue were chosen based on the following criteria: (i) the residue should be present on a stable secondary-structure element rather than a flexible loop region, (ii) the residue should be in the interior of the protein where it would be unlikely to interfere with crystal contacts, thereby enhancing the likelihood of the mutant crystallizing under the same conditions as the wild-type SH2 domain, and (iii) the mutation should not alter the cis–trans prolyl isomerization of Itk SH2. Based on the above criteria, ten potential sites (Leu253, Val264, Arg265, Val275, Ser276, Ile315, Leu318, Ile319, Gln320 and Leu333) were identified for the introduction of a methionine residue into Itk SH2 (Fig. 1 ▶ b). Each of these ten residues was individually mutated to methionine and subjected to further characterization. The Itk SH2 V264M, S276M, I315M and Q320M single mutants were expressed as insoluble proteins in bacteria and were not pursued further. The Itk SH2 L253M, R265M, L318M and L333M single mutants were either unstable upon purification or failed to crystallize under the same crystallization conditions as the wild-type SH2 domain. The Itk SH2 V275M and I319M single mutants crystallized under the same crystallization conditions as the wild-type protein; however, the Itk SH2 I319M crystals diffracted poorly. The Itk SH2 V275M mutant was therefore selected for further analysis.
To ensure that the V275M mutation did not alter the cis–trans prolyl isomerization of Itk SH2, a 1H–15N heteronuclear single-quantum coherence (HSQC) spectrum of the Itk SH2 V275M mutant was compared with that of the wild-type SH2 domain (Fig. 1 ▶ c). Cis–trans isomerization about the Asn286–Pro287 imide bond gives rise to doubled resonances for 35 of the 109 residues that are detected in the 1H–15N HSQC spectrum of Itk SH2 (Mallis et al., 2002 ▶). The 1H–15N HSQC spectrum of the Itk SH2 V275M mutant indicates that the domain remains folded (as judged by chemical shift dispersion) and shows the same peak doubling as the wild-type SH2 domain. The relative intensities of each pair of cis/trans resonances are unchanged in the Itk SH2 V275M mutant spectrum (Fig. 1 ▶ c), indicating that the V275M mutation does not alter proline isomerization within Itk SH2 and is an ideal candidate for SeMet labeling, crystallization and data acquisition.
SeMet-labeled crystals of the Itk SH2 V275M mutant diffracted to 2.5 Å resolution. The structure of the Itk SH2 V275M mutant was solved using SOLVE/RESOLVE (Terwilliger, 2000 ▶; Terwilliger & Berendzen, 1999 ▶). The initial model generated by SOLVE/RESOLVE consisted of the main-chain core of Itk SH2 with three central β-strands flanked by two helices. The loop regions as well as the N- and C-termini of Itk SH2 were absent from the model. Both of the SeMet sites were identified and their positions aligned well with the location of the corresponding residues within the NMR structure of Itk SH2. The complete structure of the Itk SH2 V275M mutant was built by iterative rounds of manual building and refinement. The structure of wild-type Itk SH2 (native data set) was solved by molecular replacement using the structure of the Itk SH2 V275M mutant as a model (see §2).
3.2. The crystal structure of Itk SH2 consists of a domain-swapped dimer
The Itk SH2 crystal form reveals two SH2 domains that exhibit the classical SH2 fold (Marengere & Pawson, 1994 ▶; Waksman et al., 1992 ▶) in a dimeric domain-swapped arrangement (Fig. 2 ▶ a). Instead of following the typical β-meander of the monomeric SH2 domain (Fig. 1 ▶ a), the EF loop and F strand of each SH2 monomer adopt an extended conformation that results in a detached C-terminal B helix (Figs. 2 ▶ a and 2 ▶ b). The canonical fold of the SH2 domain is reconstituted by the B helix of the symmetry-related dimer. The classical SH2-domain fold in the Itk SH2 crystal structure is therefore composed of two polypeptide chains: a core made up of residues Asn223–Ala307 from one chain and residues Glu308–Asp346 from the other chain (Fig. 2 ▶ c). As a result, the N- and C-termini are located on opposite sides of the SH2 domain separated by a distance of 57 Å, unlike the classical monomeric SH2-domain fold in which the N- and C-termini are located on the same side of the domain separated by a distance of 20 Å. The dimerization interface is extensive and consists primarily of hydrophobic residues throughout the domain (Fig. 2 ▶ d). The structure of the phosphotyrosine binding pocket is unperturbed in the Itk SH2 dimer and is occupied by a citrate molecule in a manner that mimics bound phosphotyrosine in solution (Pletneva et al., 2006 ▶; Figs. 2 ▶ a and 2 ▶ e).
Figure 2.

(a) Structure of the Itk SH2 domain-swapped dimer. The monomer units are colored black and gray, citrate is shown in red and the CD loops of each monomer (containing Pro287) are boxed and labeled. (b) The asymmetric unit of the SH2-domain crystal structure is a monomer with an extended EF strand and dislocated B helix. (c) Primary sequence of Itk SH2 showing the site of swapping in the dimer structure (Ala307–Glu308). Upper case indicates the sequence of Itk SH2 and lower case indicates the vector-derived sequence. The amino acids colored red indicate those for which side-chain interactions across the dimer interface are observed. (d) Two views of Itk SH2 dimer interface observed in the crystal structure. The surface of one monomer is depicted (gray) and the opposing monomer is represented using ribbons to illustrate the dimer interface. The residues that are oriented toward the opposing monomer in the dimer structure are shown as red sticks [the same as those highlighted in (c)]. (e) Citrate in the phosphotyrosine (pY) binding pocket in the Itk SH2 crystal structure. Arg246 and Arg265 (conserved pY binding residues) and the bound citrate molecule are shown as a 2F o − F c electron-density map contoured at 1σ.
3.3. Itk SH2 is predominantly a monomer in solution
To assess whether the dimerized Itk SH2 observed in the crystal structure can be detected in solution, we analyzed the purified protein using several methods. Gel-filtration analysis of Itk SH2 resulted in a single peak that eluted with a molecular radius consistent with that of a monomer (Fig. 3 ▶ a). Further gel-filtration analysis of Itk SH2 after incubation at room temperature for 3 d also showed a single peak consistent with a monomer (data not shown). NMR data showed average 1H line widths of ∼21 Hz (with a range of 19–23 Hz) in the 1H–15N HSQC spectrum of Itk SH2 (300 µM) that are consistent with the molecular weight of the monomeric species (Fig. 3 ▶ b). The solution behavior of the Itk SH2 domain was also analyzed by analytical ultracentrifugation (Figs. 3 ▶ c and 3 ▶ d). Sedimentation-equilibrium data were fitted to various models: single-component monomer, single-component dimer and monomer–dimer equilibrium models. The best fit of the data was obtained using a single-component monomer model (Fig. 3 ▶ c). The molecular mass of Itk SH2 calculated from these data is 12.69 kDa (standard deviation 6.05 × 10−3), which is close to the theoretical mass of 13.52 kDa for monomeric Itk SH2 (Figs. 3 ▶ c and 3 ▶ d). Thus, while Itk SH2 crystallizes as a dimer, we do not detect SH2 dimerization in solution under the conditions tested and therefore must conclude at this time that the dimeric arrangement is an artifact of the crystallization process.
Figure 3.

(a) Size-exclusion chromatography of Itk SH2. The elution profile of Itk SH2 (red trace) is compared with the gel-filtration protein standards (blue trace). The standard peaks (1–4, indicated by arrows) correspond to albumin (65 kDa), ovalbumin (45 kDa), chymotrypsinogen A (25 kDa) and ribonuclease A (13.7 kDa) respectively. (b) NMR line-width analysis of Itk SH2. A section of the 1H–15N HSQC spectrum of Itk SH2 is shown with the one-dimensional 1H slice above. (c, d) Sedimentation-equilibrium analysis of Itk SH2. The data (three scans at 1 h interval each) were obtained at rotor speeds of 39 600, 46 400 and 56 900 rev min−1 at protein concentrations of 16, 27 and 38 µM. The solid lines (red) in (c) represent the global fit of the data to a single-component (monomer) model. Fit residuals are shown at the top. (d) Plot of the natural logarithm of the absorbance versus radius2. Only a single data set (38 µM sample spun at 46 400 rev min−1) is shown for clarity. The calculated molecular mass of Itk SH2 is 12.69 kDa, which is consistent with that of a monomeric species.
3.4. Proline isomerization: uncertainty in the conformation of the Itk SH2 Asn286–Pro287 imide bond
The initial motivation for this work was to assess the structural features of Itk SH2 that would be evident using X-ray crystallography rather than NMR spectroscopy. Specifically, we wished to address whether cis–trans isomerization of the Asn286–Pro287 imide bond would be identified in diffraction data obtained from a crystal of Itk SH2. Our findings suggest that if X-ray crystallography were the technique of choice used for initial structural characterization of this domain, it is likely that the proline-isomerization event would have gone unnoticed. Given the modest resolution of the structure and the high B parameters for the residues surrounding Pro287, the precise conformation of the Asn286–Pro287 imide bond cannot be determined from the data. This is best illustrated by the electron-density map for the Pro287-containing CD loop region of the SH2 domain (Fig. 4 ▶ a). The three residues preceding the conformationally heterogenous Pro287 (Ser284, Glu285 and Asn286) and the 286–287 imide bond itself show poor electron density.
Figure 4.

(a) The 2F o − F c electron-density map contoured at 1σ for the CD loop region of Itk SH2 in the domain-swapped dimer structure. The electron density is poor for Ser284, Glu285, Asn286 and Pro287. (b, c) Superposition of the ensemble of NMR-derived SH2-domain structures (thin lines) with one monomer of the SH2-domain dimer structure determined by crystallography. The NMR-derived structures are shown using thinner lines for the CD loop region and the crystal structure is depicted by the wider gray line. The view focuses on the CD loop and compares the conformation in the CD loop region from the crystal structure with (b) the cis imide-bond-containing SH2-domain structures from NMR and (c) the CD loop with the trans imide-bond-containing SH2-domain conformers determined by NMR. (d) Packing interactions between symmetry-related molecules in the Itk SH2 crystal. The enlarged view focuses on the CD loop region of the Itk SH2 structure (black and gray ribbons) showing the packing of Ile282 at the SH2-domain dimer interface and Ile283 between symmetry-related dimer molecules. (e) Superimposing the trans imide-bond-containing NMR structure of Itk SH2 onto each of the four symmetry-related SH2 domains shown in (d) illustrates the steric clash that would arise in the crystal structure if the CD loop adopted the extended conformation that results from a trans prolyl imide bond at position 286–287. In addition, the interactions between CD loop isoleucines are lost in the trans CD loop conformation.
Despite our inability to directly observe the Asn286–Pro287 prolyl imide bond, we can conclude several things from this region of the Itk SH2 structure. The most dramatic difference between the NMR-derived cis and trans SH2 structures lies in the proline-containing CD loop (Fig. 1 ▶ a). Comparing the NMR ensemble of the cis and trans imide-bond-containing SH2 structures with the crystal structure shows that to the extent that it is defined by the electron density, the CD loop configuration in the crystal structure is more similar to that of the SH2 domain containing the cis imide bond at Asn286–Pro287 (Figs. 4 ▶ b and 4 ▶ c). Moreover, we find that modeling the cis imide-bond conformation at position 286–287 provides the best fit to the electron density in this region of the structure; the trans prolyl imide bond is not compatible with fitting the CD loop into the observed electron density. In addition, attempts to crystallize the all-trans Itk SH2 Pro287Ala mutant were unsuccessful.
Inspection of the Itk SH2 crystal-packing interactions reveals contacts between the CD loop regions of adjacent molecules (Fig. 4 ▶ d). The CD loop of Itk SH2 contains two hydrophobic isoleucine residues at its apex: Ile282 and Ile283. In the Itk SH2 crystal structure Ile283 makes contacts between symmetry-related dimers, while Ile282 appears to contribute to the domain-swapped dimer interface (expanded view in Fig. 4 ▶ d). Superposition of the trans imide-bond-containing Itk SH2 structure derived from NMR with the Itk SH2 in the crystal structure reveals a significant steric clash between the CD loop and the symmetry-related molecule that would result were the CD loop region to adopt the configuration associated with the trans imide bond at position 286–287 (Fig. 4 ▶ e). Moreover, the trans CD loop conformation removes Ile282 from the dimer interface. We can therefore conclude that the CD loop in the crystal structure of Itk SH2 adopts a conformation that is most similar to that observed in the cis NMR structure of the same domain. These observations, however, cannot rule in or rule out cis–trans isomerization in the Itk SH2 crystal and ultimately we must conclude that the conformational status of the prolyl imide bond at position 287 in this domain cannot be definitively determined by the diffraction data.
The Itk SH2 structure has now been solved by both NMR spectroscopy and X-ray crystallography. The NMR-derived structure reveals dynamic cis–trans-prolyl isomerization that results in divergent configurations for the large CD loop (Mallis et al., 2002 ▶). In the structure of the same domain determined using X-ray diffraction data, the most notable structural feature is the domain-swapped dimer and the absence of electron density for the region of the Asn286–Pro287 imide bond leaves the question of prolyl cis–trans isomerization unanswered by crystallography. Thus, without prior knowledge of the dynamic prolyl isomerization behavior of this domain in solution, the crystal structure of Itk SH2 by itself would not necessarily highlight Pro287 as a dynamic switch.
3.5. Generality of SH2 domain-swapped dimers
A survey of the PDB reveals two related swapped dimer structures for the SH2 domains of Grb2 (Schiering et al., 2000 ▶) and Nck (Frese et al., 2006 ▶) (Fig. 5 ▶). In a manner similar to Itk SH2, the EF loop in the Grb2 SH2-domain structure unfolds from the monomer and extends across the dimer interface (Figs. 5 ▶ a and 5 ▶ b). Additionally, the B helix of one Grb2 monomer completes the SH2-domain fold of the other monomer. The crystal structure of the Nck SH2 domain-swapped dimer differs from Itk and Grb2 in that the DE loop extends away from the body of the SH2 domain and as a result the E and F strands as well as the B helix are swapped (Fig. 5 ▶ c). It is notable that the hinge loop region that is extended in all three SH2 domain-swapped dimers (either the EF loop or the DE loop) corresponds to the region that is most devoid of regular secondary structure within the closed monomer SH2-domain structures (Fig. 1 ▶ a). This is consistent with the notion that partial unfolding of these domains might lead to the formation of the domain-swapped dimer structures observed after crystallization. In this regard, it is interesting that we find that the crystallization of Itk SH2 proceeds at a protein concentration (1.35 mg ml−1; see §2) that is about tenfold lower than typical crystallization conditions. It is possible that partial unfolding of the SH2 domain is favoured at lower protein concentrations owing to reduced molecular crowding and that this partially unfolded species leads to the swapped dimer form that is selected for upon crystallization.
Figure 5.

Structure of the Itk SH2 domain-swapped dimer (a) compared with the (b) Grb2 SH2 and (c) Nck SH2 domain-swapped dimers. In (a), (b) and (c) the extended hinge loops that deviate from the closed monomer structures to yield the domain-swapped form are labeled: the EF loop in Itk and Grb2 and the DE loop in Nck. The CD loops and BG loops of Itk SH2 (a) and the Grb2 SH2 domain (b) are labeled to illustrate the differences in the dimer interface in these otherwise related structures.
The structure of Itk SH2 solved by X-ray crystallography shows an intertwined dimer in which the B helix of one SH2 domain is exchanged with the B helix of the other. NMR spectroscopy, size-exclusion chromatography and analytical ultracentrifugation provide no evidence for dimerization of Itk SH2 in solution. Moreover, solution studies of larger domain fragments of Itk support an intermolecular interaction between the SH2 domain of one Itk molecule and the SH3 domain of another Itk molecule (Brazin et al., 2000 ▶; Min et al., 2010 ▶). The structure of the Itk SH2–SH3 heterodimer has been solved (Severin et al., 2009 ▶) and is sterically incompatible with the domain-swapped dimer structure of Itk SH2 (Fig. 6 ▶). The SH3-binding site on the SH2 domain includes the CD, BG and EF loops. The CD and BG loops are completely occluded in the dimer structure (Fig. 6 ▶ b) and the EF loop is extended, leading into the domain-swapped B helix. Our inability to detect SH2-domain dimerization in solution, combined with the fact that the SH2-mediated interactions observed both in solution (Brazin et al., 2002 ▶; Breheny et al., 2003 ▶; Severin et al., 2009 ▶) and in cellular assays (Min et al., 2010 ▶) are incompatible with the dimer structure, suggest that the domain-swapped dimer represents a species in solution that is only sparsely populated but is selected for during the crystallization process.
Figure 6.

(a) NMR-derived structure of the Itk SH2–SH3 complex. The SH2 domain is shown in black, with the EF, BG and cis CD loops labeled. The Itk SH3 domain is shown in cyan. (b) The SH2 domain-swapped dimer structure with one SH2 monomer colored black [and in the same orientation as the SH2 domain in (a)] and the other SH2 monomer colored gray. The EF, BG and CD loops are labeled to point out their involvement at the dimer interface and the resulting occlusion of the SH3-binding site on the SH2 domain by dimerization.
This study demonstrates that significantly different structural features of the same protein can be observed depending on the method used to determine the structure. Following characterization of the prolyl-isomerization event within the folded Itk SH2 using solution NMR, numerous studies (Brazin et al., 2002 ▶; Breheny et al., 2003 ▶; Colgan et al., 2004 ▶; Min et al., 2010 ▶; Severin et al., 2009 ▶) support the idea that this proline switch is likely to contribute to the physiological function of this domain within full-length Itk. Crystallization of the same domain did not clearly reveal the dynamic proline switch (likely owing to a combination of insufficient resolution, high B parameters and packing interactions that favor one loop configuration over the other) and instead leads the investigator towards pondering the significance of a domain-swapped dimer structure. This outcome also warns that the extent of prolyl isomerization within folded proteins may be underestimated in the large number of protein crystal structures in the PDB.
Supplementary Material
PDB reference: interleukin-2 tyrosine kinase Src homology 2 domain, 3s9k
Acknowledgments
We would like to thank Dr Michael Shogren-Knaak and Abdelhamid M. Azzaz for help with sedimentation-equilibrium experiments. We would also like to thank Dr Yeon-Kyun Shin and Jaeil Shin for help with size-exclusion chromatography. This work was supported by grants from the National Institutes of Health (National Institute of Allergy and Infectious Diseases, AI043957 and AI075150) to AHA and grant NS010546 to RBH. The use of the beamline at the Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under Contract No. DE-AC02-05CH11231.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Andreotti, A. H. (2003). Biochemistry, 42, 9515–9524. [DOI] [PubMed]
- Brazin, K. N., Fulton, D. B. & Andreotti, A. H. (2000). J. Mol. Biol. 302, 607–623. [DOI] [PubMed]
- Brazin, K. N., Mallis, R. J., Fulton, D. B. & Andreotti, A. H. (2002). Proc. Natl Acad. Sci. USA, 99, 1899–1904. [DOI] [PMC free article] [PubMed]
- Breheny, P. J., Laederach, A., Fulton, D. B. & Andreotti, A. H. (2003). J. Am. Chem. Soc. 125, 15706–15707. [DOI] [PubMed]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed]
- Colgan, J., Asmal, M., Neagu, M., Yu, B., Schneidkraut, J., Lee, Y., Sokolskaja, E., Andreotti, A. & Luban, J. (2004). Immunity, 21, 189–201. [DOI] [PubMed]
- DeLano, W. L. (2002). PyMOL http://www.pymol.org.
- Demeler, B. (2005). Analytical Ultracentrifugation, edited by D. J. Scott, S. E. Harding & A. J. Rowe, pp. 210–230. Cambridge: Royal Society of Chemistry.
- Eckert, B., Martin, A., Balbach, J. & Schmid, F. X. (2005). Nature Struct. Mol. Biol. 12, 619–623. [DOI] [PubMed]
- Fischer, G. & Aumüller, T. (2003). Rev. Physiol. Biochem. Pharmacol. 148, 105–150. [DOI] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Frese, S., Schubert, W. D., Findeis, A. C., Marquardt, T., Roske, Y. S., Stradal, T. E. & Heinz, D. W. (2006). J. Biol. Chem. 281, 18236–18245. [DOI] [PubMed]
- Jones, T. A., Zou, J.-Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed]
- Joseph, R. E., Ginder, N. D., Hoy, J. A., Nix, J. C., Honzatko, R. B. & Andreotti, A. H. (2011). Acta Cryst. F67, 269–273. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (1976). Acta Cryst. A32, 922–923.
- Koo, B.-K., Park, C.-J., Fernandez, C. F., Chim, N., Ding, Y., Chanfreau, G. & Feigon, J. (2011). J. Mol. Biol. 411, 927–942. [DOI] [PMC free article] [PubMed]
- Laue, T. M., Shah, B. D., Ridgeway, T. M. & Pelletier, S. L. (1992). Analytical Ultracentrifugation in Biochemistry and Polymer Science, edited by S. E. Harding, A. J. Rowe & J. C. Horton, pp. 90–125. Cambridge: Royal Society of Chemistry.
- Lu, K. P. (2000). Prog. Cell Cycle Res. 4, 83–96. [DOI] [PubMed]
- Lummis, S. C., Beene, D. L., Lee, L. W., Lester, H. A., Broadhurst, R. W. & Dougherty, D. A. (2005). Nature (London), 438, 248–252. [DOI] [PubMed]
- Mallis, R. J., Brazin, K. N., Fulton, D. B. & Andreotti, A. H. (2002). Nature Struct. Biol. 9, 900–905. [DOI] [PubMed]
- Marengere, L. E. & Pawson, T. (1994). J. Cell Sci. Suppl. 18, 97–104. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Storoni, L. C. & Read, R. J. (2005). Acta Cryst. D61, 458–464. [DOI] [PubMed]
- McRee, D. E. (1999). J. Struct. Biol. 125, 156–165. [DOI] [PubMed]
- Min, L., Wu, W., Joseph, R. E., Fulton, D. B., Berg, L. & Andreotti, A. H. (2010). J. Immunol. 184, 4228–4235. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nelson, C. J., Santos-Rosa, H. & Kouzarides, T. (2006). Cell, 126, 905–916. [DOI] [PubMed]
- OuYang, B., Pochapsky, S. S., Dang, M. & Pochapsky, T. C. (2008). Structure, 16, 916–923. [DOI] [PMC free article] [PubMed]
- Pastorino, L., Sun, A., Lu, P.-J., Zhou, X. Z., Balastik, M., Finn, G., Wulf, G., Lim, J., Li, S.-H., Li, X., Xia, W., Nicholson, L. K. & Lu, K. P. (2006). Nature (London), 440, 528–534. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Pletneva, E. V., Sundd, M., Fulton, D. B. & Andreotti, A. H. (2006). J. Mol. Biol. 357, 550–561. [DOI] [PubMed]
- Read, R. J. (2001). Acta Cryst. D57, 1373–1382. [DOI] [PubMed]
- Sarkar, P., Reichman, C., Saleh, T., Birge, R. B. & Kalodimos, C. G. (2007). Mol. Cell, 25, 413–426. [DOI] [PMC free article] [PubMed]
- Sarkar, P., Saleh, T., Tzeng, S. R., Birge, R. B. & Kalodimos, C. G. (2011). Nature Chem. Biol. 7, 51–57. [DOI] [PMC free article] [PubMed]
- Schiering, N., Casale, E., Caccia, P., Giordano, P. & Battistini, C. (2000). Biochemistry, 39, 13376–13382. [DOI] [PubMed]
- Severin, A., Joseph, R. E., Boyken, S., Fulton, D. B. & Andreotti, A. H. (2009). J. Mol. Biol. 387, 726–743. [DOI] [PMC free article] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Storoni, L. C., McCoy, A. J. & Read, R. J. (2004). Acta Cryst. D60, 432–438. [DOI] [PubMed]
- Strong, M., Sawaya, M. R., Wang, S., Phillips, M., Cascio, D. & Eisenberg, D. (2006). Proc. Natl Acad. Sci. USA, 103, 8060–8065. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed]
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed]
- Waksman, G., Kominos, D., Robertson, S. C., Pant, N., Baltimore, D., Birge, R. B., Cowburn, D., Hanafusa, H., Mayer, B. J., Overduin, M., Resh, M. D., Rios, C. B., Silverman, L. & Kuriyan, J. (1992). Nature (London), 358, 646–653. [DOI] [PubMed]
- Weiss, M. S., Jabs, A. & Hilgenfeld, R. (1998). Nature Struct. Biol. 5, 676. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: interleukin-2 tyrosine kinase Src homology 2 domain, 3s9k