

Abstract
Recent findings suggest that the relaxin-3 neural network may represent a new ascending arousal pathway able to modulate a range of neural circuits including those affecting circadian rhythm and sleep/wake states, spatial and emotional memory, motivation and reward, the response to stress, and feeding and metabolism. Therefore, the relaxin-3 receptor (RXFP3) is a potential therapeutic target for the treatment of various CNS diseases. Here we describe a novel selective RXFP3 receptor positive allosteric modulator (PAM), 3-[3,5-Bis(trifluoromethyl)phenyl]-1-(3,4-dichlorobenzyl)-1-[2-(5-methoxy-1H-indol-3-yl)ethyl]urea (135PAM1). Calcium mobilization and cAMP accumulation assays in cell lines expressing the cloned human RXFP3 receptor show the compound does not directly activate RXFP3 receptor but increases functional responses to amidated relaxin-3 or R3/I5, a chimera of the INSL5 A chain and the Relaxin-3 B chain. 135PAM1 increases calcium mobilization in the presence of relaxin-3NH2 and R3/I5NH2 with pEC50 values of 6.54 (6.46 to 6.64) and 6.07 (5.94 to 6.20), respectively. In the cAMP accumulation assay, 135PAM1 inhibits the CRE response to forskolin with a pIC50 of 6.12 (5.98 to 6.27) in the presence of a probe (10 nM) concentration of relaxin-3NH2. 135PAM1 does not compete for binding with the orthosteric radioligand, [125I] R3I5 (amide), in membranes prepared from cells expressing the cloned human RXFP3 receptor. 135PAM1 is selective for RXFP3 over RXFP4, which also responds to relaxin-3. However, when using the free acid (native) form of relaxin-3 or R3/I5, 135PAM1 doesn't activate RXFP3 indicating that the compound's effect is probe dependent. Thus one can exchange the entire A-chain of the probe peptide while retaining PAM activity, but the state of the probe's c-terminus is crucial to allosteric activity of the PAM. These data demonstrate the existence of an allosteric site for modulation of this GPCR as well as the subtlety of changes in probe molecules that can affect allosteric modulation of RXFP3.
Introduction
RXFP3, also known as GPCR 135 [1] or the SALPR [2], is the cognate receptor for relaxin-3 (Relaxin-3). Relaxin-3 and related receptors will be referred to by their IUPHAR terminology [3]. RXFP3 is a class A, Gi/o linked GPCR with an extensive distribution in the central nervous system (CNS), particularly in areas involved in memory, sensory and emotional processing [4]. The CNS distribution of RXFP3 is conserved in mice and macaques [5], [6]. RXFP4, the INSL5 receptor, is the closest homolog to RXFP3 [7].
Relaxin-3 is a member of the insulin superfamily, which includes insulin, insulin-like growth factor, the relaxin hormone (H1/H2 Relaxin) and insulin-like peptides (INSL-3, 4, 5 and 6). In addition to RXFP3, relaxin-3 is capable of high affinity interaction with RXFP4. However, RXFP4 and its native ligand (INSL5) are thought to be more active in the periphery [7]. RXFP4 is a pseudogene in the rat [8] and does not appear to be expressed in the mouse brain [9]. While Relaxin-3 shares part of its name with H1/H2 Relaxin, it has a distinct distribution and roles compared to the hormonal H1/H2 Relaxin. H1/H2 Relaxin acts through RXFP1 [10] and has important roles in collagen remodeling, impacting diverse physiological processes from pregnancy [11] to asthma [12] and heart failure [13]. Relaxin-3 is highly and focally expressed in the nucleus incertus of the hind brain of the mouse [6], [14], rat [1], and an equivalent area of the macaque [5]. Apart from the Relaxin-3 distribution, behavioral data suggest that Relaxin-3 has a role in stress/anxiety [15], cognition [16] and appetite regulation [17], [18]. The tissue distribution and function of Relaxin-3 and RXFP3 indicate potential therapeutic application of RXFP3 modulators to treat stress/anxiety, cognitive disorders and metabolic diseases [19]. In vivo agonist stimulation of RXFP3 in rodents increases hippocampal theta emissions and improves performance in behavioral cognitive assays [16], [20].
Selective orthosteric, high affinity agonist (R3/I5) and antagonist (R3(Δ23–27)R/I5) peptides have been discovered for RXFP3 [21], [22]. The selective agonist (R3/I5) pairs the INSL-5 A chain with Relaxin-3's B chain, yielding a selective, high affinity, full RXFP3/4 agonist. Residues 23 through 27 of the Relaxin-3 B chain are necessary for agonist activity and removing those resides from R3/I5 creates a competitive antagonist (R3(Δ23–27)R/I5). While these peptide modulators of RXFP3/4 have been useful, they must be administered i.c.v. Small molecule drugs for these receptors have yet to be described in the literature.
GPCR agonists and antagonists can act either orthosterically, at the same site as the native agonist, or allosterically at a separate site on the receptor. Positive allosteric activity can take the form of allosteric agonism (activity in the absence of an orthosteric agonist) or positive allosteric modulation (PAM; requires presence of an orthosteric agonist) [23], [24], [25]. Positive allosteric modulators have several theoretical advantages over direct agonists as therapeutic agents. As PAM compounds modify the efficacy of an orthosteric agonist and lack activity on their own, these compounds are expected to have an effect only in tissues where and when the endogenous agonist is released. The ability of PAM compounds to exhibit biological activity in a regulated manner enhances their safety profile and makes them attractive tools over traditional therapeutic agents [26].
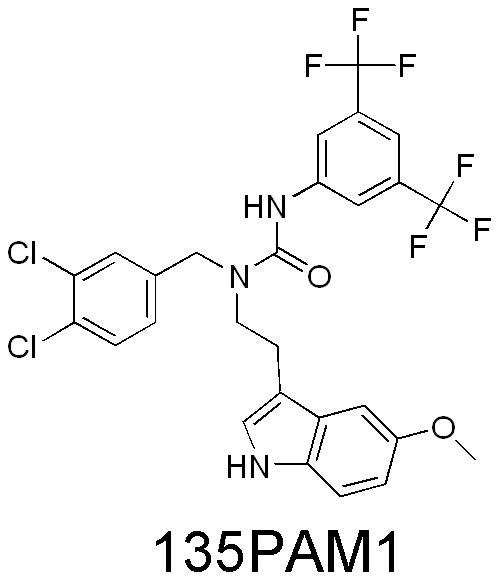
In the case of positive allosteric modulation, the allosteric activity is measured in the presence of an orthosteric agonist, referred to as a probe. Examples of PAM compounds with probe-specific activities have been described in the literature [27]. Here we describe a RXFP3 PAM compound (135PAM1, Figure 1) which is a striking example of probe selectivity. In this case, one can exchange the entire A-chain of the probe peptide while retaining PAM activity, but the state of the probe's c-terminus is crucial to allosteric activity of the PAM.
Figure 1. Structure of 135PAM1 (3-[3,5-Bis(trifluoromethyl)phenyl]-1-(3,4-dichlorobenzyl)-1-[2-(5-methoxy-1H-indol-3-yl)ethyl]urea).
Materials and Methods
Materials
All cell culture supplies, Fluo-3 AM calcium dye and Pluronic acid F-127 were obtained from Invitrogen (Carlsbad, CA). Probenecid and forskolin were obtained from Sigma (St. Louis, MO). Chlorophenol red-β-D-galactopyranoside (CPRG) was purchased from Roche Applied Science (Indianapolis, IN). C-terminally amidated Relaxin-3 (relaxin-3NH2) and R3/I5 (R3/I5NH2) were obtained from Dr. John Wade at the Howard Florey Institute (Melbourne, Australia). The C-terminal free acid forms of Relaxin-3 and R3/I5 were purchased from Peprotech (Rocky Hill, NJ) and Phoenix Pharmaceuticals (Burlingame, CA), respectively. For these studies both the A and B chains of the peptide were amidated (i.e. Relaxin-3NH2) or left in the free acid form (i.e. Relaxin-3OH). Amidated R3/I5 (R3/I5NH2) was radioiodinated with Na125I using the chloramine T methodology as previously described [28]. 135PAM1 (3-[3,5-Bis(trifluoromethyl)phenyl]-1-(3,4-dichlorobenzyl)-1-[2-(5-methoxy-1H-indol-3-yl)ethyl]urea) was synthesized and prepared at Johnson & Johnson Pharmaceutical Research and Development, L.L.C.
Cell Lines
Stable cell lines expressing RXFP3 and RXFP4 were created as previously described [1], [8], [29]. Clonal, stable cell lines were made using the receptor genes of interest, cloned into pCIneo (Promega, Madison, WI). For functional assays measuring inhibition of the cAMP pathway, the receptor constructs were transfected into SK-N-MC cells (ATCC, Manassas, VA) expressing a CRE reporter linked to β-galactosidase expression. Other assays were performed using stable clones of HEK-293 cells (ATCC, Manassas, VA) coexpressing the receptor gene of interest (RXFP3 or RXFP4) and chimeric Gα protein GqI5 [30].
Ca2+ Mobilization Assay
Intracellular Ca2+assays were used to assess the effect of 135PAM1 to either increase the response to a probe quantity of an agonist (∼EC20) or to measure changes in an agonist's EC50 at various compound doses. In either case, adherent HEK-293 cells expressing GqI5 were grown to confluency in DMEM media supplemented with 10% fetal bovine serum, 50 iu/ml penicillin, 50 µg/ml streptomycin, 1 mM Na Pyruvate, 10 mM HEPES, and 600 µg/ml G418. On the day before the assay the cells were detached with 10 mM trypsin/EDTA and seeded on black poly-D-lysine-coated 96-well plates (BD Bioscience, San Jose, CA) at a density of 35,000 cells/well, then incubated overnight at 37°C, 5% CO2. On the next day, the culture medium was discarded and replaced with a calcium dye solution, containing 4 µM Fluo-3 (AM), 2.5 mM probenecid and 0.04% Pluronic acid F-127 in DM:F12 medium. Cells were incubated in the dye solution for 60 min at room temperature or 30 min at 37°C. Ligand-stimulated Ca2+ mobilization was monitored using FLIPR Tetra High Throughput Cellular Screening System (Molecular Devices, Sunnyvale, CA).
An initial assay assessed the effect of 135PAM1 to increase the effect of an EC20 dose of Relaxin-3 or R3/I5 (each as the c-terminal acid or amide). In this case, the cells were incubated with a dose-response of 135PAM1 (0 to 20 µM) for 10 min at room temperature, then Relaxin-3 or R3/I5 was added by the FLIPR Tetra (Molecular Devices, Sunnyvale, CA) as Ca2+responses were recorded.
Shifts in the dose response curve of the orthosteric agonist were assayed using the HEK-293 cell lines co-expressing GqI5. In this case, the cells were incubated with 135PAM1 at 0, 0.2, 2 and 20 µM for 10 min at room temperature. The FLIPR Tetra then added full dose-response curves of the orthosteric agonist (H3 relaxin or R3/I5; 0 to 200 nM) at each concentration of 135PAM1 while recording data.
Results of 3 or more triplicate assays were plotted and calculated using GraphPad Prism version 5.03 (GraphPad, La Jolla, CA). The allosteric site Kb was calculated from agonist curve shift assays using the Christopoulos/Kenakin equation [23]. This calculation accounts for the limiting character of the shift in the orthosteric agonist's EC50 due to allosteric modulation. Results were analyzed together after normalization and are reported with confidence intervals in parenthesis.
cAMP Reporter Assay
Alterations in cAMP response element (CRE) activity were used to assess the functional effects of peptide agonists and 135PAM1 using methods previously described [1], [8], [29]. For these assays, stable clones of SK-N-MC cells (ATCC, Manassas, VA) were selected to express the receptor of interest (RXFP3 or RXFP4) along with a reporter gene linking CRE activity to β-galactosidase expression. CRE activity was stimulated with forskolin in cells expressing RXFP3 or RXFP4 and then inhibition of the CRE activity by agonist was tested with and without 135PAM1. In this reporter assay CRE activity is reflected in β-galactosidase expression. After incubations with compound and/or peptide agonist for 6 hours at 37 C the medium on the cells was discarded. The cells were then lysed and the assay was developed as previously described using the colored substrate chlorophenol red-β-D-galactopyranoside (CPRG) [1].
As described above for Ca2+ assays, CRE activity was measured to assess dose related effects of 135PAM1 on peptide agonists at probe concentrations (EC20) and to assay 135PAM1-dependent shifting of the agonist dose response curves. In both cases the cells were grown in MEM supplemented with 10% fetal bovine serum, 50 iu/ml penicillin, 50 µg/ml streptomycin, 2 mM L-Glutamine, 1× MEM Non-Essential Amino Acids (Gibco/InVitrogen, Carlsbad, CA), 1 mM Sodium Pyruvate, and 600 µg/ml G418. At confluency, the cells were detached and seeded in 96-well plates at a cell density of 50,000 cells/well for incubation overnight at 37°C, 5% CO2. On the following day, the media was discarded and cells were pre-incubated with 135PAM1 at the indicated concentration for 10 min at room temperature. Immediately thereafter, relaxin3 or R3/I5 was added as indicated and incubated for 10 min at room temperature. The cells were then stimulated with forskolin (5 µM, final concentration) and incubated at 37°C, 5% CO2 for 6 hours. Following washes with assay buffer, the cells were lysed and β-galactosidase activity was visualized by incubation with CPRG for up to 30 min at room temperature and absorbance was read at 570 nm. Results of 3 or more triplicate functional assays were analyzed together after normalization by non-linear regression (Prism, GraphPad, San Diego, CA) and are reported with confidence intervals in parenthesis.
Radioligand Receptor Binding Assay
Membrane homogenates were prepared from confluent 15 cm tissue culture dishes of HEK-293 cells expressing GqI5 (see above for Ca2+ functional assays). Cells were removed from the tissue culture dishes with DPBS+5 mM EDTA (pH 7.4), then centrifuged and stored at −80 C. On the day of the assay 14 ml of homogenization buffer (50 mM Tris HCl+5 mM EDTA pH 7.5+Complete Protease Inhibitor cocktail (Roche, South San Francisco, CA) was added to the cell pellet, which was then homogenized for 15 seconds using a Handishear homogenizer (Virtis). The homogenate was spun at 500 xg for 5 minutes at 4 C, then the supernatant fraction was spun at 27000 xg for 30 minutes at 4 C. The resulting membrane pellet was resuspended in 16 ml of homogenization buffer with Compete Protease Inhibitors, briefly homogenized using the Handishear (Virtis), and then kept on wet ice.
Membrane homogenates were used to measure homologous or heterologous binding displacement of [125I]R3I5NH2. Peptides and tracer were diluted in assay buffer (50 mM Tris HCl+5 mM EDTA pH 7.5) with 0.5% BSA added. Reactions were set up in a total volume of 100 µl. The PAM compound was diluted in 50 C assay buffer as a 5× concentrate and added to the binding reaction promptly to avoid precipitation. [125I]R3I5(amide) was added to a net concentration of 14 pM. Binding reactions were incubated for 90 minutes at room temperature, then filtered through GF/C membranes pretreated with polyethyleneimine using a 96 well Brandel apparatus (Brandel, Gaithersberg, MD). The filter plates were dried, then Microscint (Perkin Elmer, Shelton, CT) was added and the plates were counted on a Top Count (Perkin Elmer, Shelton, CT). Data were evaluated using GraphPad Prism 5.01 (GraphPad Software, San Diego, CA). Results of 3 or more triplicate functional assays were analyzed by non-linear regression (GraphPad Prism). Results were analyzed together after normalization and are reported with confidence intervals in parenthesis. Calculation of a cooperativity value between 135PAM and the radioligand peptide was calculated using the allosteric modulator equation based on the ternary complex model in GraphPad Prism.
Results
135PAM1 is a RXFP3 positive allosteric modulator with unique probe dependence
135PAM1's positive allosteric activity was demonstrated in functional assays using probe quantities of the orthosteric agonists and in dose response assays showing shifts in orthosteric agonist pEC50 values with increasing quantities of compound. Functional positive allosteric activity was retained following substitution of the entire A-chain of relaxin-3 (e.g. R3/I5NH2), however c-terminal amidation of the RXFP3 orthosteric agonist peptide was required for PAM activity of 135PAM1.
First, ascending doses of 135PAM1 were shown to increase calcium mobilization elicited by a probe concentration of Relaxin-3 (amide or free-acid) or R3I5 (amide or free-acid) (Figure 2). HEK293 cells co-expressing RXFP3 and Gqi5 were incubated with increasing concentrations of 135PAM1, followed by addition of relaxin-3 or R3/I5 at the EC20 (the EC20 was determined using a separate plate on the day of the assay). 135PAM1 potentiated the RXFP3-mediated calcium response to amidated relaxin-3 and R3/I5 (Fig. 2 A and C), but not to free acid analogs of either peptide (Fig. 2 B and D). As shown in Fig. 2A, 135PAM1 lacked agonist activity in the absence of a RXFP3 orthosteric agonist. 135PAM1 was more potent at RXFP3 in the presence of relaxin-3NH2 (pEC50 6.55 [6.46 to 6.64]) than R3/I5NH2 (pEC50 6.07 [5.94 to 6.20]). The EMax of 135PAM1 in the presence of Relaxin-3NH2 at its EC20 was significantly less than the EMax of the peptide (P<0.01; Relaxin-3NH2 EMax = 25811 AU [24571–27051], 135PAM1 EMax with probe = 22362 AU [21642–25221]). Similarly, the EMax of 135PAM1 with R3/I5NH2 at its EC20 was significantly less than the EMax of R3/I5NH2 (P<0.03; R3/I5NH2 EMax = 23086 AU [22257–23914], 135PAM1 EMax with probe = 21194 AU [19389–22999].
Figure 2. 135PAM1 increases the intracellular Ca2+ response to amidated, but not free acid RXFP3 agonists in cells coexpressing RXFP3 and GqI5.

Intracellular Ca2+ responses by HEK-293 cells coexpressing RXFP3 and GqI5 were measured in response to escalating concentrations of 135PAM1 using probe (EC20) concentrations of Relaxin-3NH2 (A), Relaxin-3OH (B), R3/I5NH2 (C), or R3/I5OH (D).
135PAM1 also shifted the relaxin-3NH2 and R3I5NH2 concentration-response curves in a limiting manner that is indicative of allosteric activity (Figure 3). For these experiments, cells from the same clone of HEK-293 cells expressing RXFP3 and GqI5 were preincubated with fixed concentrations of 135PAM1 (0, 0.2, 2.0, 20 µM) and then stimulated with increasing concentrations of relaxin-3NH2 or R3/I5NH2. 135PAM1 elicited a similar dose-dependent leftward shift in the relaxin-3NH2 (1.4–2.3 fold) and R3/I5NH2 (1.3–2.6 fold) concentration-response curves (Fig. 3 A and B), but did not significantly shift free agonist peptide dose response curves (Fig. 3C and 3D). An allosteric ternary complex model estimated the RXFP3 pK b value of 135PAM1 to be 6.45 (6.16 to 6.74) using the relaxin-3NH2 probe or 6.02 (5.87 to 6.18) for the R3/I5NH2 probe.
Figure 3. 135PAM1 shifts the concentration response curves of Relaxin-3NH2 and R3/I5NH2.

HEK-293 cells coexpressing RXFP3 and GqI5 were incubated with fixed concentrations of 135PAM1 (0, 0.2, 2 and 20 µM) 10 min before the addition of increasing concentrations of Relaxin-3NH2 (A), R3I5NH2 (B), Relaxin-3OH (C) or R3I5OH (D).
RXFP3 is a Gi/o linked GPCR [31], [32], therefore a cAMP response element (CRE) reporter assay was used to confirm the positive allosteric modulator activity of 135PAM1 in bioassays avoiding use of a chimeric G-protein. Results of these studies were consistent with those obtained above using GqI5.
Similar to the Ca2+ mobilization assays described above, CRE activity was first evaluated using escalating concentrations of 135PAM1 and probe concentrations of relaxin-3. When added to 10 nM Relaxin-3NH2, 135PAM1 inhibited the CRE response to 5 µM forskolin with a pIC50 of 6.12 (5.98 to 6.27), but had no effect on the CRE response to Relaxin-3OH (see Fig. 4 A and C). The EMax observed for inhibition of the CRE response to 5 µM forskolin by 135PAM1 at 30 µM was less than the EMax of Relaxin-3NH2, however the dose response curve had not yet reached its asymptote and testing of 135PAM1 at a higher concentration was not possible due to solubility limitations (Fig. 4A). While calculation of the 135PAM1 EMax is impractical in this case, the results are similar to those observed above for Ca2+ measurements in HEK293 cells co-expressing RXFP3 and Gqi5.
Figure 4. 135PAM1 dose-dependently augments the inhibition of CRE activity of probe (EC20) concentrations of Relaxin-3NH2 in cells expressing RXFP3, but not RXFP4.

As RXFP3 and RXFP4 are Gi/o-linked receptors, experiments analogous to Fig. 2 were performed without the use of a chimeric G protein, comparing cAMP response element (CRE) activity in cells expressing RXFP3 (panels A and C) to closely related receptor RXFP4 (panels B and D). RXFP3 or RXFP4 receptors were expressed in SK-N-MC cells co-expressing a CRE construct linked to β-galactosidase expression. Forskolin (5 µM) was used to stimulate CRE activity and ascending doses of 135PAM1 were added to probe (EC20) concentrations of Relaxin-3NH2 (A and B) or Relaxin-3OH (C and D).
135PAM1 also shifted Relaxin-3NH2 (Fig. 5A) and R3/I5NH2 (Fig. 5B) inhibition of forskolin-stimulated CRE responses to the left, increasing the efficacy of these peptide agonists. An allosteric ternary complex model estimated the RXFP3 pK b value of 135PAM1 to be 5.59 (5.37 to 5.80) when shifting the relaxin-3NH2 curve or 5.05 (4.70 to 5.40) when shifting the R3/I5NH2 curve.
Figure 5. Shifting of amidated RXFP3 agonist concentration response curves by 135PAM1 in cells lacking chimeric G proteins.

SK-N-MC cells coexpressing RXFP3 and a reporter construct linking CRE activity to β-galactosidase were incubated with fixed concentrations of 135PAM1 (0, 0.2, 2 and 20 µM) and increasing concentrations of Relaxin-3NH2 (A) or R3I5NH2 (B).
135PAM1 has no affinity at the orthosteric binding site of the RXFP3 receptor
We assessed the ability of 135PAM1 to compete for binding with the orthosteric radioligand, [125I] R3I5 (amide), at the orthosteric site using membranes prepared from RXFP3-HEK293-Gqi5 cells (Fig. 6). 135PAM1 failed to displace 14 pM [125I] R3I5 (amide) at concentrations of up to 20 µM (Fig. 6). In contrast, the orthosteric agonist, R3I5 (amide), displaced [125I] R3I5 (amide) with a pIC50 of 8.76 (8.61 to 8.91) (Fig. 6). At higher concentrations of 135PAM1 an increase in binding was observed, consistent with positive allosterism [33]. Further calculation of these data for 135PAM1 showed positive cooperativity with an α of 1.4 [23].
Figure 6. 135PAM1 lacks affinity at the orthosteric binding site of RXFP3 receptor.

135PAM1 did not displace [125I] R3/I5NH2 at concentrations of up to 20 µM, but instead increased total binding. R3/I5NH2 displaced the tracer with a pIC50 of 8.76 (8.91 to 8.61).
135PAM1 is selective for RXFP3
Receptor selectivity of 135PAM1 was explored using RXFP4, which has the highest known sequence identity (43%) compared to RXFP3 [29]. Relaxin-3 and R3/I5 are full RXFP4 agonists. While Relaxin-3 in either the free acid or c-terminally amidated forms inhibited the CRE response to 5 µM forskolin in cells expressing RXFP4, 135PAM1 (at up to 20 µM) had no effect on either analog of relaxin-3 at EC20 concentrations (Fig. 4B and 4D).
Ancillary pharmacology was further evaluated using broad profiling screen at CEREP and in house, evaluating a total of 60 GPCRs, ion channels and transporters in radioligand binding assays, resulted in only four significant activities (dopamine transporter 83% at 10 µM, norepinephrine transporter 59% at 10 µM, serotonin 5-HT1A 74% at 10 µM and sodium channel site 2 83% inhibition at 10 µM). In addition, the compound was tested against OX2R and NPSR in an in vitro functional assay (FLIPR). In these 2 assays, when incubated in the presence of EC20 concentrations of either orexin A or NPS, 135PAM1 did not show any PAM activity at concentrations up to 20 µM.
Discussion
Neuropeptides have long been recognized to have therapeutic potential. Successful uses of peptides have been primarily in peripheral applications, for example, in the tachykinin, somatostatin, angiotensin, calcitonin gene-related peptide (CGRP), orexin, or glucagon-like peptide1 fields [34]. Manipulation of native peptide agonists to take advantage of key domains has yielded receptor antagonists such as α-helical CRF(9–41) [35] and R3(Δ23–27)R/I5 [21]. R3/I5, the RXFP3/4 agonist used in these studies, was similarly constructed to retain the domain needed for RXFP3/4 activity while eliminating the domain needed for RXFP1/2 activity [22]. Given the complex nature of receptor–ligand interactions at the orthosteric site of GPCRs, it is not surprising that the number of such ligands, especially brain penetrant agonists, is very limited. Many synthetic neuropeptide analogs exist, but most of them are peptides, with inherent limitations (i.e. bioavailability, stability, brain penetration). In the last decade, allosteric modulation of GPCRs has offered great opportunities for receptors for which the chemical space is limited. Negative allosteric modulators of CRF, CGRP, GLP1 or ghrelin receptors have been discovered [36]. When it comes to agonists, almost no neuropeptide receptor PAMs are described. L-692,429, a ghrelin receptor PAM, is one of the very few peptide receptor PAMs to be described [37]. Additionally, low molecular weight ago-allosteric compounds acting on GLP1 receptors exist [38].
Recent data point to the therapeutic potential of an RXFP3 agonist or positive modulator [39], however, selective, systemically active, nonpeptide relaxin-3 receptor ligands have not yet been discovered [39]. Here, we describe the in vitro characterization of the first RXFP3 receptor PAM, 135PAM1. 135PAM1 was discovered in an antagonist screen of more than 100,000 compounds when negative inhibition of R3/I5NH2 activity on RXFP3 was noticed. Approximately 0.1% of the compounds tested in the screen displayed the negative inhibition and 135PAM1 proved to be a selective RXFP3 positive allosteric modulator. This low-molecular-weight ligand moderately potentiates the functional responses of amidated Relaxin-3 or amidated R3/I5 in calcium mobilization and cAMP assays in cell lines expressing the cloned human RXFP3 receptor. 135PAM1 does not compete for binding with orthosteric radioligand [125I]R3/I5NH2 but instead increases its binding and shows positive cooperativity, consistent with PAM activity. However, 135PAM1 lacks similar activities when using the free acid form of relaxin-3 (the native form) or R3/I5 instead of the amidated form, indicating that the compound is subject to probe dependency related to the state of the probe's c-terminus. This compound demonstrates a site for allosteric modulation of RXFP3 and shows the importance of following such compounds using a free acid probe.
Positive allosteric modulation of RXFP3 by 135PAM1 has been shown using a chimeric GqI5 protein to shift signaling to intracellular Ca2+ and in assays measuring native Gi/o signaling. When given with probe concentrations of Relaxin-3NH2 or R3/I5NH2, 135PAM1 has an Emax slightly lower than either peptide (fig. 2A, 2C and 4A). The PAM compound shifts the concentration response of either amidated agonist peptide in a dose related fashion (fig. 3 and 5). The apparent pKb for the allosteric site is higher when using the Gqi5-linked functional assay (pKb = 6.45 for the relaxin-3NH2 shift; Fig. 3A) than in the native Gi/o-linked assay (pKb = 5.6 for the relaxin-3NH2shift; Fig. 5A). The pKb is high enough in the Gqi5-linked functional assay to show saturation of the PAM effect (Fig. 3A/B), however the limited solubility of 135PAM1 makes it impractical to demonstrate saturability of that effect in the native Gi/o-linked assay (Fig. 5A).
In addition to the Ca2+ mobilization assay, we utilized a CRE reporter assay to evaluate the pharmacology of the RXFP3 positive allosteric modulator, 135PAM1. RXFP3 is functionally coupled to the inhibition of cAMP through the inhibition of adenylate cyclase when coupling to Gi/o proteins. We observed that increasing concentrations of both Relaxin-3NH2 and R3I5NH2 inhibit CRE activation and that 135PAM1 potentiates this effect. However, CRE can be transactivated by multiple signaling pathways, including cAMP and ERK [40]. Van der Westhuizen et al demonstrated that Relaxin-3 induces ERK1/2 phosphorylation via activation of Gi/o proteins [41]. Taking this into consideration, Relaxin-3 and related peptides could induce the activation of the CRE response in our assay via ERK1/2. However, since the van der Westhuizen's group reported maximum P-ERK1/2 levels for just minutes following Relaxin-3 administration and our CRE reporter assay incubates for 6 hours at 37 C, we speculate any effect of ERK signaling observed was minimal.
The lack of affinity of 135PAM1 for the orthosteric site of RXFP3 and the lack of effect of 135PAM1 on baseline cAMP or calcium suggests 135PAM1 is a positive allosteric modulator of RXFP3 without intrinsic activity. Also, receptor selectivity of 135PAM1 at RXFP3 has been demonstrated by comparison with cells expressing RXFP4, a GPCR with high homology to RXFP3 [29]. The selectivity of the effect of 135PAM1 at RXFP3 was evidenced by its lack of effect on Relaxin-3 or R3/I5 (amidated or free acid) responses in cells stably expressing RXFP4 receptors (Fig. 4B and 4D). In addition to RXFP4, 135PAM1 was tested for activity against 60 other receptors and channels, with no remarkable activity reported.
Initial studies demonstrated that the amidated and free-acid forms of Relaxin-3 were equipotent [42]. However, recent results both here and elsewhere (Ross Bathgate, personal communication) have shown amidated RXFP3 agonists (Relaxin-3 and R3/I5) to be roughly 10 fold less efficacious (lower pEC50) than their free acid analogs. Early work with native free acids of these insulin-like peptides used recombinant material made in eukaryotic cells, which was purified to homogeneity [1], [22]. Insulin-like peptides have since become synthetically tractable [43] and such methods have allowed for modifications such as c-terminal amidation of the peptide chains. 135PAM1 was discovered using amidated peptide, as amidation of peptides tends to increase solubility, increase stability and prior data indicated amidation of RXFP3 agonists did not alter potency [42]. All materials described here were made synthetically and were checked by LCMS. Molecular masses were correct and consistent with both chains of both peptides being free acids or amidated, as indicated. The reason for the observed differential potencies of c-terminal amides versus acid peptides remains unclear.
While allosteric modulators with probe dependency have been described in the literature [27], [44], [45], 135PAM1 is a unique example. Allosteric activity is retained while substituting the entire A-chain of Relaxin-3NH2 with the A-chain of INSL-5NH2 (yielding R3/I5NH2), yet 135PAM1 is inactive when the native free acid forms of these peptides are used as probes. Perhaps the most cited examples of probe dependent allosteric modulators are for M4 acetylcholine receptors [45], [46] and the CCR5 chemokine receptor [47]. In the case of the M4 receptor, LY2033298 shows greater cooperativity with oxotremorine than with acetylcholine or xanomeline. Oxotremorine, acetylcholine and xanomeline are small molecules with substantial chemical differences. In the chemokine receptor example, 873140 shows probe dependent allosteric antagonism at CCR5 using a site common to four other known inhibitors [47]. However, the various endogenous ligands used as probes for chemokine receptors such as CCR5 are peptides with substantial sequence variability [48]. In either case the probe agonists for the receptor in question are far more variable than the differential c-terminal amidation shown here.
Probe selective allosteric modulators of the GLP-1 receptor are in some ways comparable to the probe dependence described here. Structurally similar flavonoids show positive cooperativity with GLP-1 fragments and exendin-4, but not oxyntomodulin [49]. A hydroxyl group is crucial to positive cooperativity, but this differs from the RXFP3 case in that the needed hydroxyl group is on the allosteric modulator, as opposed to the probe.
Here, we have shown the free acid and amide forms of Relaxin-3 display different potencies at the RXFP3 receptor and described 135PAM1, a novel positive allosteric modulator of RXFP3. This RXFP3 PAM is selective to RXFP3 over RXFP4 and a panel of other receptors, shows activity in assays measuring GqI5-linked changes in intracellular Ca2+ or native Gi/o linked cAMP, and shows probe selectivity. 135PAM1 shows a probe selectivity dependent on the c-terminal amidation state of the peptide agonists, unfortunately favoring c-terminal amides which have not been shown to occur in nature. 135PAM1 is the first publicly disclosed positive allosteric modulator of RXFP3. Since 135PAM1 lacks activity when used with the native, free acid form of relaxin3 and has severe solubility limitations, the compound has very limited utility. 135PAM1 would lack in vivo activity when given alone, but could be used to potentiate effects of exogenous Relaxin-3NH2 or R3/I5NH2. This example demonstrates the subtlety of differences in probe agonists which can lead to probe dependence.
Acknowledgments
The authors thank Dr. Anindya Bhattacharya for his helpful advice on GPCR pharmacology and Dr. Ross Bathgate from the Howard Florey Institute for useful discussion concerning changes in agonist pEC50 over time.
Footnotes
Competing Interests: The authors have read the journal's policy and have the following conflicts: Johnson & Johnson Pharmaceutical Research and Development funded this study. Authors are employees of Johnson & Johnson Pharmaceutical Research & Development, LLC or were employees of that firm at the time the research was performed. Studies described here were performed with materials described in Patents/Applications US 7893197 and US 2005074814. This does not alter the authors′ adherence to all the PLoS ONE policies on sharing data and materials.
Funding: There are no current external funding sources for this study.
References
- 1.Liu C, Eriste E, Sutton S, Chen J, Roland B, et al. Identification of Relaxin-3/INSL7 as an Endogenous Ligand for the Orphan G-protein-coupled Receptor GPCR135. Journal of Biological Chemistry. 2003;278:50754–50764. doi: 10.1074/jbc.M308995200. [DOI] [PubMed] [Google Scholar]
- 2.Boels K, Hermans-Borgmeyer I, Schaller HC. Identification of a mouse orthologue of the G-protein-coupled receptor SALPR and its expression in adult mouse brain and during development. Dev Brain Res. 2004;152:265–268. doi: 10.1016/j.devbrainres.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Bathgate RA, Ivell R, Sanborn BM, Sherwood OD, Summers RJ. International union of pharmacology LVII: recommendations for the nomenclature of receptors for relaxin family peptides. Pharmacological Reviews. 2006;58:7–31. doi: 10.1124/pr.58.1.9. [DOI] [PubMed] [Google Scholar]
- 4.Sutton SW, Bonaventure P, Kuei C, Roland B, Chen J, et al. Distribution of G-Protein-Coupled Receptor (GPCR)135 Binding Sites and Receptor mRNA in the Rat Brain Suggests a Role for Relaxin-3 in Neuroendocrine and Sensory Processing. Neuroendocrinology. 2004;80:298–307. doi: 10.1159/000083656. [DOI] [PubMed] [Google Scholar]
- 5.Ma S, Sang Q, Lanciego JL, Gundlach AL. Localization of relaxin-3 in brain of Macaca fascicularis: identification of a nucleus incertus in primate. J Comp Neurol. 2009;517:856–872. doi: 10.1002/cne.22197. [DOI] [PubMed] [Google Scholar]
- 6.Smith CM, Shen PJ, Banerjee A, Bonaventure P, Ma S, et al. Distribution of relaxin-3 and RXFP3 within arousal, stress, affective, and cognitive circuits of mouse brain. J Comp Neurol. 2010;518:4016–4045. doi: 10.1002/cne.22442. [DOI] [PubMed] [Google Scholar]
- 7.Liu C, Kuei C, Sutton S, Chen J, Bonaventure P, et al. INSL5 is a high affinity specific agonist for GPCR142 (GPR100). Journal of Biological Chemistry. 2005;280:292–300. doi: 10.1074/jbc.M409916200. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Kuei C, Sutton SW, Bonaventure P, Nepomuceno D, et al. Pharmacological characterization of relaxin-3/INSL7 receptors GPCR135 and GPCR142 from different mammalian species. Journal of Pharmacology and Experimental Therapeutics. 2005;312:83–95. doi: 10.1124/jpet.104.073486. [DOI] [PubMed] [Google Scholar]
- 9.Sutton SW, Bonaventure P, Kuei C, Nepomuceno D, Wu J, et al. G-Protein-Coupled Receptor (GPCR)-142 Does Not Contribute to Relaxin-3 Binding in the Mouse Brain: Further Support that Relaxin-3 Is the Physiological Ligand for GPCR135. Neuroendocrinology. 2005;82:139–150. doi: 10.1159/000091267. [DOI] [PubMed] [Google Scholar]
- 10.Hsu Sheau Y, Nakabayashi K, Nishi S, Kumagai J, Kudo M, et al. Activation of orphan receptors by the hormone relaxin. Science. 2002;295:671–674. doi: 10.1126/science.1065654. [DOI] [PubMed] [Google Scholar]
- 11.Sherwood OD. Relaxin's physiological roles and other diverse actions. Endocr Rev. 2004;25:205–234. doi: 10.1210/er.2003-0013. [DOI] [PubMed] [Google Scholar]
- 12.Royce SG, Miao YR, Lee M, Samuel CS, Tregear GW, et al. Relaxin reverses airway remodeling and airway dysfunction in allergic airways disease. Endocrinology. 2009;150:2692–2699. doi: 10.1210/en.2008-1457. [DOI] [PubMed] [Google Scholar]
- 13.Dschietzig T, Teichman S, Unemori E, Wood S, Boehmer J, et al. Intravenous recombinant human relaxin in compensated heart failure: a safety, tolerability, and pharmacodynamic trial. J Card Fail. 2009;15:182–190. doi: 10.1016/j.cardfail.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 14.Bathgate RAD, Samuel CS, Burazin TCD, Layfield S, Claasz AA, et al. Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3) gene: novel members of the relaxin peptide family. Journal of Biological Chemistry. 2002;277:1148–1157. doi: 10.1074/jbc.M107882200. [DOI] [PubMed] [Google Scholar]
- 15.Banerjee A, Shen PJ, Ma S, Bathgate RA, Gundlach AL. Swim stress excitation of nucleus incertus and rapid induction of relaxin-3 expression via CRF1 activation. Neuropharmacology. 2010;58:145–155. doi: 10.1016/j.neuropharm.2009.06.019. [DOI] [PubMed] [Google Scholar]
- 16.Ma S, Olucha-Bordonau FE, Hossain MA, Lin F, Kuei C, et al. Modulation of hippocampal theta oscillations and spatial memory by relaxin-3 neurons of the nucleus incertus. Learn Mem. 2009;16:730–742. doi: 10.1101/lm.1438109. [DOI] [PubMed] [Google Scholar]
- 17.Hida T, Takahashi E, Shikata K, Hirohashi T, Sawai T, et al. Chronic intracerebroventricular administration of Relaxin-3 increases body weight in rats. Journal of Receptors and Signal Transduction. 2006;26:147–158. doi: 10.1080/10799890600623373. [DOI] [PubMed] [Google Scholar]
- 18.McGowan BM, Stanley SA, Ghatei MA, Bloom SR. Relaxin-3 and its role in neuroendocrine function. Ann N Y Acad Sci. 2009;1160:250–255. doi: 10.1111/j.1749-6632.2008.03796.x. [DOI] [PubMed] [Google Scholar]
- 19.van der Westhuizen ET, Halls ML, Samuel CS, Bathgate RA, Unemori EN, et al. Relaxin family peptide receptors–from orphans to therapeutic targets. Drug Discov Today. 2008;13:640–651. doi: 10.1016/j.drudis.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 20.Ma S, Kastman H, Olucha-Bordonau FE, Capogna M, Hossain MA, et al. P809.824 Relaxin-3 Receptor Activation in the Central Amygdala Enhances Fear Extinction in the Rat: Implications for Relaxin-3 Control of Emotion.; 2010; San Diego, CA, USA.
- 21.Kuei C, Sutton S, Bonaventure P, Pudiak C, Shelton J, et al. R3(BD23-27)R/I5 Chimeric Peptide, a Selective Antagonist for GPCR135 and GPCR142 over Relaxin Receptor LGR7: In Vitro and In Vivo characterization. Journal of Biological Chemistry. 2007;282:25425–25435. doi: 10.1074/jbc.M701416200. [DOI] [PubMed] [Google Scholar]
- 22.Liu C, Chen J, Kuei C, Sutton S, Nepomuceno D, et al. Relaxin-3/insulin-like peptide 5 chimeric peptide, a selective ligand for G protein-coupled receptor (GPCR)135 and GPCR142 over leucine-rich repeat-containing G protein-coupled receptor 7. Molecular Pharmacology. 2005;67:231–240. doi: 10.1124/mol.104.006700. [DOI] [PubMed] [Google Scholar]
- 23.Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 24.Conn PJ, Christopoulos A, Lindsley CW. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kenakin T. Allosteric theory: taking therapeutic advantage of the malleable nature of GPCRs. Curr Neuropharmacol. 2007;5:149–156. doi: 10.2174/157015907781695973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burford NT, Watson J, Bertekap R, Alt A. Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem Pharmacol. 2011;81:691–702. doi: 10.1016/j.bcp.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Keov P, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology. 2011;60:24–35. doi: 10.1016/j.neuropharm.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Vale W, Vaughan J, Jolley D, Yamamoto G, Bruhn T, et al. Assay of Growth Hormone-releasing Factor. Methods in Enzymology: Academic Press; 1986. pp. 389–401. [DOI] [PubMed] [Google Scholar]
- 29.Liu C, Chen J, Sutton S, Roland B, Kuei C, et al. Identification of Relaxin-3/INSL7 as a Ligand for GPCR142. Journal of Biological Chemistry. 2003;278:50765–50770. doi: 10.1074/jbc.M308996200. [DOI] [PubMed] [Google Scholar]
- 30.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. Substitution of three amino acids switches receptor specificity of Gq alpha to that of Gi alpha. Nature. 1993;363:274–276. doi: 10.1038/363274a0. [DOI] [PubMed] [Google Scholar]
- 31.Matsumoto M, Kamohara M, Sugimoto T, Hidaka K, Takasaki J, et al. The novel G-protein coupled receptor SALPR shares sequence similarity with somatostatin and angiotensin receptors. Gene. 2000;248:183–189. doi: 10.1016/s0378-1119(00)00123-2. [DOI] [PubMed] [Google Scholar]
- 32.Van der Westhuizen Emma T, Sexton Patrick M, Bathgate Ross AD, Summers Roger J. Responses of GPCR135 to human gene 3 (H3) relaxin in CHO-K1 cells determined by microphysiometry. Ann N Y Acad Sci FIELD Full Journal Title:Annals of the New York Academy of Sciences. 2005;1041:332–337. doi: 10.1196/annals.1282.053. [DOI] [PubMed] [Google Scholar]
- 33.Leach K, Sexton PM, Christopoulos A. Enna SJ, editor. Quantification of Allosteric Interactions at G Protein-Coupled Receptors Using Radioligand Binding Assays. Current Protocols in Pharmacology. 2011. pp. 1.22.21–21.22.41. March 2011 ed: Wiley Online Library. [DOI] [PubMed]
- 34.Hokfelt T, Bartfai T, Bloom F. Neuropeptides: opportunities for drug discovery. Lancet Neurol. 2003;2:463–472. doi: 10.1016/s1474-4422(03)00482-4. [DOI] [PubMed] [Google Scholar]
- 35.Rivier J, Rivier C, Vale W. Synthetic competitive antagonists of corticotropin-releasing factor: effect on ACTH secretion in the rat. Science. 1984;224:889–891. doi: 10.1126/science.6326264. [DOI] [PubMed] [Google Scholar]
- 36.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 37.Holst B, Brandt E, Bach A, Heding A, Schwartz TW. Nonpeptide and peptide growth hormone secretagogues act both as ghrelin receptor agonist and as positive or negative allosteric modulators of ghrelin signaling. Mol Endocrinol. 2005;19:2400–2411. doi: 10.1210/me.2005-0059. [DOI] [PubMed] [Google Scholar]
- 38.Knudsen LB, Kiel D, Teng M, Behrens C, Bhumralkar D, et al. Small-molecule agonists for the glucagon-like peptide 1 receptor. Proc Natl Acad Sci U S A. 2007;104:937–942. doi: 10.1073/pnas.0605701104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith CM, Ryan PJ, Hosken IT, Ma S, Gundlach AL. Relaxin-3 systems in the brain - the first 10 years. Journal of Chemical Neuroanatomy. 2011 doi: 10.1016/j.jchemneu.2011.05.013. In press. [DOI] [PubMed] [Google Scholar]
- 40.Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- 41.van der Westhuizen ET, Werry TD, Sexton PM, Summers RJ. The relaxin family peptide receptor 3 activates extracellular signal-regulated kinase 1/2 through a protein kinase C-dependent mechanism. Molecular Pharmacology. 2007;71:1618–1629. doi: 10.1124/mol.106.032763. [DOI] [PubMed] [Google Scholar]
- 42.Rosengren KJ, Lin F, Bathgate RAD, Tregear GW, Daly NL, et al. Solution Structure and Novel Insights into the Determinants of the Receptor Specificity of Human Relaxin-3. Journal of Biological Chemistry. 2006;281:5845–5851. doi: 10.1074/jbc.M511210200. [DOI] [PubMed] [Google Scholar]
- 43.Wade JD, Lin F, Hossain MA, Shabanpoor F, Zhang S, et al. The chemical synthesis of relaxin and related peptides. Ann N Y Acad Sci. 2009;1160:11–15. doi: 10.1111/j.1749-6632.2009.03951.x. [DOI] [PubMed] [Google Scholar]
- 44.Kenakin T. G protein coupled receptors as allosteric proteins and the role of allosteric modulators. J Recept Signal Transduct Res. 30:313–321. doi: 10.3109/10799893.2010.503964. [DOI] [PubMed] [Google Scholar]
- 45.Suratman S, Leach K, Sexton P, Felder C, Loiacono R, et al. Impact of species variability and ‘probe-dependence’ on the detection and in vivo validation of allosteric modulation at the M4 muscarinic acetylcholine receptor. Br J Pharmacol. 2011;162:1659–1670. doi: 10.1111/j.1476-5381.2010.01184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leach K, Davey AE, Felder CC, Sexton PM, Christopoulos A. The role of transmembrane domain 3 in the actions of orthosteric, allosteric, and atypical agonists of the M4 muscarinic acetylcholine receptor. Mol Pharmacol. 2011;79:855–865. doi: 10.1124/mol.111.070938. [DOI] [PubMed] [Google Scholar]
- 47.Watson C, Jenkinson S, Kazmierski W, Kenakin T. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric noncompetitive HIV entry inhibitor. Mol Pharmacol. 2005;67:1268–1282. doi: 10.1124/mol.104.008565. [DOI] [PubMed] [Google Scholar]
- 48.Shimizu N, Gojobori T. How can human and simian immunodeficiency viruses utilize chemokine receptors as their coreceptors? Gene. 2000;259:199–205. doi: 10.1016/s0378-1119(00)00432-7. [DOI] [PubMed] [Google Scholar]
- 49.Wootten D, Simms J, Koole C, Woodman OL, Summers RJ, et al. Modulation of the glucagon-like peptide-1 receptor signaling by naturally occurring and synthetic flavonoids. J Pharmacol Exp Ther. 2011;336:540–550. doi: 10.1124/jpet.110.176362. [DOI] [PubMed] [Google Scholar]