

SUMMARY
The Arabidopsis ACCELERATED CELL DEATH 2 (ACD2) protein protects cells from programmed cell death (PCD) caused by endogenous porphyrin-related molecules like red chlorophyll catabolite or exogenous protoporphyrin IX. We previously found that during bacterial infection, ACD2, a chlorophyll breakdown enzyme, localizes to both chloroplasts and mitochondria in leaves. Additionally, acd2 cells show mitochondrial dysfunctions. In plants with acd2 and ACD2+ sectors, ACD2 functions cell autonomously, implicating a pro-death ACD2 substrate as cell non-autonomous in promoting spreading PCD. ACD2 targeted solely to mitochondria can reduce the accumulation of an ACD2 substrate that originates in chloroplasts, indicating that ACD2 substrate molecules are likely mobile within cells. Two different light-dependent reactive oxygen bursts in mitochondria play prominent and causal roles in the acd2 PCD phenotype. Finally, ACD2 can complement acd2 when targeted to mitochondria or chloroplasts, respectively, as long as it is catalytically active; the ability to bind substrate is not sufficient for ACD2 to function in vitro or in vivo. Together the data suggest that ACD2 localizes dynamically during infection to protect cells from pro-death mobile substrate molecules, some of which may originate in chloroplasts, but have major effects on mitochondria.
Keywords: Cell death, Mitochondria, Chloroplast, Hydrogen peroxide, Singlet oxygen, Red Chlorophyll Catabolite
INTRODUCTION
The Arabidopsis ACCELERATED CELL DEATH 2 (ACD2) protein, also known as Red Chlorophyll Catabolite (RCC) Reductase has multiple functions. acd2 mutants were first identified as being compromised for cell death regulation due to their spontaneous spreading cell death phenotype (Greenberg et al., 1994). Later, the ACD2 protein was identified as a component of the chlorophyll breakdown pathway (Rodoni et al., 1997; Wuthrich et al., 2000). In mature leaves, ACD2 localizes to chloroplasts, whereas in young seedlings and in roots, ACD2 is found in both chloroplasts (or plastids) and mitochondria (Mach et al., 2001; Yao and Greenberg, 2006). After Pseudomonas syringae infection or protoporphyrin IX (PPIX) treatment of mature leaves, ACD2 is induced around cell death sites and is found in chloroplasts and mitochondria (Yao and Greenberg, 2006). Since the mitochondrial form of ACD2 is larger than the chloroplast form, it is likely that unprocessed ACD2 is independently targeted to and processed in mitochondria and chloroplasts, respectively. Modulation of the levels of ACD2 strongly influences cell death caused by P. syringae and PPIX treatment: loss of ACD2 results in excessive cell death whereas its over production is cytoprotective (Greenberg et al., 1994; Mach et al., 2001; Yao et al., 2004; Yao and Greenberg, 2006).
ACD2 is involved in the conversion of RCC, a chlorophyll degradation pathway intermediate, to primary fluorescent chlorophyll catabolite (pFCC; Rodoni et al., 1997; Wuthrich et al., 2000). Details of the biochemical mechanism of ACD2 in the conversion of RCC to pFCC remain unclear; possibly ACD2 functions as a chaperone in the catalytic reaction that converts RCC to pFCC (Rodoni et al., 1997; Wuthrich et al., 2000; Oberhuber and Krautler, 2002, Pruzinska et al., 2007). From the crystal structure of ACD2, it was hypothesized that glutamic acid 154 and aspartic acid 291 are the possible substrate binding and/or catalytic sites (Sugishima et al., 2009, 2010). Excised leaves of acd2 mutants accumulate RCC and RCC-like pigments after dark incubation for several days, which promotes their accumulation (Pruzinska et al., 2007). Dark incubation also protects the pigments from light-induced fragmentation.
As compared to many other cell death mutants, the acd2 mutant is somewhat unusual in that the cell death in each leaf starts spontaneously and propagates to consume the whole leaf (Greenberg et al., 1994). The propagation of cell death lesions in acd1 mutant is similar to acd2, however, the onset of lesions in acd2 occurs earlier in development compared to acd1. Cell death in acd2 is light dependent and involves the production of hydrogen peroxide (H2O2; Mach et al., 2001; Yao and Greenberg, 2006). Assuming some RCC/RCC-like pigments or that of other substrates can accumulate in the light, their photo-activation may lead to singlet oxygen (1O2) production that could also contribute to cell death. Indeed, RCC accumulation in dark-incubated acd2 leaves is correlated with increased 1O2 generation after leaves are exposed to light (Pruzinska et al., 2007). Various chlorophyll precursors and their degradation intermediates also generate 1O2 in light, which may contribute to cell death phenotypes in several mutants (Greenberg and Ausubel, 1993; Hu et al., 1998; Ishikawa et al., 2001; Pruzinska et al., 2003; op den Camp et al., 2003; Pruzinska et al., 2007, Mur et al., 2010).
Mitochondria play a key role in cellular metabolism and also are important players in the regulation of programmed cell death (PCD, Moller, 2001; Jones, 2000; Lam et al., 2001). One of the early events in apoptotic cell death is the mitochondrial membrane permeability transition (MPT) that is induced by multiple independent pathways (Crompton, 1999; Moller, 2001) and occurs before cells exhibit apoptotic features (Arpagaus et al., 2002; Tiwari et al., 2002; Yao et al., 2004). Although the release of cytochrome c has been documented during plant PCD (Balk and Leaver, 2001; Tiwari et al., 2002), it is not always correlated with a MPT and cell death in plants (Yu et al., 2002; Yao et al. 2004). We previously characterized cell death events in acd2 protoplasts, which die in response to light with an apoptotic morphology that includes chromatin condensation and the induction of DNA fragmentation (Yao et al., 2004). Exogenous application of PPIX to acd2 protoplasts accelerates cell death with apoptotic features. After exposure of acd2 protoplasts to light, the mitochondria lose their membrane potential, accumulate H2O2, change their morphology and the cells finally die. Although acd2 cells also accumulate H2O2 in chloroplasts, this later accumulating pool of H2O2 does not contribute to cell death induction (Yao and Greenberg, 2006).
Here, we address the possible reason and role, respectively, for spreading PCD and mitochondrial dysfunctions in acd2, as well as the sites and mechanism of action of ACD2. Our data suggest an important role for ACD2’s enzymatic activity in protecting mitochondria from substrate molecules that may move between organelles and cells, and cause mitochondrial ROS that ultimately leads to cell death.
RESULTS
Disruption of the mitochondrial H2O2 partially suppresses cell death in acd2
Since chloroplast-derived H2O2 does not contribute to the acd2 cell death phenotype (Yao and Greenberg, 2006), the mitochondrial pool may be important. To test this, we increased the H2O2 scavenging capacity in acd2 mitochondria by targeting the anti-oxidant enzymes thylakoid ascorbate peroxidase (tAPX, Jespersen et al., 1997) and human catalase (HsCAT, Schriner et al., 2005) to acd2 mitochondria. As shown in Figure 1a, using purified mitochondrial fractions from the acd2/m-tAPX and acd2/m-HsCAT plants, we detected m-tAPX and m-HsCAT proteins, respectively, along with the mitochondrial marker Peroxiredoxin II F (PRX II F, Finkemeier et al., 2005). We did not detect HsCAT in chloroplasts (Figure S1), indicating that the targeting to mitochondria was robust (tAPX is normally present in chloroplasts, Jespersen et al., 1997).
Figure 1. Targeting anti-oxidant enzymes to mitochondria increases the survival of acd2 cells.
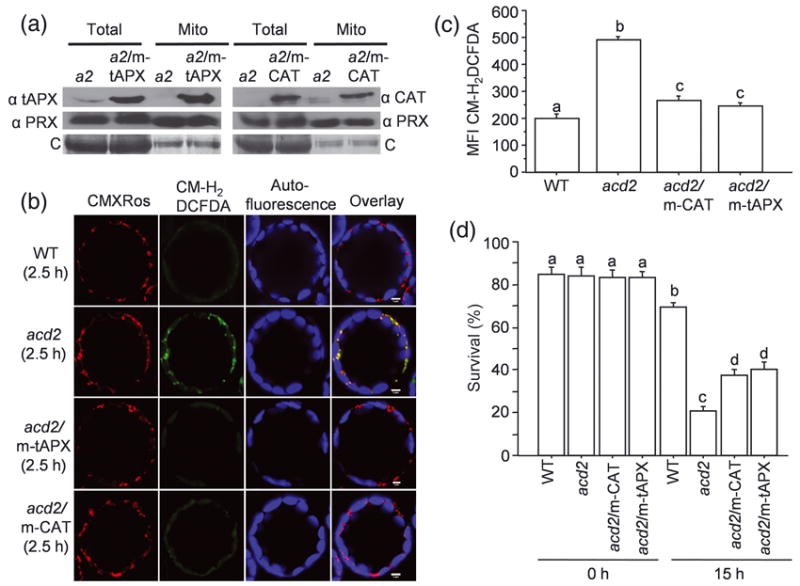
(a) Total proteins and mitochondrial proteins were isolated from acd2 (a2), a2/m-tAPX and a2/m-CAT plants and tAPX, catalase (CAT) and mitochondrial marker peroxiredoxin (PRX) II F proteins were detected by Western blot analysis. tAPX and CAT were targeted to mitochondria in the a2/m-tAPX and a2/m-CAT plants, respectively. C= Coomassie staining of the membranes. This experiment was done three times with similar results.
(b) Protoplasts isolated from 20 d-old plants were exposed to light for 2.5 h and double-stained with CM-H2DCFDA to detect H2O2 and CMXRos to mark mitochondria. Fifty to sixty protoplasts were examined under laser scanning confocal microscope (LSCM) and representative protoplasts are shown. Staining with CMXRos validated that CM-H2DCFDA-positive organelles were mitochondria. This experiment was done three times with similar results.
(c, d) The mean fluorescence intensity (MFI) of CM-H2DCFDA-stained protoplasts determined using flow cytometry (c). Protoplasts were exposed to light for 15 h and the percentage (%) of surviving cells was determined by FDA staining (d). Results are from a single analysis, representative of three independent experiments that showed similar results. The error bars represent SD (n=3). Letters indicate different values using Fisher’s Protected Least Significant Difference (PLSD) test (P < 0.004).
acd2/m-tAPX and acd2/m-HsCAT protoplasts showed very little mitochondrial H2O2 after light exposure, as compared to acd2 (Figure 1b). The mean fluorescence intensity (MFI) of the H2O2 indicator dye CM-H2DCFDA in light-exposed cells in acd2/m-tAPX and acd2/m-CAT protoplasts was reduced compared to acd2, but increased relative to wild-type protoplasts (Figure 1c). Reducing the mitochondrial H2O2 pool significantly improved the survival of acd2 protoplasts exposed to light, although wild-type protoplasts still showed the best survival (Figure 1d). Additionally, in whole plants, reduction of mitochondrial H2O2 partially rescued the acd2 cell death phenotype (Table S1).
Mitochondrial-derived 1O2 correlates with the light-induced death of acd2 cells
Since reduction in mitochondrial H2O2 did not completely rescue the acd2 cell death phenotype, we assessed the contribution of other ROS. Superoxide levels were not different in acd2 and wild-type protoplasts (Figure S2). However, 1O2 accumulates in acd2 leaves, possibly due to RCC accumulation (Pruzinska et al., 2007). To discern whether RCC can directly generate 1O2 after light exposure, we made spin-trap EPR measurements using TEMP (2, 2, 6, 6-tetramethylpiperidine) as a 1O2 trap. Figure 2a shows the light-induced 1O2 production from RCC (and PPIX, a positive control) as revealed from the EPR spectra of nitroxy radical 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO), which is formed by the reaction of 1O2 with TEMP. RCC or PPIX solutions incubated in the dark did not produce 1O2 (Figure 2a). The TEMPO intensity increased in a time-dependent manner (Figure S3).
Figure 2. Singlet oxygen (1O2) production and its contribution to the acd2 phenotype.
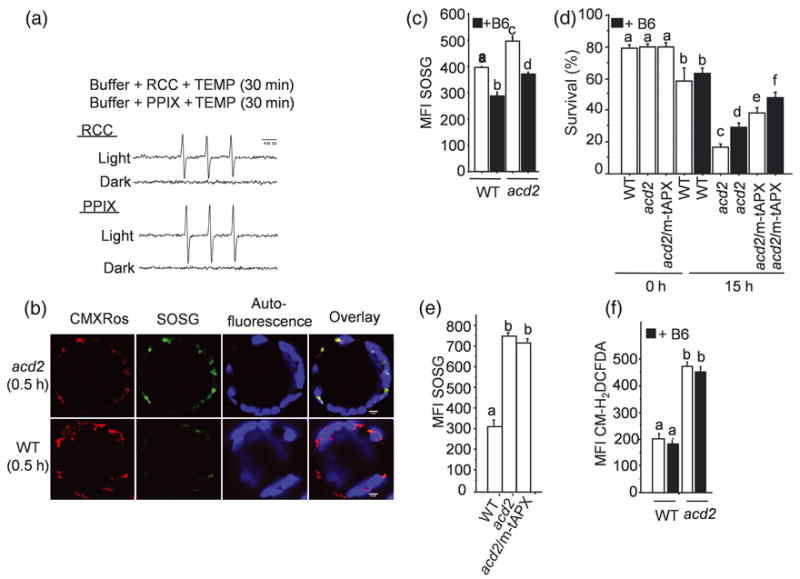
(a) 1O2 formation from red chlorophyll catabolite (RCC) and protoporphyrin IX (PPIX) after light exposure as detected by spin-trap EPR measurement. The characteristic EPR spectrum represents the TEMPO formation; TEMPO was formed by the reaction of TEMP (1O2 trap) and 1O2. Note there was no 1O2 production from RCC or PPIX in the dark. Results are from a single analysis, representative of two independent experiments that showed similar results.
(b) Protoplasts were incubated with SOSG dye to detect 1O2 and exposed to light for 0.5 h and double-stained with CMXRos. Images were assessed by LSCM. Note that the strong SOSG (green) signal from acd2 protoplasts after light exposure, which overlapped with the CMXRos signals. This experiment was done three times with similar results.
(c) The MFI of SOSG-stained protoplasts isolated from WT and acd2 leaves in the absence and presence of vitamin B6 as measured by flow cytometry.
(d) Protoplasts were treated with and without vitamin B6 for 15 h under light. Percentage (%) of cell survival was determined by FDA staining. Note the cell viability of acd2/m-tAPX was further increased in presence of B6.
(e, f) The MFI of SOSG-stained protoplasts after light exposure (0.5 h) (e) and the MFI of CM-H2DCFDA-stained light-exposed (2.5 h) protoplasts in presence and absence of vitamin B6 (f). Results (c-f) are from a single analysis, representative of three independent experiments that showed similar results. The error bars represent SD (n=3). Letters indicate different values using Fisher’s PLSD test (P < 0.002).
The cellular site of 1O2 generation in protoplasts assessed by laser scanning confocal microscope after staining them with the fluorescent probe Singlet Oxygen Sensor Green (SOSG; Flors et al., 2006; Gollmer et al., 2011), co-localized with the mitochondrial marker, MitoTracker Red CMXRos, in acd2 (Figure 2b). However, the SOSG signal in wild-type protoplasts was too low to detect a specific colocalization pattern. The SOSG MFI of light-exposed acd2 protoplasts was also higher compared to wild-type protoplasts (Figure 2c). Chloroplast-generated 1O2 can also contribute to cell death events (op den Camp et al., 2003), a process suppressed by loss of chloroplast-localized Executer proteins or by certain photoreceptors (Wagner et al., 2004; Danon et al., 2006; Lee et al., 2007). However, mutations in executers or photoreceptors did not alter cell death in acd2 (Figure S4). Thus, chloroplast-generated 1O2 does not appear to contribute to the acd2 PCD phenotype.
Application of vitamin B6 (Pyridoxine), a 1O2 quencher (Bilski et al., 2000; Danon et al., 2005), significantly reduced the MFI of SOSG in protoplasts (Figure 2c) and resulted in the increased cell survival of acd2 but not wild type (Figure 2d). The difference in B6’s effect on survival is likely due to the highly concentrated 1O2 found in acd2, but not in wild-type mitochondria. B6 further increased the cell survival of acd2/m-tAPX also (Figure 2d). This indicates that both 1O2 and H2O2 contribute to the acd2 PCD-phenotype. Interestingly, 1O2 production in acd2/m-tAPX protoplasts, assessed by quantifying the SOSG MFI, was unchanged relative to acd2 (Figure 2e). Similarly, the production of H2O2 by acd2 in the presence of vitamin B6, assessed by quantifying the CM-H2DCFDA MFI, was also unchanged relative to acd2 alone (Figure 2f). Thus, 1O2 and H2O2 are independently generated.
ACD2 targeted to chloroplasts or mitochondria reduces the acd2 cell death phenotype
During pathogen and PPIX treatment, ACD2 shifts from chloroplast localization to both chloroplasts and mitochondria (Yao and Greenberg, 2006). This, along with the mitochondrial dysfunction of acd2 cells suggests a role for ACD2 in mitochondria and the possibility that pro-death ACD2 substrate molecules are mobile within and/or between cells. To understand this further, we targeted ACD2 solely to the mitochondria or to the chloroplasts of acd2. Using total protein, we detected ACD2 (Figure 3a), which was significantly increased in acd2/m-ACD2 and acd2/c-ACD2 plants relative to wild type. m-ACD2 and c-ACD2 were correctly targeted to chloroplasts and mitochondria, respectively (Figure 3a). Chloroplasts from the mitochondria-targeted plants (acd2/m-ACD2) and mitochondria from the chloroplast-targeted plants (acd2/c-ACD2) did not contain detectable levels of ACD2 (Figure 3a). The purity of the organelles was verified using mitochondria- and chloroplast-specific markers (Figure 3a).
Figure 3. ACD2 either in mitochondria or in chloroplasts significantly suppresses the acd2 cell death phenotype.
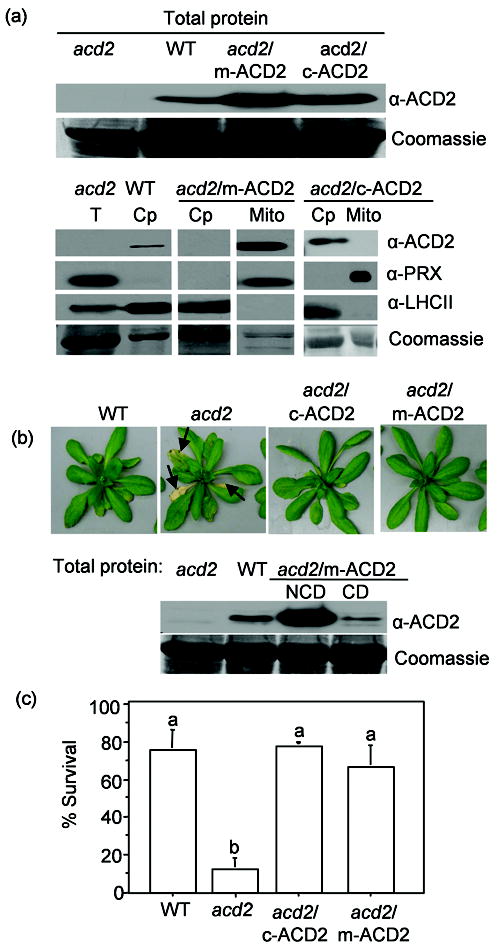
Transgenic acd2 carrying ACD2 targeting to mitochondria (acd2/m-ACD2) or chloroplasts (acd2/c-ACD2) were generated and characterized. Experiments were performed with at least two independent transgenic lines per construct; data from representative lines are presented.
(a) The ACD2 level in total protein (T) extracts (upper panel) and in chloroplasts (Cp) and mitochondria (Mito) isolated from the indicated plants was detected by Western blot analysis (lower panel). Organelle purity was assessed using antibodies against PRX II F and LHCII (chloroplasts). Signals of ACD2, PRX II F and LHC II, for all samples are from single exposures of one continuous membrane. This experiment was done three times with similar results.
(b) Phenotypes of 31-d-old plants of the indicated genotype. By day 31, about 20% of acd2/m-ACD2 showed a mild cell death phenotype (see text). ACD2 protein content in the healthy leaves of 21-d-old acd2/m-ACD2 plants that showed mild cell death (CD) and did not show cell death (NCD) on day 21 (lower panel). This experiment was done three times with similar result.
(c) Protoplasts were exposed to light for 15 h and percentage (%) of live cells was determined. This experiment was done four times with similar results and the error bars represent SD (n = 2). Letters indicate different values using Fisher’s PLSD test (P < 0.001).
The acd2/c-ACD2 plants displayed complete and stable rescue of the acd2 cell death phenotype. Most acd2/m-ACD2 plants (> 79 % of plants) were also fully rescued. A minority of acd2/m-ACD2 plants showed cell death later in development relative to acd2, which was accompanied by reduced levels of ACD2 (Figure 3b, Table S2). (Some acd2/c-ACD2 lines had similar behavior to the acd2/m-ACD2 lines, but these were not characterized further.) It is unclear why ACD2 levels decreased with age in some transgenic lines. Light-exposed acd2/c-ACD2 and acd2/m-ACD2 protoplasts showed significantly higher cell viability (Figure 3c) and very little mitochondrial H2O2 and 1O2 compared to acd2 (Figure S5). Thus, c-ACD2 and m-ACD2 significantly rescued the acd2 PCD phenotype and suppressed mitochondrial ROS.
Cytoprotective function of ACD2 in chloroplasts or mitochondria correlates with its enzymatic activity
Structural analysis of ACD2 indicates possible roles for Glu154 and Asp291 in substrate binding and/or enzyme catalysis (Sugishima et al., 2009). Therefore, we tested whether the enzymatic activity and/or substrate binding is important for the cytoprotective role of ACD2. We generated a variant of ACD2 (ACD2**) in which both Glu154 (Glu154Ala; E154A) and Asp291 (Asp291His; D291H) residues were altered. Interestingly, RCC bound equally well to ACD2** and ACD2, as assessed by tryptophan fluorescence quenching (Figure 4a). In contrast, a coupled PAO (pheophorbide a oxygenase) /ACD2 enzyme assay using pheophorbide a as substrate with PAO isolated from bell pepper fruits and ACD2 variants indicated that ACD2** and the E154A variant completely lost their enzymatic activities, since no enzymatic product, pFCC, was produced (Figure 4b).
Figure 4. Biochemical characterization of ACD2.
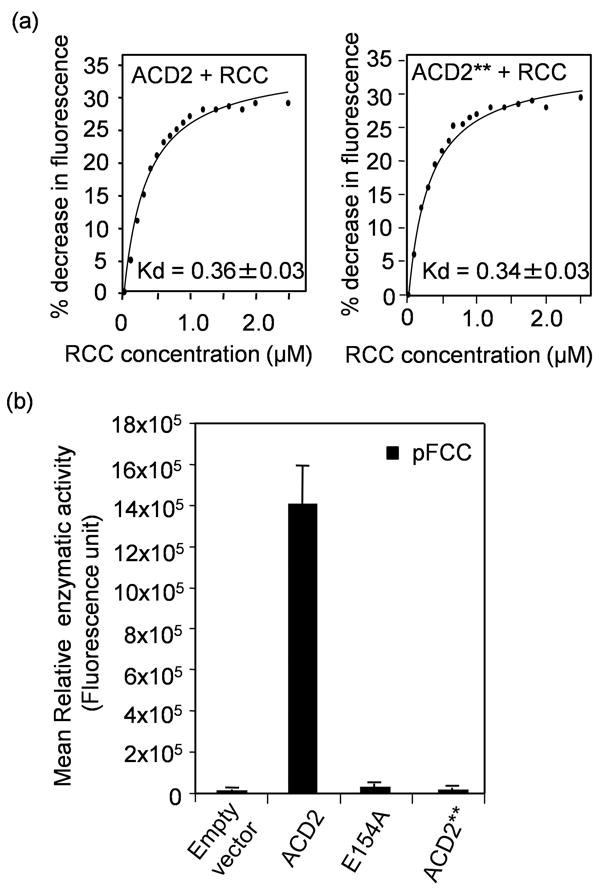
(a) Quenching of endogenous fluorescence of purified wild-type ACD240-320 and ACD2**40-320 (E154AD291H) protein, respectively, by RCC. The percentage (%) decrease in intrinsic fluorescence of the respective proteins caused by RCC binding was plotted against the concentration of RCC and the Kd was found to be 0.36 ± 0.03 μM and 0.34 ± 0.03 μM, respectively. This experiment was done twice with similar results.
(b) Activity of purified recombinant ACD2 or ACD2** or ACD2E154A proteins was assessed in a coupled assay using purified PAO and co-factors (Pruzinska et al., 2007) and pFCC was measured by HPLC. As a negative control, the vector protein alone (pQE 30 in E. coli M15) was used. Error bars represent SD (n=3). This experiment was done twice with similar results.
To discern the functional consequences of a loss of enzyme activity but not RCC binding, we targeted ACD2** to acd2 chloroplasts (acd2/c-ACD2**) and mitochondria (acd2/m-ACD2**), respectively (Figure 5a). Targeting ACD2** either to chloroplasts or mitochondria did not alter the protoplast viability (Figure 5b) or the cell death initiation or progression relative to acd2 in any of the 72 plants analyzed from each targeted line. Mitochondrial H2O2 and 1O2 were also not altered in acd2/m-ACD2** or acd2/c-ACD2** compared to acd2 (Figure S6). Thus, the cytoprotective function of ACD2 is dependent on its catalytic activity.
Figure 5. Targeting ACD2** to chloroplasts or mitochondria does not rescue the acd2 cell death phenotype.
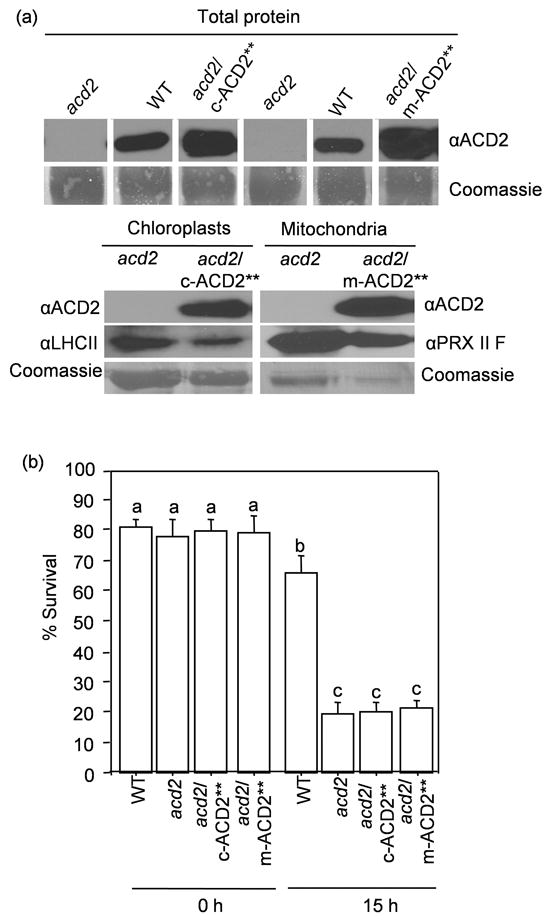
(a) The ACD2 level in total protein extracts (upper panel) and in chloroplasts and mitochondria isolated from the indicated plants was detected by Western blot analysis (lower panel). Signals for ACD2 for chloroplast-targeted as well as for mitochondria-targeted plants with its controls are from one continuous membrane (upper panel). Both chloroplast and mitochondrial organelle fractions were qualitatively checked by using chloroplast or mitochondria marker antibody (LHC II and PRX II F, respectively; lower panel). This experiment was done twice with similar results.
(b) Protoplasts were exposed to light for 15 h. Percentage (%) of surviving cells in the population was determined. This experiment was done three times with similar results. The error bars represent SD (n=3). Bars with the same letters indicate the lack of differences in values using Fisher’s PLSD test (P>0.4).
Targeting ACD2 to either chloroplasts or mitochondria reduces the accumulation of chlorophyll catabolites in acd2
Cell death in acd2 correlates with the accumulation with RCC and RCC-like pigments, which can accumulate in dark-incubated leaves (Pruzinska et al., 2007). Production of c-ACD2 or m-ACD2 in acd2 resulted in reduced RCC in the dark-incubation assay, albeit c-ACD2 was more effective at reducing RCC than m-ACD2 (Figure 6a). In contrast, RCC levels were high in acd2/c-ACD2** and acd2/m-ACD2** plants that were not rescued (Figure 6a). The ability of m-ACD2 to cause reduced RCC accumulation is likely due to the movement of RCC from chloroplasts to mitochondria (see discussion).
Figure 6. HPLC analysis of chlorophyll catabolites.
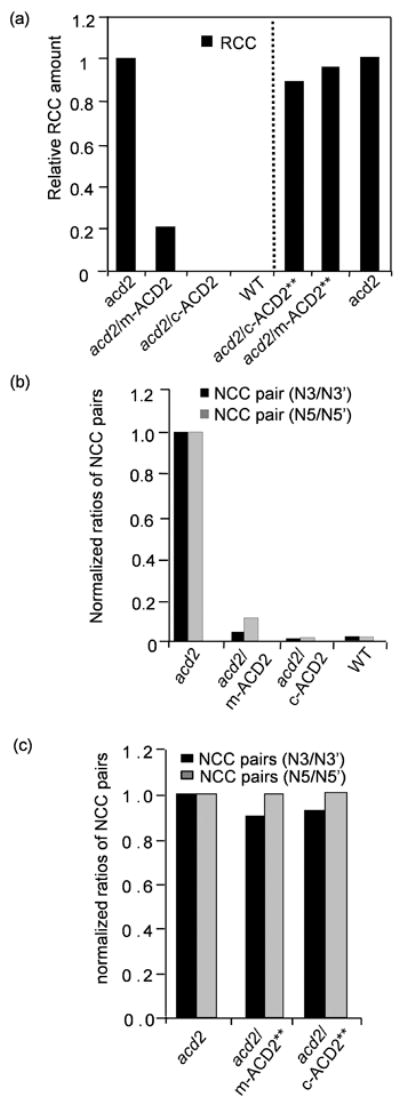
(a) RCC levels in extracts of detached leaves incubated for 4 d in dark were quantified by HPLC. Note the relatively higher level of RCC that accumulated in acd2 leaves. RCC was undetectable in WT and acd2/c-ACD2 leaves. The amount of RCC was significantly decreased in acd2/m-ACD2 leaves. RCC levels were high in acd2/c-ACD2** and acd2/m-ACD2** leaves. RCC quantification in acd2/c-ACD2** and acd2/m-ACD2** leaves was done separately from acd2/m-ACD2 and acd2/c-ACD2 extraction.
(b,c) Ratio of NCC stereoisomers in acd2/c-ACD2, acd2/m-ACD2 leaves or in acd2/c-ACD2** or acd2/m-ACD2** leaves normalized to the content in acd2. Note the ratios of NCC stereoisomers were significantly reduced in acd2/c-ACD2, acd2/m-ACD2 plants indicative of ACD2 activity in respective organelles (b). These experiments (a-c) were done twice with similar results.
RCC is converted to non-fluorescent chlorophyll catabolite (NCC) isomers either enzymatically or non-enzymatically. The ratio of isomers is diagnostic of the mechanism of NCC formation. NCC3, NCC5 and their respective epimers (NCC3’ and NCC5’) occur in acd2 leaves due to the non-enzymatic conversion of RCC (Pruzinska et al. 2007). Complementation of acd2 with ACD2 results in loss of NCC3’ and NCC5’ (Pruzinska et al., 2007). As shown in Figure 6b, the ratios of NCC stereoisomers (NCC-3’/NCC-3; NCC-5’/NCC-5) were reduced in acd2/m-ACD2 and acd2/c-ACD2 plants, but not in the acd2/c-ACD2** and acd2/m-ACD2** plants (Figure 6c). Thus, ACD2 shows RCC reductase-like activity, which depends on the catalytic site, when targeted to either chloroplasts or mitochondria.
Genetic evidence that an ACD2 substrate(s) or a signal that induces the substrate can move between cells
Suppression of acd2 PCD by mitochondria-targeted ACD2 is consistent with a pro-death ACD2 substrate migrating from chloroplasts to mitochondria. Possibly, such a pro-death ACD2 substrate(s) (or an inducer of such substrates) could also move from cell to cell explaining the spreading of PCD phenotype in acd2. Alternatively, an ACD2 substrate might initiate cell death, but spreading might be caused by a separate mobile pro-death signal that indiscriminately affects acd2 or ACD2+ cells.
To distinguish between these possible models, we created mosaic plants with acd2 sectors next to ACD2+ sectors. We generated acd2 seeds containing a cassette flanked by LOX recombination sites with ACD2 linked to a gene encoding nuclear-localized GFP that could be excised by CRE recombinase after a transient heat shock (Figure S7a). acd2 plants carrying this cassette were complemented (Figure S7b). Non-heat shocked leaves showed GFP throughout the leaves (Figure 7a). A brief heat shock resulted in the creation of various sizes of random sectors lacking GFP and hence ACD2 (Figure 7b). By visualizing the nuclear-localized GFP, we tracked the presence of ACD2 relative to regions of dying cells.
Figure 7. ACD2 acts cell autonomously in mosaic plants.
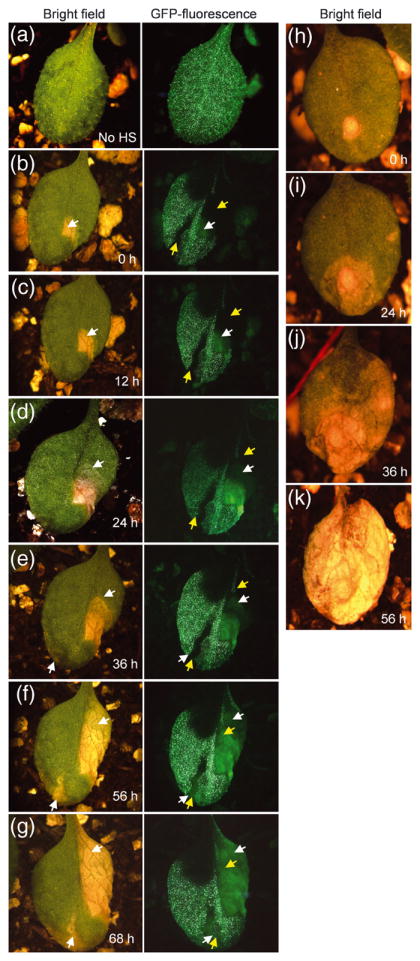
Homozygous acd2 plants carrying both heat shock inducible CRE recombinase and LOX recombination sites flanking genes encoding ACD2 and nuclear-GFP were used to study cell death patterns. The initiation and spreading of cell death lesions were followed in single leaves over 10-12 h time intervals. This experiment was done three times with similar results. Twenty-six leaves were analyzed. Lesions in panels “b” and “h” were arbitrarily assigned time 0 h.
(a) Non heat-shocked (No HS) control plant showed GFP throughout the leaf.
(b) Leaves showing GFP− sectors after heat-shock treatment. The yellow colored arrow marks the GFP− sector area. The white arrow marks the initiation of cell death. Note the cell death areas under GFP channel gave autofluorescence that was lighter in color compared to GFP and also lacked the nuclear-localization seen with GFP fluorescence.
(c) and (d) Note the spreading of cell death lesions (follow the spreading of the lesion in the bright field image and the autofluorescence area in the GFP− sectors). However, the spreading of the lesion was contained in the area with GFP.
(e) A second lesion started in the other GFP− sector (follow the white arrow in bright field as well as GFP channel).
(f) and (g) Spreading of the new lesion in the GFP− sector (f). (g) Note that the leaf is alive after 68 h of light exposure.
(h - k) Initiation and spreading of cell death in acd2. Note that the cell death spreads continuously from its starting point to consume the whole leaf. Although panel k shows a completely dead leaf at 56 h, this leaf was dead by 48 h.
In twenty-six mosaic leaves, cell death from leaves on different plants started within a sector lacking ACD2 (Figure 7b) and spread rapidly to areas that lacked ACD2 (Figure 7c-f), but not to GFP+/ACD2+ sectors (Figure 7c-f). When a new lesion started, it occurred in another sector that lacked GFP (and ACD2) and extended to cells that lacked GFP (Figure 7e,f). Mosaic leaves retained living cells even 5-7 days after the initiation of a lesion(s). In contrast, in 10 acd2 leaves closely examined, cell death spread continuously in all directions once it started and consumed the whole leaf within 3 days (Figure 7h-k). Thus ACD2 acts cell autonomously in whole leaves and can suppress the effects of possible mobile pro-PCD substrate molecules.
In planta PPIX levels are not affected in acd2 plants although PPIX binds to ACD2
Exogenous application of PPIX can initiate and accelerate the PCD events in acd2 (Yao et al., 2004, Yao and Greenberg, 2006). To test whether endogenous PPIX levels are modulated by ACD2, we analyzed its steady state level in acd2 leaves using HPLC. There was no difference in the amount of PPIX or other chlorophyll precursors in 15- or 20-days-old wild-type and acd2 leaves (Figure 8 a,b).
Figure 8. ACD2 does not affect the steady state level of protoporphyrin IX in planta, although ACD2 binds to protoporphyrin IX in vitro.
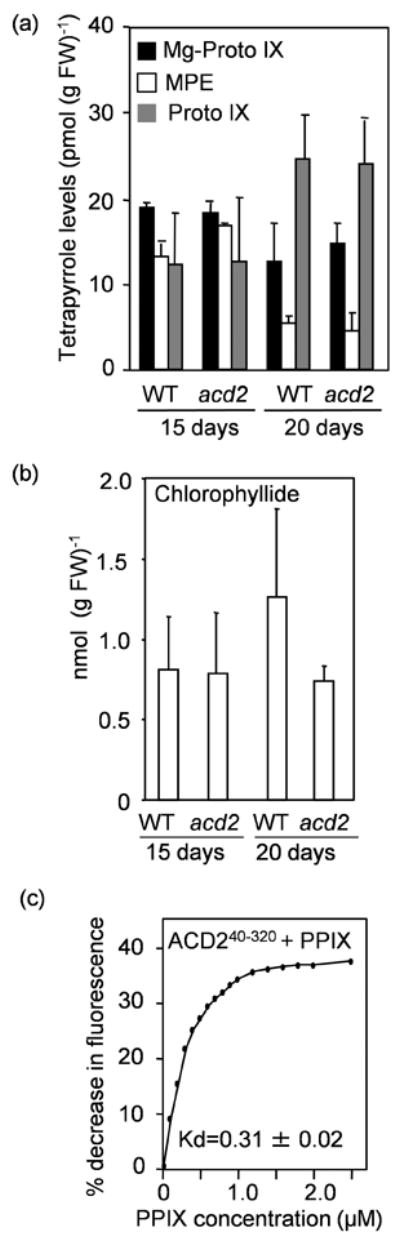
Steady-state levels of chlorophyll precursors were measured from leaves of WT and acd2 plants grown for 15 days or 20 days. This experiment was done twice with similar results. Error bars represent SD (n=2).
(a) Mg-protoporphyrin IX (Mg-Proto IX), Mg-protoporphyrin IX monomethyl ester (MPE) and protoporphyrin IX (PPIX) contents in WT and acd2 plants.
(b) Chlorophyllide contents in WT and acd2 plants.
(c) Quenching of ACD2 protein fluorescence by PPIX. The percentage (%) decrease in intrinsic fluorescence of purified ACD2 protein, due to PPIX binding, was plotted against the concentration of PPIX and the Kd was found to be 0.31 ± 0.02 μM. This experiment was done three times with similar results.
Interestingly, plants that over-express ACD2 show less PPIX-induced cell death (Yao and Greenberg, 2006). This might result from the direct binding of PPIX to ACD2. Indeed, recombinant ACD2 (Figure S8) binds to PPIX with a binding constant of 0.31±0.02 μM (Figure 8c), as determined using a tryptophan fluorescence quenching assay. Additionally, when ACD2 was purified from E. coli, a fraction of the respective proteins already harbored bound PPIX (Figures S9). Thus, ACD2 might protect plants from PPIX by a mechanism that involves direct binding.
DISCUSSION
In leaves, ACD2 is predominantly a chloroplast-localized protein. However, a major consequence of its loss in acd2 plants is manifested in the production of early mitochondrial ROS, which we showed here significantly contributes to cell death. The fact that infection induces ACD2 to localize to mitochondria as well as chloroplasts was a first hint that ACD2 may also function directly in mitochondria (Yao and Greenberg, 2006). Indeed, targeting ACD2 solely to mitochondria can largely rescue the acd2 cell death phenotypes, mitochondrial ROS and the accumulation of the ACD2 substrate RCC. Since targeting ACD2 solely in chloroplasts also rescues acd2 phenotypes, we propose that the dynamic localization found in wild-type plants serves as a fail-safe mechanism to suppress spreading cell death during times of infection. In this “fail-safe” model (Figure S10), infection causes release of a pro-death ACD2 substrate(s) from chloroplasts. Most substrate molecules can be removed by ACD2 in chloroplasts, but some may escape chloroplasts and affect mitochondria through metabolite channeling or the close proximity of chloroplasts and mitochondria (Yao and Greenberg, 2006). By targeting some ACD2 to mitochondria during infection, the substrate molecules can be removed to minimize lethal bursts of 1O2 and H2O2. A likely candidate for a pro-death ACD2 substrate is RCC, which we showed can produce light-dependent 1O2 in vitro.
The above model invokes movement of an ACD2 substrate(s) out of chloroplasts and requires that ACD2 has an enzymatic function in mitochondria. Movement of substrate is plausible, since RCC has been found in vacuoles (Pruzinska et al., 2007) and the main early dysfunction in acd2 occurs in mitochondria (Yao et al., 2004; Yao and Greenberg, 2006). Mosaic analysis also supports the possibility that a pro-death ACD2 substrate is mobile. Cells containing ACD2 can stem the spread of cell death initiated in acd2 cells of mosaic leaves, implying that a pro-death ACD2 substrate is mobile. However, we cannot rule out that acd2 cells produce a signal that moves to its neighbors where it then induces the ACD2 substrate. Our model is also consistent with the phenotype of ACD2-overexpressing plants. Such plants show reduced cell death spreading during infection, indicating that in wild-type plants the amount of ACD2 is limiting (Yao and Greenberg, 2006).
An alternative “threshold” model (Figure S10) can also explain our observation that ACD2 targeted to either chloroplasts or mitochondria rescues acd2 mutants. In this model, different pro-death ACD2 substrates originate in both organelles. The accumulation of both substrates is necessary to trigger cell death by surpassing a pro-death threshold; therefore targeting ACD2 to either organelle can suppress cell death. This model does not rule out the possibility that substrates may migrate from one organelle to another. Indeed, RCC levels are reduced in ACD2-mitochondrial-targeted plants, implying that the RCC, which originates from chlorophyll in chloroplasts, can migrate from chloroplasts to mitochondria. We previously found that ACD2 is targeted to both mitochondria and plastids in roots. acd2 root cells, which have no chlorophyll, also have mitochondrial ROS and show cell death (Yao and Greenberg, 2006), indicating there is likely at least one non-RCC substrate for ACD2 possibly arising from structurally similar bilin-derived molecules. Indeed, ACD2 is related to ferredoxin-dependent bilin reductases such as phytochromobilin reductase and shares with them a few highly conserved amino acids (Frankenberg et al., 2001). A prediction of this model is that mitochondrial-derived substrates should accumulate in acd2.
Distinguishing between these models will require the development of improved analytical and fractionation methods optimized for the analysis of metabolites that may be labile.
How might ACD2 protect mitochondria from chloroplast-derived substrate molecules? Considering its enzymatic mechanism in leaves, ACD2 converts RCC to pFCC without any cofactors to effect the transfer of electrons to RCC (Rodoni et al., 1997; Wuthrich et al., 2000). Rather the electron transfer is thought to occur from ferredoxin (Oberhuber and Krautler, 2002). It is likely that ACD2 can use a mitochondrial ferredoxin to support catalysis of RCC (or some other ACD2 substrate). The activity of ACD2 inside mitochondria correlates with the RCC reductase activity; its binding to RCC is not sufficient to rescue the PCD phenotype.
In addition to reducing RCC levels, ACD2 can protect against cell death and mitochondrial H2O2 induced by exogenous application of PPIX (Yao and Greenberg, 2006). Interestingly, plants have a translocator in their mitochondrial outer membrane that can transport PPIX into mitochondria (Lindemann et al., 2004). Additionally, PPIX treatment also induces mitochondrial targeting of ACD2 (Yao and Greenberg, 2006). We showed here that ACD2 can bind PPIX as well as its known substrate RCC. However, loss of ACD2 does not affect steady state levels of PPIX. ACD2 may affect the ability of PPIX to become photo-activated in vivo, similar to water soluble chlorophyll binding protein, which binds to free chlorophyll molecules and reduces 1O2 production (Schmidt et al., 2003). Interestingly, recombinant ACD2 co-elutes with a small fraction of PPIX, which suggests the possibility of ACD2 binding to PPIX in planta. It is also possible that ACD2 may affect PPIX mobility after binding to it, which might lead to decreased ROS production inside mitochondria.
How might the accumulation or localized release of ACD2 substrate molecules contribute to cell death? Analysis of acd2 mutant protoplasts has strongly implicated mitochondrial ROS as contributing to cell death. Interestingly, mitochondrial ROS occurs in at least two light-dependent waves, an early 1O2 burst and a later, independently-generated, H2O2 burst. 1O2 could result from the direct photoactivation of an ACD2 substrate; indeed, in vitro RCC can generate 1O2. Although 1O2 quenchers are present in plants, they mainly reside in chloroplasts (Triantaphylides and Havaux, 2009; Krieger-Liszkay and Trebst, 2006), leaving mitochondria vulnerable. H2O2 generation might result from a different mechanism. Chlorophyll degradation intermediates may directly produce H2O2 inside the mitochondria by inhibiting the mitochondrial electron transport chain. Indeed, a pheophorbide a derivative inhibits the electron transport chain and produces H2O2 in a light-dependent manner in human cell lines (Kim et al., 2004). The defense signal salicylic acid (SA) that accumulates in acd2 plants (Greenberg et al., 1994) may also contribute to H2O2 production. High SA can block mitochondrial electron transport flow resulting in ROS production (Norman et al., 2004).
Production of various kinds of ROS in different organelles may contribute to cell death through different pathways. Although acd2 mutants accumulate chloroplastic H2O2, this H2O2 pool forms later than the mitochondrial pool and does not contribute to acd2 cell death (Yao and Greenberg, 2006). Might other chloroplast ROS contribute to cell death in acd2? A possible candidate could be 1O2. However, no 1O2 was detected in acd2 chloroplasts. Additionally, triple mutants of acd2exe1exe2 and acd2cry1cry2 did not show cell death suppression, even though exe1exe2 and cry1 mutations block cell death due to chloroplast 1O2 accumulation in flu mutants that accumulate a photo-activatable chlorophyll precursor (Wagner et al., 2004; Danon et al., 2006; Lee et al., 2007).
In summary, this study highlights the involvement and importance of mitochondrial events in cell death control in plants and suggests that the location of ROS within cells can cause cell death by different mechanisms.
EXPERIMENTAL PROCEDURES
Materials
All Arabidopsis thaliana plant materials used herein were in the Columbia background. The acd2-2, phyA-211, phyB, cry1, cry2 were described previously (Greenberg et al., 1994; Neff et al., 1998; Lin et al., 1995, 1998). T-DNA insertion lines for executer1 (exe1) and executer2 (exe2) (SALK_002088 and SALK_012127, respectively) were obtained from Arabidopsis Biological Resource Center. We generated and validated double and triple mutants carrying acd2 and other mutants by crossing plants and using appropriate primers (Table S3). Plants were grown in soil (1:1 mix of C2 (Courad Fafard, Inc) and Metromix 200 (Sun Gro Horticulture)) in a growth room with a light intensity of 288 μmoles photons m-2 s-1 (from 400 watt metal halide and 400 watt sodium bulbs) and a 16 h light / 8 h dark photoperiod at 22°C ± 2°C and 50-60% relative humidity.
The primers used in this study are shown in Table S3. Protoporphyrin IX (PPIX)-disodium salt was from Frontier Scientific. TEMP, NATA (N-acetyltryptophanamide) and vitamin B6 (pyridoxine) was obtained from Sigma-Aldrich. CM-H2DCFDA (5-and- 6-chloromethyl-2’, 7’-dichlorodihydrofluorescein diacetate, acetyl ester), SOSG (singlet oxygen sensor green), MitoTracker Red CMXRos (CMXRos) were obtained from Molecular Probes.
Generation of constructs and transgenic plants
The AttAPX cDNA (Jespersen et al., 1997) lacking the chloroplast transit peptide sequences (78 aa; 234 bp) was PCR amplified from an Arabidopsis cDNA pool, restriction digested by BamHI and EcoRI and ligated into the pCB302-2 (Xiang et al., 1999) vector under the control of 35S promoter to generate the construct for mitochondrial targeting. The vector pCB302-2 carries the ß-ATPase transit peptide sequence for mitochondria targeting (Xiang et al., 1999). Similarly, the human catalase cDNA lacking the peroxisome-targeting signal was PCR amplified from the pCAGGScatalase plasmid (Schriner et al., 2005) using specific primers. The resulting PCR product was digested by SmaI and ligated into pCB302-2 to generate the construct for mitochondrial targeting. ACD2 cDNA fragment lacking 39 amino acids of chloroplast transit peptide sequences (ACD240-320) and the point mutated ACD240-320 (Glu154>Ala154, and Asp291>His291; see Methods S1) fragments were PCR amplified from the pBI121-ACD2 plasmid (Mach et al., 2001) using different pairs of primer sets. The resulting PCR products were digested with BamHI and EcoRI and ligated into pCB302-1 or pCB302-2 to generate the constructs to target chloroplasts or mitochondria, respectively. The vector pCB302-1 carries the Rubisco small subunit transit peptide sequence for chloroplast targeting (Xiang et al., 1999). Two sets of constructs were used for the mosaic analysis experiment (See Methods S1). All the constructs were introduced into Agrobacterium (GV3101), which were used to transform Arabidopsis using the floral dip method (Clough and Bent, 1998). The homozygous lines were screened in T2 generation, confirmed by Western blot analysis and used for further analysis.
Protoplast preparation, treatments and live imaging of ROS production
Leaf protoplasts were isolated in the dark from 20-days-old plants, exposed to light (100 μmoles photons m-2 s-1, at 22°C ± 2°C) and the percentage of surviving protoplasts was determined by fluorescein diacetate (500 ng/mL) staining using a hemocytometer (Hausser Scientific Company) as described (Yao et al., 2004).
Live imaging of ROS production from protoplasts
Confocal images were collected by using a laser-scanning confocal microscope (Leica TCS SP2 AOBS) with a 63X (numerical aperture 1.4) glycerol objective. After the light treatment, protoplasts were double stained with CMXRos (500 nM) and CM-H2DCFDA (1 μM) to label the mitochondria and to detect the H2O2, respectively. For 1O2 detection, protoplasts were incubated with the SOSG (100 μM) in dark for one hour, then washed with the protoplast suspension buffer and exposed to light for 30 min, double stained with CMXRos. The CM-H2DCFDA and SOSG signals were visualized with excitation at 488 nm (emission: 498-532 nm). CMXRos signals were visualized with excitation at 543 nm (emission: 495-635 nm), and chloroplast autofluorescence (488 nm excitation) was visualized at 738 to 793 nm.
Flow cytometry
Flow cytometry (FACScan, Becton–Dickinson) was used to determine the mean fluorescence intensity (MFI) of cells stained with the fluorescent dyes H2DCFDA and SOSG. A solid state 488 nm laser was used for triggering and light scatter parameters. Forward scatter, which gives a rough estimate of cell size, and side scatter, which correlates with internal complexity/granularity, was measured. From these parameters a ‘live’ gate was made to exclude cell debris. Light-treated protoplasts were incubated with 5 μM of H2DCFDA and 100 μM of SOSG at room temperature in the dark for 30 min and 1 h respectively, washed twice with protoplast re-suspension solution and then subjected to flow analysis. Unstained protoplasts were used as autofluorescence controls. For each sample, 5000 protoplasts were gated and the MFI was measured using the FlowJo software (Tree Star, Inc.)
Cellular fractionation
Chloroplasts and mitochondria were isolated from 18-20-days-old Arabidopsis plants. Intact chloroplasts were purified using Percoll gradients as described by Lamppa (1995) with modification. Instead of one Percoll gradient, two Percoll gradients were run to purify the chloroplasts. Intact mitochondria were purified on Percoll gradients as described (Meyer and Millar, 2008). Organelle purity and/or contamination were assessed by Western blot analysis using organelle-specific marker antibodies.
Protein extraction and Western blot analysis
Protein extraction and Western blot analysis were performed as described previously (Mach et al., 2001; See Methods S1).
Binding assays
Intrinsic fluorescence measurements were performed at 22°C on a FluoroMax-3 spectrofluorimeter (Horiba Jobin Yvon Inc. Japan) using an excitation wavelength of 296 nm. The emission spectra were collected from 310 nm to 460 nm. The proteins (ACD2 and ACD2**; 40 - 320 aa) were diluted to a concentration of 0.1 μM in a buffer containing 50 mM Na2HPO4 (pH 8.0) and 25 mM NaCl. Stock solutions of PPIX and RCC were prepared as described (Shepherd et al., 2007; Sugishima et al., 2010). Quenching experiments with PPIX or RCC were performed by addition of small aliquots of concentrated stock solutions to protein samples. To correct for the increased sample absorption due to the increased concentration of the pigments in the sample, we measured the influence of these compounds on the fluorescence emission of the model compound NATA (0.1 μM) (λex = 296 nm and λem = 332 nm) at each pigment concentration. The effect of pigments was linear with respect to concentration and the relevant values were subtracted from those obtained with ACD2 and ACD2**. The data were analyzed as described by Solomaha and Palfrey (2005).
Pigment analysis
Chlorophyll biosynthetic intermediates, from leaves of wild-type and acd2 plants were extracted and the chromatographic separation was performed by HPLC as described (Papenbrock et al., 2000).
Excised leaves were incubated in dark for 4-5 days and their tetrapyrrolic chlorophyll catabolites were extracted and chromatographic separation was performed by HPLC as described (Pruzinska et al., 2005, 2007).
Enzyme assays
The coupled PAO/ACD2 activity assay was done as described (Hortensteiner et al., 1995; Wuthrich et al., 2000; Pruzinska et al., 2005).
RCC synthesis
RCC was synthesized and validated as described (Kräutler et al., 1997; Sugishima et al., 2010).
Spin-trapping of 1O2 by TEMP
Spin-trapping assays were performed in 10 mM sodium phosphate pH 7.8 buffer with pigments (20 μM RCC, 5 μM of PPIX), 100 mM TEMP and methanol (ultrapure), final concentration 3.5% (v/v). Samples were illuminated (350 μmol of photons m-2 s-1) for a given time and their electron-paramagnetic resonance (EPR) signals were measured with a Bruker ESP 300 spectrometer as described (Schmidt et al., 2003).
Supplementary Material
Figure S10. How & where does ACD2 function to protect cells?
Figure S2. Superoxide levels in acd2 and wild-type protoplasts.
Figure S3. Singlet oxygen production by red chlorophyll catabolite (RCC).
Figure S4. Mutations in Executers and photoreceptors do not affect the survival of light-treated acd2 protoplasts.
Figure S6. Targeting of ACD2** either to chloroplasts or mitochondria does not reduce ROS generation in acd2 protoplasts.
Figure S7. Cre-Lox construct and characterization of the acd2 transgenic plants.
Figure S8. Expression and purification of ACD2 from E.coli.
Figure S9. PPIX is present in methanol extract of recombinant ACD240-320.
Figure S1. Plants with HsCAT targeted to mitochondria do not accumulate HsCAT in chloroplasts.
Figure S5. ACD2 in mitochondria or chloroplasts significantly suppresses the ROS production in acd2 protoplasts.
Methods S1. Protein extraction and Western blot analysis; purification of recombinant ACD2; generation of constructs and transgenic plants for mosaic analysis; superoxide levels in protoplasts isolated from wild type and acd2 leaves.
Table S1. Leaf cell death phenotype in acd2/m-tAPX and acd2/m-HsCAT plants.
Table S2. Impact of targeting ACD2 to different organelles on the cell death phenotype. of acd2.
Table S3. Primers used in this study.
Acknowledgments
We thank Jocelyn Malamy, Gayle Lamppa, Eric Ottesen, Mark Shepherd, Robert M. Larkin, Christian Fankhauser, Junichi Taira and Greenberg lab members for helpful discussions. We thank Vytas Bindokas, Christine Labno, Ryan Duggan, Elena Solomaha and Eugene Barth for technical assistance. We thank Ron Mittler, Karl-Josef Dietz, Roberto Bassi, Peter Rabinovitch, Leslie E. Sieburth, Patricia Zambryski, Joanne Chory, Chentao Lin for reagents. This work was supported by grants from the United States Department of Agriculture (2006-35100-17265) and National Institutes of Health (R01 GM54292) to J. T. G. and by grants from the Swiss National Science Foundation and the Swiss National Center of Competence in Research Plant Survival to S. H.
References
- Arpagaus S, Rawyler A, Braendle R. Occurrence and characteristics of the mitochondrial permeability transition in plants. J Biol Chem. 2002;277:1780–1787. doi: 10.1074/jbc.M109416200. [DOI] [PubMed] [Google Scholar]
- Balk J, Leaver C. The PET1-CMS mitochondrial mutation in sunflower is associated with premature programmed cell death and cytochrome c release. Plant Cell. 2001;13:1803–1818. doi: 10.1105/TPC.010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilski P, Li MY, Ehrenshaft M, Daub M, Chignell CF. Vitamin B6 (pyridoxine) and its derivatives are efficient singlet oxygen quenchers and potential fungal antioxidants. Photochem Photobiol. 2000;71:129–134. doi: 10.1562/0031-8655(2000)071<0129:sipvbp>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998;16:735–743. doi: 10.1046/j.1365-313x.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Danon A, Miersch O, Felix G, Camp RG, Apel K. Concurrent activation of cell death-regulating signaling pathways by singlet oxygen in Arabidopsis thaliana. Plant J. 2005;41:68–80. doi: 10.1111/j.1365-313X.2004.02276.x. [DOI] [PubMed] [Google Scholar]
- Danon A, Coll NS, Apel K. Cryptochrome-1-dependent execution of programmed cell death induced by singlet oxygen in Arabidopsis thaliana. Proc Natl Acad Sci USA. 2006;103:17036–17041. doi: 10.1073/pnas.0608139103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkemeier I, Goodman M, Lamkemeyer P, Kandlbinder A, Sweetlove LJ, Dietz KJ. The mitochondrial type II peroxiredoxin F is essential for redox homeostasis and root growth of Arabidopsis thaliana under stress. J Biol Chem. 2005;280:12168–12180. doi: 10.1074/jbc.M413189200. [DOI] [PubMed] [Google Scholar]
- Flors C, Fryer MJ, Waring J, Reeder B, Bechtold U, Mullineaux PM, Nonell S, Wilson MT, Baker NR. Imaging the production of singlet oxygen in vivo using a new fluorescent sensor, Singlet Oxygen Sensor Green. J Exp Bot. 2006;57:1725–1734. doi: 10.1093/jxb/erj181. [DOI] [PubMed] [Google Scholar]
- Frankenberg N, Mukougawa K, Kohchi T, Lagarias JC. Functional genomic analysis of the HY2 family of ferredoxindependent bilin reductases from oxygenic photosynthetic organisms. Plant Cell. 2001;13:965–978. doi: 10.1105/tpc.13.4.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollmer A, Arnbjerg J, Blaikie FH, Pedersen BW, Breitenbach T, Daasbjerg K, Glasius M, Ogilby PR. Singlet Oxygen Sensor Green: photochemical behavior in solution and in a mammalian cell. Photochem Photobiol. 2011;87:671–679. doi: 10.1111/j.1751-1097.2011.00900.x. [DOI] [PubMed] [Google Scholar]
- Greenberg JT, Ausubel FM. Arabidopsis mutants compromised for the control of cellular damage during pathogenesis and aging. Plant J. 1993;4:327–341. doi: 10.1046/j.1365-313x.1993.04020327.x. [DOI] [PubMed] [Google Scholar]
- Greenberg JT, Guo A, Klessig DF, Ausubel FM. Programmed cell death in plants, a pathogen-triggered response activated coordinately with multiple defense functions. Cell. 1994;77:551–563. doi: 10.1016/0092-8674(94)90217-8. [DOI] [PubMed] [Google Scholar]
- Hortensteiner S, Vicentini F, Matile P. Chlorophyll breakdown in senescent cotyledons of rape, Brassica napus L.: enzymatic cleavage of pheophorbide a in vitro. New Phytol. 1995;129:237–246. doi: 10.1111/j.1469-8137.1995.tb04293.x. [DOI] [PubMed] [Google Scholar]
- Hu G, Yalpani N, Briggs SP, Johal GS. A porphyrin pathway impairment is responsible for the phenotype of a dominant disease lesion mimic mutant of maize. Plant Cell. 1998;10:1095–1105. doi: 10.1105/tpc.10.7.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa A, Okamoto H, Iwasaki Y, Asahi T. A deficiency of coproporphyrinogen III oxidase causes lesion formation in Arabidopsis. Plant J. 2001;27:89–99. doi: 10.1046/j.1365-313x.2001.01058.x. [DOI] [PubMed] [Google Scholar]
- Jespersen HM, Kjaersgard IV, Ostergaard L, Welinder KG. From sequence analysis of three novel ascorbate peroxidase from Arabidopsis thaliana to structure, function and evolution of seven type of ascorbate peroxidase. Biochem J. 1997;326:305–310. doi: 10.1042/bj3260305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones A. Does the plant mitochondria integrate cellular stress and regulate programmed cell death? Trends Plant Sci. 2000;5:225–230. doi: 10.1016/s1360-1385(00)01605-8. [DOI] [PubMed] [Google Scholar]
- Kim CS, Lee CH, Lee PH, Han S. Inactivation of mitochondrial electron transport by photosensitization of a pheophorbide a derivative. Mol Cells. 2004;17:347–352. [PubMed] [Google Scholar]
- Krautler B, Muhlecker W, Anderl M, Gerlach B. Breakdown of chlorophyll: partial synthesis of a putative intermediary catabolite. Helv Chim Acta. 1997;80:1355–1362. [Google Scholar]
- Krieger-Liszkay A, Trebst A. Tocopherol is the scavenger of singlet oxygen produced by the triplet states of chlorophyll in the PSII reaction center. J Exp Bot. 2006;57:1677–1684. doi: 10.1093/jxb/erl002. [DOI] [PubMed] [Google Scholar]
- Lam E, Kato N, Lawton M. Programmed cell death, mitochondria and plant hypersensitive response. Nature. 2001;411:848–853. doi: 10.1038/35081184. [DOI] [PubMed] [Google Scholar]
- Lamppa GK. In vitro import of proteins into chloroplasts. In: Maliga P, Klsessig DF, Cashmore AR, Gruissem W, Varner JE, editors. Methods in Plant Molecular Biology. Cold Spring Harbor Lab Press; Plainview, NY: 1995. pp. 141–172. [Google Scholar]
- Lee KP, Kim C, Landgraf F, Apel K. Executer1- and Executer2-dependent transfer of stress-related signals from the plastid to nucleus of Arabidopsis thaliana. Proc Natl Acad Sci USA. 2007;104:10270–10275. doi: 10.1073/pnas.0702061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Robertson DE, Ahmad M, Raibekas AA, Jorns MS, Dutton PL, Cashmore AR. Association of flavin adenine dinucleotide with the Arabidopsis blue light receptor CRY1. Science. 1995;269:968–970. doi: 10.1126/science.7638620. [DOI] [PubMed] [Google Scholar]
- Lin C, Yang H, Guo H, Mockler T, Chen J, Cashmore AR. Enhancement of blue-light sensitivity of Arabidopsis seedlings by a blue light receptor cryptochrome 2. Proc Natl Acad Sci USA. 1998;95:2686–2690. doi: 10.1073/pnas.95.5.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemann P, Koch A, Degenhardt B, Hause G, Grimm B, Papadopoulos V. A novel Arabidopsis thaliana protein is a functional peripheral-type benzodiazepine receptor. Plant Cell Physiol. 2004;45:723–733. doi: 10.1093/pcp/pch088. [DOI] [PubMed] [Google Scholar]
- Mach JM, Castillo AR, Hoogstraten R, Greenberg JT. The Arabidopsis accelerated cell death gene ACD2 encodes red chlorophyll catabolite reductase and suppresses the spread of disease symptoms. Proc Natl Acad Sci USA. 2001;98:771–776. doi: 10.1073/pnas.021465298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer EH, Millar AH. Isolation of mitochondria from plant culture. Methods Mol Biol. 2008;425:163–169. doi: 10.1007/978-1-60327-210-0_15. [DOI] [PubMed] [Google Scholar]
- Moller IM. Plant mitochondria and oxidative stress: electron transport, NADPH turnover, and metabolism of reactive oxygen species. Annu Rev Plant Physiol Plant Mol Biol. 2001;52:561–591. doi: 10.1146/annurev.arplant.52.1.561. [DOI] [PubMed] [Google Scholar]
- Mur LA, Aubry S, Mondhe M, Kingston-Smith A, Gallagher J, Timms-Taravella E, James C, Papp I, Horteinsteiner S, Thomasm H, Ougham H. Accumulation of chlorophyll catabolites photosensitizes the hypersensitive response elicited by Pseudomonas syringae in Arabidopsis. New Phytol. 2010;188:161–174. doi: 10.1111/j.1469-8137.2010.03377.x. [DOI] [PubMed] [Google Scholar]
- Neff MM, Neff JD, Chory J, Pepper AE. dCAPS, a simple technique for the genetic analysis of single nucleotide polymorphisms: experimental applications in Arabidopsis thaliana genetics. Plant J. 1998;14:387–392. doi: 10.1046/j.1365-313x.1998.00124.x. [DOI] [PubMed] [Google Scholar]
- Norman C, Howell KA, Milar AH, Whelan JM, Day DA. Salicylic acid is an uncoupler and inhibitor of mitochondrial electron transport. Plant Physiol. 2004;134:492–501. doi: 10.1104/pp.103.031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhuber M, Krautler B. Breakdown of chlorophyll: Electrochemical bilin reduction provides synthetic access to fluorescent chlorophyll catabolites. Chem Bio Chem. 2002;3:104–107. doi: 10.1002/1439-7633(20020104)3:1<104::AID-CBIC104>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- op den Camp RG, Przybyla D, Ochsenbein C, Laloi C, Kim C, Danon A, Wagner D, Hideg E, Gobel C, Feussner I, Nater M, Apel K. Rapid induction of distinct stress responses after release of singlet oxygen in Arabidopsis. Plant Cell. 2003;15:2320–2332. doi: 10.1105/tpc.014662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenbrock J, Mock H, Tanaka R, Kruse E, Grimm B. Role of Magnesium Chelatase activity in the early steps of the tetrapyrrole biosynthetic pathway. Plant Physiol. 2000;122:1161–1170. doi: 10.1104/pp.122.4.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruzinska A, Anders I, Tanner G, Roca M, Hortensteiner S. Chlorophyll breakdown: Pheophorbide a oxygenase is a Rieske-type iron-sulfur protein, encoded by the accelerated cell death 1 gene. Proc Natl Acad Sci USA. 2003;100:15259–15264. doi: 10.1073/pnas.2036571100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruzinska A, Tanner G, Aubry S, Anders I, Moser S, Muller T, Ongania K-H, Krautler B, Youn J-Y, Liljegren SJ, Hortensteiner S. Chlorophyll breakdown in senescent Arabidopsis leaves: Characterization of chlorophyll catabolites and of chlorophyll catabolic enzymes involved in the degreening reaction. Plant Physiol. 2005;139:52–63. doi: 10.1104/pp.105.065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruzinska A, Anders I, Aubry S, Schenk N, Tapernoux-Luthi E, Muller T, Krautler B, Hortensteiner S. In vivo participation of Red Chlorophyll Catabolite Reductase in chlorophyll breakdown. Plant Cell. 2007;19:369–387. doi: 10.1105/tpc.106.044404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodoni S, Muhlecker W, Anderl M, Krautler B, Moser D, Thomas H, Matile P, Hortensteiner S. Chlorophyll breakdown in senescent chloroplasts. Cleavage of pheophorbide a in two enzymic steps. Plant Physiol. 1997;115:669–676. doi: 10.1104/pp.115.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Fufezan C, Krieger-Liszkay A, Satoh H, Paulsen H. Recombinant water-soluble chlorophyll protein from Brassica oleracea var. Botrys binds various chlorophyll derivatives. Biochemistry. 2003;42:7427–7433. doi: 10.1021/bi034207r. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, Coskun PE, Ladiges W, Wolf N, Van Remmen H, Wallace DC, Rabinovitch PS. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Shepherd M, Heath MD, Poole RK. NikA binds heme: a new role for an Escherichia coli periplasmic nickel-binding protein. Biochemistry. 2007;46:5030–5037. doi: 10.1021/bi700183u. [DOI] [PubMed] [Google Scholar]
- Solomaha E, Palfrey HC. Conformational changes in dynamin on GTP binding and oligomerization reported by intrinsic and extrinsic fluorescence. Biochem J. 2005;391:601–611. doi: 10.1042/BJ20050707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugishima M, Kitamori Y, Noguchi M, Kohchi T, Fukuyama K. Crystal structure of red chlorophyll catabolite reductase: enlargement of the ferredoxin-dependent bilin reductase family. J Mol Biol. 2009;389:376–87. doi: 10.1016/j.jmb.2009.04.017. [DOI] [PubMed] [Google Scholar]
- Sugishima M, Okamoto Y, Noguchi M, Kohchi T, Tamiaki H, Fukuyama K. crystal structures of the substrate-bound forms of red chlorophyll catabolite reductase: implications for site-specific and stereospecific reaction. J Mol Biol. 2010;402:879–891. doi: 10.1016/j.jmb.2010.08.021. [DOI] [PubMed] [Google Scholar]
- Tiwari BS, Belenghi B, Levine A. Oxidative stress increased respiration and generation of reactive oxygen species, resulting in ATP depletion, opening of mitochondrial permeability transition, and programmed cell death. Plant Physiol. 2002;128:1271–1281. doi: 10.1104/pp.010999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantaphylides C, Havaux M. Singlet oxygen in plants: production, detoxification and signaling. Trends Plant Sci. 2009;14:219–228. doi: 10.1016/j.tplants.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Wagner D, Przybyla D, op den Camp R, Kim C, Landgraf F, Lee KP, Wursch M, Laloi C, Nater M, Hideg E, Apel K. The genetic basis of singlet oxygen-induced stress responses of Arabidopsis thaliana. Science. 2004;306:1183–1185. doi: 10.1126/science.1103178. [DOI] [PubMed] [Google Scholar]
- Wuthrich KL, Bovet L, Hunziker PE, Donnison IS, Hortensteiner S. Molecular cloning, functional expression and characterisation of RCC reductase involved in chlorophyll catabolism. Plant J. 2000;21:189–198. doi: 10.1046/j.1365-313x.2000.00667.x. [DOI] [PubMed] [Google Scholar]
- Xiang C, Han P, Lutziger I, Wang K, Oliver DJ. A mini binary vector series for plant transformation. Plant Mol Biol. 1999;40:711–717. doi: 10.1023/a:1006201910593. [DOI] [PubMed] [Google Scholar]
- Yu X, Perdue T, Heimer Y, Jones A. Mitochondrial involvement in tracheary element programmed cell death. Cell Death Differ. 2002;9:189–198. doi: 10.1038/sj.cdd.4400940. [DOI] [PubMed] [Google Scholar]
- Yao N, Eisfelder B, Marvin J, Greenberg JT. The mitochondrion—An organelle commonly involved in programmed cell death in Arabidopsis thaliana. Plant J. 2004;40:596–610. doi: 10.1111/j.1365-313X.2004.02239.x. [DOI] [PubMed] [Google Scholar]
- Yao N, Greenberg JT. Arabidopsis ACCELERATED CELL DEATH2 modulates programmed cell death. Plant Cell. 2006;18:397–411. doi: 10.1105/tpc.105.036251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S10. How & where does ACD2 function to protect cells?
Figure S2. Superoxide levels in acd2 and wild-type protoplasts.
Figure S3. Singlet oxygen production by red chlorophyll catabolite (RCC).
Figure S4. Mutations in Executers and photoreceptors do not affect the survival of light-treated acd2 protoplasts.
Figure S6. Targeting of ACD2** either to chloroplasts or mitochondria does not reduce ROS generation in acd2 protoplasts.
Figure S7. Cre-Lox construct and characterization of the acd2 transgenic plants.
Figure S8. Expression and purification of ACD2 from E.coli.
Figure S9. PPIX is present in methanol extract of recombinant ACD240-320.
Figure S1. Plants with HsCAT targeted to mitochondria do not accumulate HsCAT in chloroplasts.
Figure S5. ACD2 in mitochondria or chloroplasts significantly suppresses the ROS production in acd2 protoplasts.
Methods S1. Protein extraction and Western blot analysis; purification of recombinant ACD2; generation of constructs and transgenic plants for mosaic analysis; superoxide levels in protoplasts isolated from wild type and acd2 leaves.
Table S1. Leaf cell death phenotype in acd2/m-tAPX and acd2/m-HsCAT plants.
Table S2. Impact of targeting ACD2 to different organelles on the cell death phenotype. of acd2.
Table S3. Primers used in this study.