

Abstract
Formaldehyde is a reactive chemical that is commonly used in the production of industrial, laboratory, household, and cosmetic products. The causal association between formaldehyde exposure and increased incidence of cancer led the International Agency for Research on Cancer to classify formaldehyde as a carcinogen. Formaldehyde-induced DNA-protein crosslinks (DPCs) elicit responses involving nucleotide excision repair (NER) and homologous recombination (HR) repair pathways; however, little is known about the cellular and genetic changes that subsequently lead to formaldehyde-induced genotoxic and cytotoxic effects. Herein, investigations of genes that modulate the cytotoxic effects of formaldehyde exposure revealed that of five NER-deficient Chinese Hamster Ovary (CHO) cell lines tested, XPF- and ERCC1-deficient cells were most sensitive to formaldehyde treatment as compared to wild-type cells. Cell cycle analyses revealed that formaldehyde-treated XPF-deficient cells exhibited an immediate G2/M arrest that was associated with altered cell ploidy and apoptosis. Additionally, an elevated number of DNA double-strand breaks (DSBs), chromosomal breaks and radial formation were also observed in XPF-deficient cells following formaldehyde treatment. Formaldehyde-induced DSBs occurred in a replication-dependent, but an XPF-independent manner. However, delayed DSB repair was observed in the absence of XPF function. Collectively, our findings highlight the role of an XPF-dependent pathway in mitigating the sensitivity to formaldehyde-induced DNA damage as evidenced by the increased genomic instability and reduced cell viability in an XPF-deficient background. In addition, centrosome and microtubule abnormalities, as well as enlarged nuclei, caused by formaldehyde exposure are also demonstrated in a repair-proficient cell line.
Keywords: DNA-protein crosslink, formaldehyde, DSBs, nucleotide excision repair, XPF
1. Introduction
Formaldehyde is a common environmental and occupational pollutant. Recently, formaldehyde exposure has raised significant concern due to mounting evidence suggesting its carcinogenic potential and serious effects on human health [1]. While data supporting the carcinogenic potential of formaldehyde remain controversial, cellular and cytogenetic studies consistently demonstrate that formaldehyde induces mutations and chromosomal damage in both mammalian and Escherichia coli (E. coli) cells [2–5]. In both cellular and human studies, formaldehyde exposure is associated with increased levels of chromosomal aberrations, sister chromatid exchange and micronuclei formation [6–11]. Increased ring and dicentric chromosomes were also observed in lymphocytes of workers exposed to formaldehyde [12].
In order to mechanistically understand the basis of formaldehyde-induced genomic instability, the formation, persistence, tolerance, and processing of the DNA damage caused by exposure to formaldehyde must be elucidated. It is believed that the primary genotoxic DNA lesions generated following formaldehyde exposure are DNA-protein crosslinks (DPCs). DPCs have been detected in cultured mammalian cells, in the respiratory tract of rats, and in human populations following exogenous formaldehyde exposure [2, 13–15]. The mutagenic potency and mechanisms of DPC-induced genotoxicity remain unclear. Advancements in the creation of synthetic and site-specific DPC-containing DNAs are improving the experimental assessment of these mechanistic details, including the elucidation of DNA repair pathways that are important for cell survival following exposure to DPC-inducing agents such as formaldehyde [16–18]. Several studies have investigated the relative contribution of the homologous recombination (HR) and nucleotide excision repair (NER) pathways in repair and tolerance of DPCs at the cellular level [19–23]. A formaldehyde cytotoxicity screen of the non-essential yeast gene deletion library demonstrated that under low dose, chronic exposure conditions, HR is the primary pathway that confers resistance to formaldehyde-induced DNA lesions; while following acute, high dose exposure, the NER pathway becomes critical for cell survival [21]. Specifically, it was shown that the rad1 (yeast homolog of human XPF), rad4 (yeast homolog of human XPC), and rad14 (yeast homolog of human XPA) genes of the NER pathway are critical for survival. Interestingly, in the mammalian cell study by Nakano et al (2009), the formaldehyde sensitivities of both Rad51- (HR-related) and XPF- (NER- and interstrand crosslink (ICL) repair-related) deficient cells were similar. Together, these data suggest that the role of HR and XPF in the prevention of formaldehyde-induced cytotoxicity is conserved from yeasts to mammals [21].
Despite the evidence that NER and HR are important for survival following DPC-induction, the molecular events leading to the cytotoxicity and genotoxicity are not well understood. This study was designed to investigate the genotoxic events induced by formaldehyde in DNA repair-deficient and proficient cell lines. We show that the function of XPF is critical in preventing formaldehyde-induced alterations that may lead to genome instability and cell death.
2. Materials and methods
2.1. Cells and culture conditions
AA8 (wild-type), UV20 (ERCC1-deficient), UV5 (XPD-deficient), UV24 (XPB-deficient), UV135 (XPG-deficient), and UV41 (XPF-deficient) Chinese hamster ovary (CHO) cells (purchased from ATCC) were used in this study. Rad51D (Rad51-deficient) cells were a kind gift from John Hinz (Washington State University, WA) and Paul Wilson (Brookhaven National Laboratory, NY). Cells were grown in DMEM supplemented with 10% fetal bovine serum and antibiotics (ampicillin and streptomycin, Gibco) at 37°C in a 5% CO2 incubator. Cells were arrested at the G1/S phase boundary by treatment with a replication elongation inhibitor, aphidicolin (1µg/ml) for 17 hr. Synchronized cells were allowed a 2 hr recovery prior to formaldehyde treatment to facilitate the progression of cells into S phase. For all experiments, subconfluent cultures of cells were treated for 4 hr with various concentrations of formaldehyde (Fisher Scientific) and harvested at times as indicated.
2.2. Survival assays
For survival assays, 200–300 cells were plated in a 60 mm plate and grown overnight at 37°C prior to formaldehyde treatment. After 7 days, the plates were fixed, stained with methylene blue diluted in methanol (4 g/L), and the colonies were counted.
2.3. Cell cycle analyses
Cells were harvested, fixed in ice-cold 70% ethanol, and stained with propidium iodide (PI) (Invitrogen) prior to measuring DNA content by using a FACSCalibur instrument (Becton Dickinson) (Flow Cytometry Core, OHSU). Aggregated cells were excluded from the PI-stained cell suspensions by passing the cell suspensions through the strainer-capped tubes (BD Falcon) just before analyses. The results were analyzed using FlowJo software (Tree Star).
2.4. Western blot analyses
Cells were harvested and lysed in a lysis buffer (50 mM Tris–HCl, pH 8.0, 0.5% Nonidet P-40, 5 mM EDTA, 150 mM NaCl, 1 mM Pepstatin, and 1 mM PMSF). Approximately 50 µg of total cell lysates were run on a SDS–PAGE gel, followed by transfer onto PVDF membranes that were then immunoblotted independently with the following primary antibodies overnight at 4°C: mouse monoclonal anti-cyclin B1 (Millipore, 1:1000 dilution), rabbit polyclonal anti-cdk1/cdc2 (Millipore, 1:1000 dilution), and mouse monoclonal anti-GAPDH (Sigma, 1:10,000 dilution). After incubation with appropriate secondary antibodies conjugated to horseradish peroxidise, proteins were detected by the enhanced chemiluminescence detection system (Western LightningTM Plus-ECL from Perkin Elmer).
2.5. Apoptosis assays
The percentage of cells undergoing apoptosis was determined using an Annexin-V assay following the manufacturer’s instructions (Invitrogen). Samples were analyzed by flow cytometry and the percentage of apoptotic cells was determined by scoring for the fraction of cells that stained negative for PI but positive for Annexin-V.
2.6. Immunofluorescence studies
Cells were fixed in ice-cold methanol:acetone solution (3:1) at −20°C for 10 min, washed with either PBS (for H2AX staining) or PBG buffer (PBS containing 50 mM glycine), permeabilized with PBS-T (PBS with 0.2% Triton-X100) for 10 min, and blocked with 0.5 % BSA. Cells were then stained with either anti-mouse γH2AX (Upstate; 1:1000 dilution), anti-rabbit γ-tubulin (Sigma; 1:10,000 dilution), or anti-mouse β-tubulin (Sigma; 1:1000 dilution) for 1.5 hr to visualize γH2AX foci, centrosomes and microtubules, respectively. Appropriate fluorochrome-conjugated secondary antibodies (Alexa Fluor 594-conjugated goat anti-rabbit (Invitrogen, 1:1000 dilution) and Alexa-fluor 488-conjugated mouse (Invitrogen, 1:300 dilution)) were used for 30–45 min prior to mounting the slides with Prolong Gold Antifade Reagent with DAPI (Invitrogen). Cells were visualized with a 40X objective on an Axioskop 2 microscope (Zeiss). The area and number of giant nuclei were determined using the ImageJ software.
2.7. Cytogenetic analyses
Cells were harvested, treated with hypotonic solution (75 mM KCl, 5% fetal calf serum) for 10 min, and fixed with 3:1 methanol:acetic acid. Cells were dropped onto slides for metaphase spreads and stained with Wright’s stain (Fisher Scientific) and G-banded. For each sample, fifty metaphases for radial formation and one-hundred metaphases for ploidy analyses were analyzed on a Nikon Eclipse E800 photoscope. Representative photographs were taken using CytoVision software (Applied Imaging). Assessment of karyotype was based on comparison with published CHO G-banded karyotypes [24].
3. Results
3.1. XPF- and ERCC1-deficient cells exhibit extreme sensitivity to formaldehyde treatment
Based on increased cytotoxicity following acute formaldehyde exposure in yeast NER mutants [21], we examined the effect of formaldehyde on cellular viability in several CHO cell lines with mutations in XPB (UV24), XPD (UV5), XPF (UV41), XPG (UV135), or ERCC1 (UV20) (Fig. 1). As compared to wild-type cells (AA8), XPF- and ERCC1-deficient cells demonstrated the greatest sensitivity to formaldehyde treatment; however, the XPB-, XPD-, and XPG-deficient cells were also sensitive, but to a lesser degree. While this study was in progress, Nakano et al (2009) reported similar findings on the sensitivity of XPF- and XPD-deficient cells following an acute (3 hr) formaldehyde exposure. To distinguish between the contributions of NER and HR pathways in the formaldehyde-induced DNA damage response, formaldehyde sensitivity of NER mutants was compared with a HR mutant. Rad51-deficient cells displayed less sensitivity than the ERCC1- and XPF-deficient cells but greater sensitivity than the other NER mutants (Fig. 1). Interestingly, the ERCC1- and Rad51-deficient cells exhibit similar relative survivals as reported following exposure to the cross-linking agents, mitomycin C (MMC) and cisplatin [25]. Together, the survival data suggest that the XPF/ERCC1 protein complex and Rad51 may be more critical than the other NER proteins in limiting formaldehyde cytotoxicity in mammalian cells.
Fig. 1.

An XPF/ERCC1-dependent pathway is critical in limiting formaldehyde-induced cytotoxicity. Wild-type, a HR mutant and several NER mutant cell lines were treated with formaldehyde at the indicated concentrations for 4 hr. The viability of the cells was determined by colony forming assay. Standard deviations (S.D.) were derived from three or more independent experiments. AA8: Wild-type, UV24: XPB-deficient, UV5: XPD-deficient, UV41: XPF-deficient, UV135: XPG-deficient, UV20: ERCC1-deficient, and Rad51D: Rad51-deficient.
3.2. Exposure to formaldehyde causes G2/M arrest
To determine if formaldehyde exposure resulted in altered cell cycle progression, flow cytometry analyses were carried out on both wild-type and XPF-deficient cells that were treated with formaldehyde (0–400 µM) for 4 hr followed by staining with PI. Relative to untreated cells, no obvious change was observed in formaldehyde-treated wild-type cells (Fig. 2A, first panel), whereas formaldehyde treatment (≥200 µM) caused a noticeable accumulation of the XPF-deficient cells in G2/M phase (Fig. 2B, first panel). The reversibility of G2/M blockage in XPF-deficient cells was investigated by allowing a recovery period of 24 hr or 48 hr following formaldehyde treatment. It was noted that when XPF-deficient cells were analyzed after a 24 hr or 48 hr recovery, they continued to accumulate in the G2/M phase in a dose-dependent manner (Fig. 2B, middle and last panels). In parallel, it was noteworthy that wild-type cells showed a significant G2/M blockage during the recovery phase (Fig. 2A, middle and right panels). These observations suggested that the cellular changes continue to accumulate, even after the removal of formaldehyde. The rise in the G2/M peak occurred concomitantly with a decrease in the G1 and S phase populations, as is most evident in Fig. 2B (middle panel). The ability of cells to overcome G2/M arrest appeared to be dose-dependent and a treatment for 4 hr up to 200 µM was found to be the threshold limit for wild-type cells, whereas at higher formaldehyde concentrations (≥300 µM), the effect appeared to be irreversible (Fig. 2A, last panel). In the recovered XPF-deficient cell populations, a significant rise in sub-G1 and post-G2 peaks representing apoptosis and polyploid populations was also observed at high formaldehyde exposures (≥200 µM) (Fig. 2B, last panel). Overall, the PI profile of the formaldehyde-treated XPF-deficient cells displayed a similar, but more severe and immediate G2/M arrest accompanied with a significant increase in apoptosis and polyploid populations compared to wild-type cells. In contrast, the cell cycle response of the moderately sensitive XPB-deficient cells was not significantly different than wild-type cells (Fig. S1).
Fig. 2.


Formaldehyde treatment causes cell cycle arrest and cell death that is severely pronounced upon loss of XPF function. Cell cycle progression was measured for AA8 (wild-type) (A) and UV41 (XPF-deficient) (B) cells by flow cytometry analyses of DNA content. The cells were treated with formaldehyde for 4 hr at the indicated concentrations and DNA content was measured immediately after the treatment, as well as after a 24 hr (24h-R) and 48 hr (48h-R) recovery time. The flow cytometry results have been summarized by generating overlay images representing all the samples harvested at each time point (4h, 24h-R, and 48h-R). (C) Western blot analyses were performed to study the changes in the levels of G2/M checkpoint protein, cyclin B1. AA8 and UV41 cells were treated with 150 µM and 300 µM formaldehyde and harvested at indicated time points. Anti-cyclin B1 and anti-GAPDH antibodies were used to detect the protein levels of cyclin B1 and GAPDH (the loading control), respectively. (D) Synchronized cultures of AA8 and UV41 cells were released from aphidicolin treatment, and 2 hr later treated with formaldehyde (200 µM). The cells were harvested immediately after a 4 hr formaldehyde treatment (Aph 2h-R+F200 4h), as well as after a 24 recovery following formaldehyde treatment (Aph 2h-R+F200 24h-R).
(E) Using an Annexin-V assay, the relative percentage of apoptosis was measured for AA8 and UV41 cells after a 48 hr recovery following a 4 hr formaldehyde treatment (200 µM and 400 µM). Graphic representation of the quantitative data obtained in Figs. S3A and B.
Entry or exit from G2/M is regulated by the activity of the cdc2-cyclin B complex [26, 27]. Stabilization of this complex is expected to delay mitotic exit, suggesting that the arrest is associated with the deregulation of cellular checkpoint machinery. To further characterize the potential molecular mechanisms causing the formaldehyde-induced G2/M arrest, the levels of cyclin B1 and cdc2 proteins were analyzed in cells treated with either 150 or 300 µM formaldehyde (4 hr), with or without, a 24 hr recovery. Relative to untreated cells, an elevated expression of cyclin B1 was observed in both formaldehyde-treated wild-type and XPF-deficient cells (Fig. 2C). However, no change in the levels of cdc2 was observed in either wild-type or XPF-deficient cells (data not shown). This observation indicates that formaldehyde-induced G2/M arrest is likely to be associated with stabilization of the cell cycle regulatory protein, cyclin B1.
To examine if formaldehyde-induced G2/M arrest was related to DNA replication, wild-type and XPF-deficient cells were synchronized with aphidicolin (Fig. S2A and B, respectively) followed by an acute formaldehyde treatment (200 µM) for 4 hr. Formaldehyde-induced cell cycle arrest was even more pronounced in synchronized cultures compared to unsynchronized cultures, suggesting that actively replicating cells are more sensitive to formaldehyde exposure, resulting in a G2/M arrest (Fig. 2D). Furthermore, following a 24 hr recovery, the S phase arrested and formaldehyde-treated wild-type cells were able to recover significantly from G2/M arrest (Fig. 2D, right panel), whereas minimal recovery, accompanied with cell death and ploidy changes was observed for XPF-deficient cells (Fig. 2D, right panel).
3.3. Formaldehyde-induced cell death
The cell cycle distribution following formaldehyde exposure showed that both wild-type and XPF-deficient cells exhibited a pronounced sub-G1 peak, following a recovery of 48 hr in a dose-dependent manner. To measure apoptosis, Annexin-V assays were used to identify the cells in an early apoptotic phase when the membrane phospholipid phosphatidylserine is redistributed from the cytoplasmic face to the outer leaflet, making it accessible to Annexin-V. Wild-type and XPF-deficient cells were harvested following a 48 hr recovery from a 4 hr formaldehyde treatment at the indicated concentrations. Consistent with the cell cycle analyses, high levels of apoptosis and necrosis were observed in wild-type and XPF-deficient cells during the recovery phase post formaldehyde treatment (Figs. S3A and B). Both wild-type and XPF-deficient formaldehyde-treated cells showed a dose-dependent gradual, but clear shift, of the viable population towards the quadrant representing Annexin-V positive cells (Fig. S3). Relative to wild-type cells, the higher percentage of XPF-deficient cells undergoing apoptosis (Fig. 2E) implicates XPF in preventing cell death, presumably by repairing formaldehyde-induced DNA lesions.
3.4. Enlargement of nuclei and centrosome abnormalities in formaldehyde-treated cells
To examine any effects caused by formaldehyde on the nuclear morphology, formaldehyde-treated wild-type cells were fixed and stained with a nuclear counterstain (DAPI) 4 hr post treatment and following a 48 hr recovery. The untreated cells had a mean nuclear area of 119 µm2. Relative to untreated cells, a significantly high number of cells with enlarged nuclei were observed in the cell populations that were processed immediately after a 4 hr treatment as well as those that had undergone a 48 hr recovery post formaldehyde treatment (mean nuclear area of giant nuclei: ≥ 185 µm2) (Figs. 3A and B). The enlarged nuclei exhibited active, ongoing DNA synthesis, as evidenced by continued BrdU incorporation (data not shown). Consistent with previous studies, several cells bearing micronuclei were also detected in the formaldehyde-treated populations (as indicated by an arrow in Fig. 3A).
Fig. 3.

Appearance of enlarged nuclei after formaldehyde treatment. Indirect immunofluorescence staining of untreated and formaldehyde-treated AA8 (wild-type) cells that were fixed immediately after a 4 hr treatment (300 µM) or following a 48 hr recovery (A). Cells were fixed in paraformaldehyde, permeabilized with Triton-X100 and the nuclei were stained with DAPI. (B) In order to determine the relative ratio of enlarged nuclei, approximately 400 nuclei were scored for each time point in three independent experiments. The photos were taken under 40× magnification on a Zeiss microscope (scale bar = 25 µM). The error bars indicate S.D. derived from three independent experiments.
As shown in Fig. 2, cell cycle analyses following formaldehyde exposure showed enhanced post-G2 peaks, suggestive of polyploidy. To investigate this further, formaldehyde-treated wild-type cells were also stained for centrosomes using an anti-γ tubulin antibody. It was observed that a significant number of formaldehyde-treated cells contained an abnormal number of centrosomes (Figs. 4A and B). The abnormality in the number of centrosomes began to appear immediately after the formaldehyde treatment; however, cells with giant nuclei were observed to a minor extent in formaldehyde-treated cells after 4 hr treatment and increased dramatically after a 48 hr recovery (Figs. 3A and B). Together, these data suggest that the appearance of centrosome defects precede the enlargement of nuclei in the formaldehyde-treated cells. Defects in centrosome amplification and microtubule formation have been associated with impaired cytokinesis [28, 29], that could explain the appearance of giant nuclei in response to formaldehyde exposure. When formaldehyde-treated cells were stained for microtubules using an anti-β-tubulin antibody, these analyses revealed that abnormal centrosome-containing cells were capable of undergoing both bipolar and multipolar cell divisions (Fig. 4C). Based on these observations, it was predicted that abnormal centrosome-containing cells after cell division would yield progenies with chromosomal mis-segregation defects.
Fig. 4.


Centrosome and microtubule abnormalities in cells exposed to formaldehyde. (A) Indirect immunofluorescence staining of untreated and formaldehyde-treated AA8 (wild-type) cells immunostained with antibody against centrosomes, followed by rhodamine-conjugated secondary antibody (scale bar = 25 µM). (B) The percentage of cells with normal number (≤2), and abnormal number and size (≥2 and/or large) of centrosomes are summarized in panel B. A total of 200–400 cells were counted at each time point in three independent experiments. (C) Control and formaldehyde-treated (300 uM for 4 hr followed by a 48 hr recovery) cells were immunostained with antibody against microtubules, followed by FITC-conjugated secondary antibody (scale bar = 10 µM). DAPI was used as a nuclear counterstain. Photographic images were taken under 40× magnification on a Zeiss microscope.
3.5. Formaldehyde-induced DSBs occur in a replication-dependent manner
It is plausible that the cell cycle deregulation is a secondary, rather than a primary consequence of formaldehyde treatment, where the G2/M arrest of formaldehyde-treated cells is triggered in response to aberrant DNA structures and abnormal chromosomal rearrangements occurring during DNA replication. In order to address this possibility, DNA and chromosomal breakage/radial analyses were carried out with wild-type and XPF-deficient cells under the same conditions as followed for cell cycle analyses. It has previously been shown that formaldehyde induces DNA double-strand breaks (DSBs) [19, 30]. To investigate the effect of XPF deficiency on formaldehyde-induced DSBs, DNA breaks were measured by examining the phosphorylation of histone variant (γH2AX) that accumulates at the sites of DSBs, forming foci detectable by immunostaining. DNA DSB formation was quantitated by counting the number of γH2AX foci per nucleus and the cells were categorized as having zero, one to ten, and > 10 foci per nucleus. Both wild-type and XPF-deficient cells showed an accumulation of γH2AX foci upon formaldehyde treatment (Figs. 5A and B), suggesting that formaldehyde-induced DSBs are not dependent on the XPF endonuclease. Time-course experiments (24–72 hr post formaldehyde treatment) revealed that the accumulation of γH2AX foci peaked between 24–48 hr; however, the repair kinetics of formaldehyde-induced DSBs was significantly delayed in the XPF-deficient cells compared to wild-type cells (Figs. 5A and B). No overall decline of γH2AX foci containing nuclei was observed in XPF-deficient formaldehyde-treated cells, whereas the fraction of formaldehyde-treated wild-type cells decreased by 15% in a 24 hr time frame (compare 48R and 72R). Although an increase in the number of nuclei with one to ten γH2AX foci accompanied by a decline in nuclei with >10 γH2AX foci was observed in XPF-deficient cells, we believe that the receding number of nuclei with >10 γH2AX foci may be associated with cell death rather than repair of DSBs. This observation suggests that the XPF protein may participate in repairing formaldehyde-induced DNA damage following DSB formation.
Fig. 5.
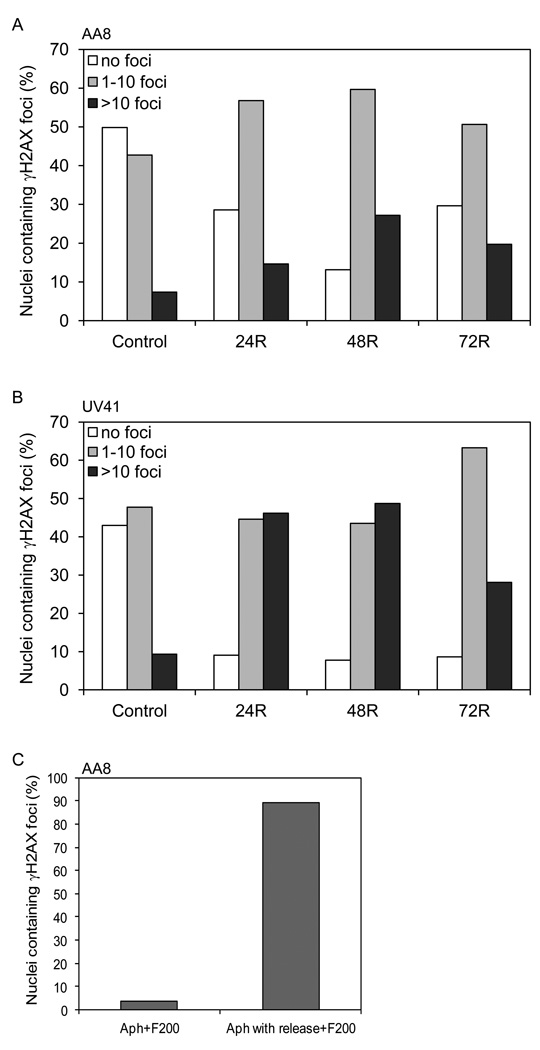
Formaldehyde-induced DSBs are generated in an XPF-independent, but replication-dependent manner. γH2AX foci formation was measured in AA8 (wild-type) (A) and UV41 (XPF-deficient) (B) cells at different time points following a recovery (24–72 hr) from formaldehyde exposure (200 µM for 4 hr). An average of two experiments is graphically represented, in which 300–500 nuclei were scored for each sample per experiment. (C) γH2AX foci formation was measured in AA8 cells treated with formaldehyde (200 µM for 4 hr) in the presence of aphidicolin or released from aphidicolin treatment and 2 hr later treated with formaldehyde (200 µM for 4 hr).
It has been previously demonstrated that DSBs induced by cross-linking agents are specifically formed in S phase cells, presumably at stalled replication forks [25]. Further experiments were performed to determine if formaldehyde-induced DSBs were also dependent on active replication elongation in a manner similar to that observed for cross-linking agents. Cells were treated with formaldehyde (200 µM) for 4 hr either in the presence of aphidicolin (an inhibitor of DNA polymerase α) or following a 2 hr release after aphidicolin treatment when most of the cells had progressed into the S phase. Accumulation of γH2AX foci was predominantly observed in the S phase arrested and formaldehyde-treated cells (Fig. 5C), suggesting that replication elongation is required for generation of formaldehyde-induced DSBs.
3.6. Exposure to formaldehyde generates cytogenetic defects
Cytogenetic analyses of wild-type cells treated with formaldehyde revealed chromosomal breaks and radial formation in a dose-dependent manner. However, consistent with the flow cytometry data, the chromosomal breaks and radials appeared only during the recovery period and not immediately after treatment (Figs. 6A, B and S4). In contrast, the XPF-deficient cells showed accumulation of breaks immediately after treatment in a dose-dependent manner. However, at 48 hr post treatment, cells showed fewer chromosomal breaks but increased radial formation, as is evident from Figs. 6A and 6B. The functional significance of the XPF gene was evident by the fact that XPF-deficient cells formed radials at a significantly higher frequency (>90%) as compared to wild-type cells (12–36%) at the same given formaldehyde concentrations (300–400 µM) (Fig. 6B). A comparison of radial formation in the XPF-deficient, XPB-deficient, and Rad51-deficient cells revealed that following formaldehyde treatment (100 µM), accumulation of radials in the XPF-deficient and Rad51-deficient cells occurred at a significantly higher frequency than the XPB-deficient cells (Fig. 6C). It was also observed that approximately 5% of Rad51-deficient cells exhibited spontaneous radial formation and unlike XPF-deficient cells, Rad51-deficient cells showed increased radials immediately after formaldehyde treatment that continued to escalate following a recovery period of 48 hr (Fig. 6C).
Fig. 6.

Formaldehyde-induced chromosomal breaks and radials are limited in the presence of a functional XPF gene. (A) A comparison of AA8 (wild-type) and UV41 (XPF-deficient) cells containing chromosomal breaks is graphically represented. Following formaldehyde treatment, AA8 and UV41 cells were harvested immediately after a 4 hr treatment or following a 48 hr recovery. (B) Percentage of AA8 and UV41 cells containing radials (post 48 hr recovery) is graphically represented. (C) A comparison of radial formation in AA8, UV24 (XPB-deficient), UV41, and Rad51D (Rad51-deficient) cells immediately after a 4 hr formaldehyde treatment (100 µM) or following a 48 hr recovery.
At higher formaldehyde concentrations, both wild-type and XPF-deficient cells also showed an accumulation of cells exhibiting aneuploidy and polyploidy (ranging from triploid to octaploid). Interestingly, karyotypic analyses showed comparable ploidy changes for both wild-type and XPF-deficient formaldehyde-treated cells (Table 1). This was surprising since the flow cytometry data suggested more ploidy change in XPF-deficient cells (Fig. 2B). While flow cytometry measures DNA content of the entire population being studied, karyotyping is performed selectively on the metaphase population derived from the dividing cells. This technical limitation of cytogenetic analyses allowed for an assessment of only the mitotic population exclusive of the non-mitotic population and could result in an underestimation of the ploidy status. Thus, it is possible that a subset of cells analyzed through flow cytometry, but not identified by karyotypic analyses, represented the population that failed to reach mitosis during the time period captured by cytogenetics. As an indirect test, this hypothesis was examined by comparing the mitotic indices for the untreated and formaldehyde-treated wild-type and XPF-deficient cells. Although the mitotic indices for the untreated wild-type and XPF-deficient cells were comparable, the mitotic indices for the XPF-deficient formaldehyde-treated cells were significantly lower than the wild-type treated cells (data not shown). Based on these cytogenetic studies, it can be concluded that a functional XPF gene is important for suppression of formaldehyde-induced chromosomal breaks, radials, and possibly ploidy alterations.
Table 1.
Ploidy analyses of AA8 and UV41 cells following formaldehyde treatment.
| Cell Line | Formaldehyde (μM) |
Treatment time (hr) |
Recovery time (hr) |
% diploid | % triploid | % tetraploid | % > tetraploid |
|---|---|---|---|---|---|---|---|
| AA8 | |||||||
| 0 | n/a | n/a | 100 | -- | -- | -- | |
| 50 | 4 | 92 | 1 | 7 | -- | ||
| 100 | 4 | 94 | 1 | 5 | -- | ||
| 200 | 4 | 92 | 2 | 6 | -- | ||
| 50 | 4 | 48 | 99 | -- | 1 | -- | |
| 100 | 4 | 48 | 99 | -- | 1 | -- | |
| 200 | 4 | 48 | 93 | -- | 5 | 2 | |
| 300 | 4 | 48 | 61 | 7 | 22 | 10 | |
| 400 | 4 | 48 | 67 | 5 | 19 | 10 | |
| UV41 | 0 | n/a | n/a | 100 | -- | -- | -- |
| 50 | 4 | 96 | -- | 4 | -- | ||
| 100 | 4 | 100 | -- | -- | -- | ||
| 200 | 4 | 91 | -- | 6 | 3 | ||
| 50 | 4 | 48 | 96 | -- | 4 | -- | |
| 100 | 4 | 48 | 99 | -- | 1 | -- | |
| 200 | 4 | 48 | 92 | 5 | 3 | -- | |
| 300 | 4 | 48 | 74 | 2 | 19 | 5 | |
| 400 | 4 | 48 | 74 | 6 | 17 | 3 | |
4. Discussion
Previous studies have begun to elucidate the DNA repair and tolerance pathways that mitigate the genotoxic events following formaldehyde exposure. Specifically, formaldehyde has been shown to elicit cellular responses involving NER and HR pathways to overcome DNA damage. One of the critical players in the NER pathway is the XPF/ERCC1 complex that makes an incision 5′ to the lesion, followed by strand displacement and XPG-mediated 3′ incision [31]. Interestingly, both biochemical and cellular studies support an additional role for XPF/ERCC1 in ICL repair [32–38], where it is proposed to work in conjunction with the HR pathway (independent of NER) to remove the ICL lesion. Consistent with previously published data [19], our study shows an increased formaldehyde sensitivity of XPF and ERCC1 mutants compared to other NER mutants, suggesting that the role of the XPF/ERCC1 complex is more critical than the other NER components for processing formaldehyde-induced DNA damage. One plausible explanation for the hypersensitive phenotype of the XPF and ERCC1 mutants, relative to other NER mutants may be that these proteins are involved in repair of both formaldehyde-induced DPCs and non-DPC lesions (such as ICLs), while the function of classical NER players remain confined to DPCs only. Alternatively, it is possible that both DPC and ICL repair mechanisms share common steps for processing and removal of the lesions. However, our studies in yeast clearly demonstrate that formaldehyde and ICL tolerance pathways are distinct but may have some common steps for processing and removal of formaldehyde-induced lesions [21].
To elucidate the role of XPF endonuclease in repair of formaldehyde-induced DPCs and examine similarities and/or differences between ICL- and DPC-induced repair mechanisms in mammalian cells, we studied the formation and repair of DSBs that may be formed during the processing of formaldehyde-induced DPCs. It has been previously demonstrated that formaldehyde induces DNA DSBs as measured by the appearance of γH2AX foci; however, the nature of these DSBs and their repair kinetics have not been well studied, particularly in an XPF-deficient background. Unlike IR-induced DNA DSBs that have a very short half-life of minutes [39], formaldehyde-induced DSBs are significantly long-lived as evidenced by the slow rate at which γH2AX foci declined in AA8 cells (an overall 15% decrease in a 24 hr period). A previous study conducted by Niedernhofer et al (2004) showed that MMC-induced DSBs occur via an ERCC1-independent mechanism; however, the DSBs persisted in the absence of ERCC1 suggesting that ERCC1 is required for the repair of MMC-induced DSBs. It is worth noting that our data on formaldehyde-induced DPCs are comparable to MMC-induced DSBs that also exhibit very slow repair kinetics as shown in ERCC1-deficent cells [40]. Thus, it is tempting to speculate that MMC and formaldehyde-induced DSBs share common structural intermediates but differ from IR-induced DSBs. Relative to wild-type cells, XPF-deficient cells exhibited a slightly higher frequency of spontaneous γH2AX foci presumably due to DSBs arising from unrepaired endogenous DNA lesions. It is shown herein that analogous to MMC-induced DSBs [40], most of the formaldehyde-induced DSBs are initiated during the S phase of the cell cycle and can be prevented by inhibiting replication elongation. The significance of the S phase-specific formaldehyde-induced DSBs may be associated with either lesion recognition, or processing of lesions resulting in a DSB at stalled replication forks. Our finding that formaldehyde-induced DSBs are generated in an XPF-independent manner favors a model in which the initial DSB is catalyzed by another endonuclease. However, when XPF function is compromised, DNA DSBs persist for longer and failure to repair these breaks eventually results in cell death. Collectively, these data support a role for XPF in the late steps of formaldehyde-induced DSB repair.
Many of the earlier studies on the formaldehyde-induced DNA damage response focused on studying the immediate effects of this agent and did not assess the cellular alterations following post treatment. This study demonstrates that the cytotoxic and genotoxic effects of formaldehyde may be a consequence of secondary lesions generated during processing of the primary adducts (i.e., DPCs converted to single-strand or double-strand breaks). This is supported by the flow cytometry and cytogenetic results demonstrating that the cell cycle changes and chromosomal rearrangements were more pronounced during recovery following formaldehyde treatment. However, in the absence of repair/tolerance mechanisms, these adverse effects were exhibited immediately and more severely. Specifically, the appearance of chromosomal breaks and radials at high formaldehyde concentrations (≥200 µM) in the wild-type background were observed only during the recovery period, whereas these effects were evident in the XPF-deficient cells immediately after treatment, even at the lowest concentration of formaldehyde (50 µM). Collectively, these findings highlight the cellular and cytogenetic abnormalities that develop following formaldehyde exposure, and also suggest that humans with compromised repair function(s) are at increased risks of formaldehyde-induced genomic instability. In mammalian cells, similar patterns of cell cycle profiles, DSB formation and repair kinetics, and chromosomal abnormalities, suggest that there is a substantial overlap between the repair mechanisms triggered by ICL-inducing agents and formaldehyde.
In addition to chromosomal breaks and radial formation, formaldehyde treatment also resulted in polyploidy and aneuploidy in both wild-type and XPF-deficient cells. Polyploidy and aneuploidy are believed to be a consequence of unequal chromosomal segregation, and it has been suggested that the presence of multiple centrosomes can subsequently promote either bipolar or multipolar cell divisions, leading to chromosomal mis-segregation and abnormalities [28]. However, viable progenies result mostly from bipolar cell divisions but not multipolar cell divisions [41]. Our data suggest that formaldehyde-treated cells accumulate centrosome abnormalities and that following bipolar cell divisions, these may result in daughter cells containing mis-segregated chromosomes as evidenced by the ploidy changes demonstrated by flow cytometry and cytogenetic analyses.
5. Conclusions
Epidemiological studies provide evidence linking formaldehyde exposure with an increased risk of leukemia [42, 43]. Humans exposed to formaldehyde appeared to have low peripheral blood cell counts and the myeloid blood progenitor cells isolated from these workers had significantly elevated leukemia-specific chromosomal changes [44]. Despite some contradictory findings, studies in rodents have suggested a correlation between formaldehyde exposure with squamous cell carcinomas, intestinal leiomyosarcoma, hemolymphoreticular neoplasms, and testicular tumors [44]. Animal studies have also suggested that formaldehyde could act as a tumor promoter or act as a co-carcinogen when administered with other substances [1, 45].
Our observation that formaldehyde-induced genomic instability is associated with centrosome abnormalities and chromosome segregation defects shed light on the possible underlying mechanisms contributing to carcinogenic risks associated with formaldehyde exposure. Considering the carcinogenic potential of formaldehyde, it is important to study the cytotoxic intermediates that are generated upon exposure to formaldehyde and understand the underlying mechanisms involved in processing of the DNA lesions. Our findings substantially extend the knowledge of the cellular and genetic changes caused by formaldehyde exposure and reveal the significance of XPF function in maintaining genomic stability and mitigating cytogenetic defects caused by formaldehyde.
Supplementary Material
Cell cycle profiles of UV24 (XPB-deficient) cells measured by flow cytometry analyses of DNA content. The cells were treated with formaldehyde for 4 hr at the indicated concentrations and DNA content was measured immediately after the treatment, as well as after a 24 hr (24h-R) and 48 hr (48h-R) recovery time. The flow cytometry results have been summarized by generating overlay images representing all the samples harvested at each time point (4h, 24h-R, and 48h-R).
Cell cycle profiles of aphidicolin-synchronized cultures of AA8 (wild-type) (A) and UV41 (XPF-deficient) (B) cells. The cells were treated with aphidicolin for 17 hr and samples were harvested immediately after the treatment (Aph 17h), as well as after a 2 hr (Aph 2h-R), 6 hr (Aph 16h-R), and 26 hr (Aph 26h-R) release post aphidicolin treatment.
Cell death in response to formaldehyde-induced DNA damage. Apoptosis was measured in formaldehyde-treated AA8 (wild-type) (A) and UV41 (XPF-deficient) (B) cells using an Annexin-V assay.
Metaphase spreads of AA8 (wild-type) and UV41 (XPF-deficient) cells showing radial formation (arrows) after formaldehyde treatment.
Acknowledgements
We thank R. Stephen Lloyd, Aaron C. Jacobs, and other members of the McCullough and Lloyd laboratories for their valuable insights and suggestions. We are thankful to John Hinz and Paul F. Wilson for providing Rad51D cells. We thank Mandy Boyd and Dorian LaTocha for assistance with flow cytometry analyses, and Stefanie Kaech Petrie and Aurelie Snyder for microscopy assistance. This research was supported by a grant from the National Institute of Health CA 106858 (AKM).
Abbreviations
- CHO
Chinese hamster ovary cells
- DPC
DNA-protein crosslink
- NER
nucleotide excision repair
- PI
propidium iodide
- G1
gap phase 1
- G2
gap phase 2
- HR
homologous recombination
- M
mitotic phase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that they have no conflict of interests.
Appendix A. Supplementary data
References
- 1.Formaldehyde. Rep Carcinog. 2011:195–205. [PubMed] [Google Scholar]
- 2.Craft TR, Bermudez E, Skopek TR. Formaldehyde mutagenesis and formation of DNA-protein crosslinks in human lymphoblasts in vitro. Mutat Res. 1987;176:147–155. doi: 10.1016/0027-5107(87)90262-4. [DOI] [PubMed] [Google Scholar]
- 3.Crosby RM, Richardson KK, Craft TR, Benforado KB, Liber HL, Skopek TR. Molecular analysis of formaldehyde-induced mutations in human lymphoblasts and E. coli. Environ Mol Mutagen. 1988;12:155–166. doi: 10.1002/em.2860120202. [DOI] [PubMed] [Google Scholar]
- 4.Ma TH, Harris MM. Review of the genotoxicity of formaldehyde. Mutat Res. 1988;196:37–59. doi: 10.1016/0165-1110(88)90027-9. [DOI] [PubMed] [Google Scholar]
- 5.Liber HL, Benforado K, Crosby RM, Simpson D, Skopek TR. Formaldehyde-induced and spontaneous alterations in human hprt DNA sequence and mRNA expression. Mutat Res. 1989;226:31–37. doi: 10.1016/0165-7992(89)90089-4. [DOI] [PubMed] [Google Scholar]
- 6.Suruda A, Schulte P, Boeniger M, Hayes RB, Livingston GK, Steenland K, Stewart P, Herrick R, Douthit D, Fingerhut MA. Cytogenetic effects of formaldehyde exposure in students of mortuary science. Cancer Epidemiol Biomarkers Prev. 1993;2:453–460. [PubMed] [Google Scholar]
- 7.Shaham J, Gurvich R, Kaufman Z. Sister chromatid exchange in pathology staff occupationally exposed to formaldehyde. Mutat Res. 2002;514:115–123. doi: 10.1016/s1383-5718(01)00334-5. [DOI] [PubMed] [Google Scholar]
- 8.Ye X, Yan W, Xie H, Zhao M, Ying C. Cytogenetic analysis of nasal mucosa cells and lymphocytes from high-level long-term formaldehyde exposed workers and low-level short-term exposed waiters. Mutat Res. 2005;588:22–27. doi: 10.1016/j.mrgentox.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Iarmarcovai G, Bonassi S, Sari-Minodier I, Baciuchka-Palmaro M, Botta A, Orsiere T. Exposure to genotoxic agents, host factors, and lifestyle influence the number of centromeric signals in micronuclei: a pooled re-analysis. Mutat Res. 2007;615:18–27. doi: 10.1016/j.mrfmmm.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Lazutka JR, Lekevicius R, Dedonyte V, Maciuleviciute-Gervers L, Mierauskiene J, Rudaitiene S, Slapsyte G. Chromosomal aberrations and sister-chromatid exchanges in Lithuanian populations: effects of occupational and environmental exposures. Mutat Res. 1999;445:225–239. doi: 10.1016/s1383-5718(99)00128-x. [DOI] [PubMed] [Google Scholar]
- 11.He JL, Jin LF, Jin HY. Detection of cytogenetic effects in peripheral lymphocytes of students exposed to formaldehyde with cytokinesis-blocked micronucleus assay. Biomed Environ Sci. 1998;11:87–92. [PubMed] [Google Scholar]
- 12.Bauchinger M, Schmid E. Cytogenetic effects in lymphocytes of formaldehyde workers of a paper factory. Mutat Res. 1985;158:195–199. doi: 10.1016/0165-1218(85)90085-0. [DOI] [PubMed] [Google Scholar]
- 13.Miller CA, 3rd, Costa M. Analysis of proteins cross-linked to DNA after treatment of cells with formaldehyde, chromate, and cis-diamminedichloroplatinum(II) Mol Toxicol. 1989;2:11–26. [PubMed] [Google Scholar]
- 14.Casanova M, Morgan KT, Steinhagen WH, Everitt JI, Popp JA, Heck HD. Covalent binding of inhaled formaldehyde to DNA in the respiratory tract of rhesus monkeys: pharmacokinetics, rat-to-monkey interspecies scaling, and extrapolation to man. Fundam Appl Toxicol. 1991;17:409–428. doi: 10.1016/0272-0590(91)90230-2. [DOI] [PubMed] [Google Scholar]
- 15.Casanova M, Morgan KT, Gross EA, Moss OR, Heck HA. DNA-protein cross-links and cell replication at specific sites in the nose of F344 rats exposed subchronically to formaldehyde. Fundam Appl Toxicol. 1994;23:525–536. doi: 10.1006/faat.1994.1137. [DOI] [PubMed] [Google Scholar]
- 16.Minko IG, Kurtz AJ, Croteau DL, Van Houten B, Harris TM, Lloyd RS. Initiation of repair of DNA-polypeptide cross-links by the UvrABC nuclease. Biochemistry. 2005;44:3000–3009. doi: 10.1021/bi0478805. [DOI] [PubMed] [Google Scholar]
- 17.Minko IG, Zou Y, Lloyd RS. Incision of DNA-protein crosslinks by UvrABC nuclease suggests a potential repair pathway involving nucleotide excision repair. Proc Natl Acad Sci U S A. 2002;99:1905–1909. doi: 10.1073/pnas.042700399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurtz AJ, Lloyd RS. 1,N2-deoxyguanosine adducts of acrolein, crotonaldehyde, and trans-4-hydroxynonenal cross-link to peptides via Schiff base linkage. J Biol Chem. 2003;278:5970–5976. doi: 10.1074/jbc.M212012200. [DOI] [PubMed] [Google Scholar]
- 19.Nakano T, Katafuchi A, Matsubara M, Terato H, Tsuboi T, Masuda T, Tatsumoto T, Pack SP, Makino K, Croteau DL, Van Houten B, Iijima K, Tauchi H, Ide H. Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J Biol Chem. 2009;284:27065–27076. doi: 10.1074/jbc.M109.019174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakano T, Morishita S, Katafuchi A, Matsubara M, Horikawa Y, Terato H, Salem AM, Izumi S, Pack SP, Makino K, Ide H. Nucleotide excision repair and homologous recombination systems commit differentially to the repair of DNA-protein crosslinks. Mol Cell. 2007;28:147–158. doi: 10.1016/j.molcel.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 21.de Graaf B, Clore A, McCullough AK. Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair (Amst) 2009;8:1207–1214. doi: 10.1016/j.dnarep.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, Buerstedde JM, Gillespie DA, Sale JE, Yamazoe M, Bishop DK, Takata M, Takeda S, Watanabe M, Swenberg JA, Nakamura J. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Res. 2007;67:11117–11122. doi: 10.1158/0008-5472.CAN-07-3028. [DOI] [PubMed] [Google Scholar]
- 23.Kumari A, Minko IG, Smith RL, Lloyd RS, McCullough AK. Modulation of UvrD helicase activity by covalent DNA-protein cross-links. J Biol Chem. 2010;285:21313–21322. doi: 10.1074/jbc.M109.078964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ray M, Mohandas T. Proposed banding nomenclature for the Chinese hamster chromosomes (Cricetulus griseus) Cytogenet Cell Genet. 1976;16:83–91. doi: 10.1159/000130559. [DOI] [PubMed] [Google Scholar]
- 25.Al-Minawi AZ, Lee YF, Hakansson D, Johansson F, Lundin C, Saleh-Gohari N, Schultz N, Jenssen D, Bryant HE, Meuth M, Hinz JM, Helleday T. The ERCC1/XPF endonuclease is required for completion of homologous recombination at DNA replication forks stalled by inter-strand cross-links. Nucleic Acids Res. 2009;37:6400–6413. doi: 10.1093/nar/gkp705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Farrell PH. Triggering the all-or-nothing switch into mitosis. Trends Cell Biol. 2001;11:512–519. doi: 10.1016/s0962-8924(01)02142-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shimada M, Komatsu K. Emerging connection between centrosome and DNA repair machinery. J Radiat Res (Tokyo) 2009;50:295–301. doi: 10.1269/jrr.09039. [DOI] [PubMed] [Google Scholar]
- 29.Thompson SL, Bakhoum SF, Compton DA. Mechanisms of chromosomal instability. Curr Biol. 2010;20:R285–R295. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noda T, Takahashi A, Kondo N, Mori E, Okamoto N, Nakagawa Y, Ohnishi K, Zdzienicka MZ, Thompson LH, Helleday T, Asada H, Ohnishi T. Repair pathways independent of the Fanconi anemia nuclear core complex play a predominant role in mitigating formaldehyde-induced DNA damage. Biochem Biophys Res Commun. 2011;404:206–210. doi: 10.1016/j.bbrc.2010.11.094. [DOI] [PubMed] [Google Scholar]
- 31.Staresincic L, Fagbemi AF, Enzlin JH, Gourdin AM, Wijgers N, Dunand-Sauthier I, Giglia-Mari G, Clarkson SG, Vermeulen W, Scharer OD. Coordination of dual incision and repair synthesis in human nucleotide excision repair. EMBO J. 2009;28:1111–1120. doi: 10.1038/emboj.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000;20:7980–7990. doi: 10.1128/mcb.20.21.7980-7990.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McHugh PJ, Sones WR, Hartley JA. Repair of intermediate structures produced at DNA interstrand cross-links in Saccharomyces cerevisiae. Mol Cell Biol. 2000;20:3425–3433. doi: 10.1128/mcb.20.10.3425-3433.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mu D, Bessho T, Nechev LV, Chen DJ, Harris TM, Hearst JE, Sancar A. DNA interstrand cross-links induce futile repair synthesis in mammalian cell extracts. Mol Cell Biol. 2000;20:2446–2454. doi: 10.1128/mcb.20.7.2446-2454.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J Biol Chem. 2000;275:26632–26636. doi: 10.1074/jbc.C000337200. [DOI] [PubMed] [Google Scholar]
- 36.Adair GM, Rolig RL, Moore-Faver D, Zabelshansky M, Wilson JH, Nairn RS. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 2000;19:5552–5561. doi: 10.1093/emboj/19.20.5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McHugh PJ, Spanswick VJ, Hartley JA. Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2001;2:483–490. doi: 10.1016/S1470-2045(01)00454-5. [DOI] [PubMed] [Google Scholar]
- 38.Wood RD. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ Mol Mutagen. 2010;51:520–526. doi: 10.1002/em.20569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NG, Beverloo HB, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol. 2004;24:5776–5787. doi: 10.1128/MCB.24.13.5776-5787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature. 2009;460:278–282. doi: 10.1038/nature08136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Freeman LE, Nakamura J, Hecht SS, Vandenberg JJ, Smith MT, Sonawane BR. Formaldehyde and leukemia: epidemiology, potential mechanisms, and implications for risk assessment. Environ Mol Mutagen. 2010;51:181–191. doi: 10.1002/em.20534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jakab MG, Klupp T, Besenyei K, Biro A, Major J, Tompa A. Formaldehyde-induced chromosomal aberrations and apoptosis in peripheral blood lymphocytes of personnel working in pathology departments. Mutat Res. 2010;698:11–17. doi: 10.1016/j.mrgentox.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 44.Zhang L, Tang X, Rothman N, Vermeulen R, Ji Z, Shen M, Qiu C, Guo W, Liu S, Reiss B, Freeman LB, Ge Y, Hubbard AE, Hua M, Blair A, Galvan N, Ruan X, Alter BP, Xin KX, Li S, Moore LE, Kim S, Xie Y, Hayes RB, Azuma M, Hauptmann M, Xiong J, Stewart P, Li L, Rappaport SM, Huang H, Fraumeni JF, Jr, Smith MT, Lan Q. Occupational exposure to formaldehyde, hematotoxicity, and leukemia-specific chromosome changes in cultured myeloid progenitor cells. Cancer Epidemiol Biomarkers Prev. 2010;19:80–88. doi: 10.1158/1055-9965.EPI-09-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Final Report on Carcinogens Background Document for Formaldehyde. Rep Carcinog Backgr Doc. 2010 i-512. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell cycle profiles of UV24 (XPB-deficient) cells measured by flow cytometry analyses of DNA content. The cells were treated with formaldehyde for 4 hr at the indicated concentrations and DNA content was measured immediately after the treatment, as well as after a 24 hr (24h-R) and 48 hr (48h-R) recovery time. The flow cytometry results have been summarized by generating overlay images representing all the samples harvested at each time point (4h, 24h-R, and 48h-R).
Cell cycle profiles of aphidicolin-synchronized cultures of AA8 (wild-type) (A) and UV41 (XPF-deficient) (B) cells. The cells were treated with aphidicolin for 17 hr and samples were harvested immediately after the treatment (Aph 17h), as well as after a 2 hr (Aph 2h-R), 6 hr (Aph 16h-R), and 26 hr (Aph 26h-R) release post aphidicolin treatment.
Cell death in response to formaldehyde-induced DNA damage. Apoptosis was measured in formaldehyde-treated AA8 (wild-type) (A) and UV41 (XPF-deficient) (B) cells using an Annexin-V assay.
Metaphase spreads of AA8 (wild-type) and UV41 (XPF-deficient) cells showing radial formation (arrows) after formaldehyde treatment.