

Abstract
Abnormal cardiac ryanodine receptor (RyR2) function is recognized as an important factor in the pathogenesis of heart failure (HF). However, the specific molecular causes underlying RyR2 defects in HF remain poorly understood. In the present study, we used a canine model of chronic HF to test the hypothesis that the HF-related alterations in RyR2 function are caused by posttranslational modification by reactive oxygen species generated in the failing heart. Experimental approaches included imaging of cytosolic ([Ca2+]c) and sarcoplasmic reticulum (SR) luminal Ca2+ ([Ca2+]SR) in isolated intact and permeabilized ventricular myocytes and single RyR2 channel recording using the planar lipid bilayer technique. The ratio of reduced to oxidized glutathione, as well as the level of free thiols on RyR2 decreased markedly in failing versus control hearts consistent with increased oxidative stress in HF. RyR2-mediated SR Ca2+ leak was significantly enhanced in permeabilized myocytes, resulting in reduced [Ca2+]SR in HF compared to control cells. Both SR Ca2+ leak and [Ca2+]SR were partially normalized by treating HF myocytes with reducing agents. Conversely, oxidizing agents accelerated SR Ca2+ leak and decreased [Ca2+]SR in cells from normal hearts. Moreover, exposure to antioxidants significantly improved intracellular Ca2+-handling parameters in intact HF myocytes. Single RyR2 channel activity was significantly higher in HF versus control because of increased sensitivity to activation by luminal Ca2+ and was partially normalized by reducing agents through restoring luminal Ca2+ sensitivity oxidation of control RyR2s enhanced their luminal Ca2+ sensitivity, thus reproducing the HF phenotype. These findings suggest that redox modification contributes to abnormal function of RyR2s in HF, presenting a potential therapeutic target for treating HF.
Keywords: ryanodine receptor, heart failure, disulfide oxidation, Ca2+-induced Ca2+ release
Heart failure (HF) is a disease state in which the muscle of the heart becomes too weak to adequately pump blood through the body. Although HF is very complex, altered intracellular Ca2+ handling, and, in particular, depletion of intracellular Ca2+ stores, is recognized as an important feature of myocytes from failing hearts.1–5 In cardiac muscle, Ca2+ release from the intracellular storage compartment, the sarcoplasmic reticulum (SR), is central to the process of excitation– contraction (EC) coupling.6 During each heart beat, in response to a small Ca2+ entry via sarcolemmal voltage-dependent Ca2+ channels, SR Ca2+ is released through specialized Ca2+-gated SR Ca2+ channels called ryanodine receptors (RyR2s) causing shortening of the contractile filaments. Evidence from human HF and various models of HF suggests that RyR2s become excessively active ie, “leaky” in the failing heart.7–9 This increase in RyR2 activity has been linked to defective modulation of the channel by SR luminal Ca2+, a mechanism that normally operates to terminate SR Ca2+ release and keep the RyR2 channels closed (ie, refractory) during diastole.10 Uncontrolled RyR2 gating is expected to result in increased diastolic SR Ca2+ leak causing a reduction of the SR Ca2+ content, thus limiting the ability of cardiac muscle to contract in systole. Consistent with this scenario, we recently found that increased SR Ca2+ leak is a major determinant of the reduced SR Ca2+ content and suppressed Ca2+ signaling in myocytes from a chronic canine model of HF.11,12
The underlying biochemical causes of HF-related abnormalities in RyR2 function are a subject of intense debate. Marks and colleagues have reported that increased phosphorylation of RyR2 by protein kinase A (on Ser2809) results in dissociation of FKBP12.6 from the RyR2 complex, causingRyR2s to become hyperactive.7,13 However, various aspects of this proposed mechanism have been questioned by others.14–16 More recently, Bers and colleagues17,18 presented data suggesting that RyR2 phosphorylation by Ca2+/calmodulin-dependent protein kinase Ca2+/calmodulin-dependent protein kinase (CaMK II on Ser2815) contributes to elevated SR Ca2+ leak in HF. However, other investigators failed to detect a stimulatory influence of CaMKII phosphorylation on SR Ca2+ release.19
Alterations in phosphorylation status is not the only type of biochemical modification that could affect RyR2s in HF. HF is accompanied by increased oxidative stress resulting from formation of reactive oxygen species (ROS) (eg, superoxide anion, hydrogen peroxide, and hydroxyl radical).20–23 These compounds are capable of reacting with reactive cysteines (ie, thiols susceptible to redox-based modifications), causing changes in RyR2 function. Sulfhydryl oxidation by various oxidizing agents has been shown to increase RyR2 open probability.24–26 Accordingly, the goal of the present study was to test the hypothesis that increased RyR2-mediated leak in the failing heart is caused by abnormal modification(s) of the RyR2s by ROS generated in the course of HF.
Materials and Methods
Chronic left ventricular dysfunction was induced by right ventricular tachypacing in a canine model for ≈4 months.27 Single myocytes were isolated as previously described.9 Cytosolic and intra-SR [Ca2+] changes were monitored by using confocal microscopy. Single-channel recordings were performed using the lipid bilayer technique. Redox balance in control and HF tissue samples were assessed by measuring levels of reduced (GSH) and oxidized (GSSG) forms of glutathione with high-performance liquid chromatography, whereas in myocytes, the fluorescent indicator 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) and confocal microscopy were used to measure ROS. The amounts of oxidized thiols in RyR2s were determined with monobromobimane (mBB).
An expanded Material and Methods section can be found in the online data supplement at http://circres.ahajournals.org.
Results
Increased Oxidative Stress and RyR2 Oxidation in HF
We assessed redox balance and oxidative stress in HF versus control myocardium. Although the reduced glutathione levels were not different, the ratio of reduced to oxidized glutathione decreased significantly in HF compared to controls confirming that oxidative stress is elevated in HF (Figure 1A and 1B). Consistent with increased oxidative stress, imaging of myocytes loaded with the ROS indicator DCFDA showed a significant increase in ROS in HF compared to control cells (Figure 1C and 1D). Importantly, incubation of HF myocytes with the cell-permeable ROS scavenger 2-mercaptopropionylglycine (MPG) restored normal ROS levels in these cells. Furthermore, measurements with mBB revealed an increased amount of oxidized thiols in RyR2s from failing hearts (Figure 1E and 1G and Figure I in the online data supplement).
Figure 1.

Increases in oxidative stress, ROS formation, and RyR2 oxidation in heart failure. A, Averages of the levels of reduced form of glutathione (GSH) measured in tissue homogenates from normal (38.9±14.6 nmol per mg tissue) and failing hearts (29.4±11.2 nmol per mg tissue). B, Averages of ratios of reduced to oxidized glutathione (GSH/GSSG) measured in tissue homogenates from normal (28.6±10.4) and failing hearts (17.7±5.1). *P<0.05 (n=9 and 8 for control and HF, respectively). C, Fluorescence of the ROS-sensitive indicator DCFDA loaded into myocytes from normal and failing hearts. Representative images of DCFDA-loaded cells from control (top) and failing hearts under basal conditions (middle) and after 30 minutes of incubation with 800 μmol/L MPG (bottom). D, Summary graph for averages of DCFDA fluorescence normalized to the signal in the presence of 10 mmol/L H2O2 (Fmax) measured in myocytes from control hearts (2.47±0.09) and myocytes from failing hearts under basal conditions (10.77±1.01) and in the presence of MPG (2.48±0.28). *P<0.05 vs control (n=21 to 32). E, Representative images of Coomassie-stained gels and mBB fluorescence intensity of RyR2 from normal and failing hearts. F, Pooled data for RyR2 content in samples from normal vs failing hearts. G, Relative free thiol content (%) of RyR2s from control vs HF samples obtained by normalizing mBB fluorescence to RyR2 level. *P<0.05 vs control (n=10).
Normalization of Ca2+ Transients in HF Myocytes by Antioxidants
Next, we performed measurements of cytosolic Ca2+ transients in intact cardiac myocytes isolated from control and failing hearts (Figure 2). Figure 2A depicts representative line-scan images and corresponding time-dependent profiles of Ca2+ transients recorded in field-stimulated control and HF myocytes under baseline conditions and after treatment of the cells with oxidizing or reducing reagents. Characteristic of HF, the amplitude of the Ca2+ transients was significantly lower in HF than in control myocytes. In control myocytes, application of the oxidizing agent 2,2′-dithiodipyridine (DTDP) depressed the amplitude of Ca2+ transients making them similar to the HF phenotype. Conversely, incubation of HF myocytes with the antioxidant MPG increased the amplitude of the Ca2+ transients rendering them similar to controls. Summary data are shown in Figure 2B. Exposure to MPG had no significant effect in control myocytes (supplemental Figure II, A and B), consistent with the notion that reversible oxidation of components of the EC coupling machinery is minimal in myocytes from healthy hearts. Thus, treatment with antioxidants improves the parameters of Ca2+ transients in HF, whereas the impact of HF on Ca2+ transients can be mimicked by oxidative agents.
Figure 2.
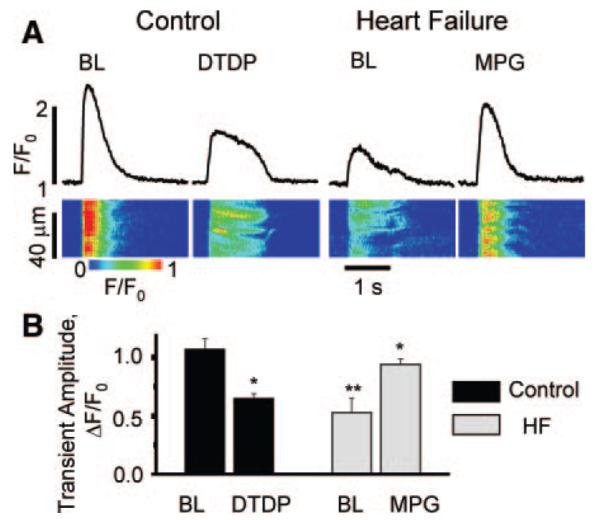
Effects of oxidizing and reducing reagents on Ca2+ transients in control and HF myocytes. A, Representative linescan images and corresponding time-dependent profiles of cytosolic Ca2+ transients recorded during field stimulation (0.3 Hz) of control (left) and HF (right) myocytes under baseline (BL) conditions and after treatment of the cells with DTDP (100 μmol/L) or reducing MPG (800 μmol/L for 30 minutes), respectively. B, Summary graph for averages of amplitudes of Ca2+ transient. Rhod-2 ΔF/F0 values were 1.07±0.09 for control, 0.65±0.04 for control+DTDP, 0.53±0.12 for HF, and 0.94±0.05 for HF+MPG (n from 5 to 9). *P<0.05 vs baseline, **P<0.05 vs control.
Normalization of [Ca2+]SR and SR Ca2+ Leak in HF Myocytes by Antioxidants
To further investigate the role of redox modifications in mediating the HF-associated alterations in intracellular Ca2+ handling, we performed measurements of intra-SR [Ca2+]in saponin-permeabilized myocytes using the low-affinity Ca2+ indicator Fluo-5N loaded to the SR. Recently, we demonstrated that [Ca2+]SR is reduced in this particular model of HF because of increased Ca2+ leak via RyR2s in the absence of significant changes in SERCA-mediated SR Ca2+ uptake.9,11 Consistent with our previous results, baseline [Ca2+]SR was substantially lower in HF than in control myocytes (Figure 3A and 3B). Furthermore, SR Ca2+ leak, assessed as the difference in [Ca2+]SR measured in the presence or absence of the RyR2 inhibitor ruthenium red (RR) (30 μmol/L), was significantly higher in HF than in control myocytes (Figure 3C). Application of the oxidizing agent DTDP (1 to 200 μmol/L) to control cells caused a rapid decline in [Ca2+]SR, and increased SR Ca2 leak. Subsequent applica tion of dithiothreitol (DTT) (1 mmol/L) caused a partial recovery of [Ca2+]SR and normalization of leak in DTDP-treated cells. These results are documented in Figure 3A, 3B, and 3D. At the same time, treatment of [Ca2+]SR-depleted HF cells with DTT (0.01 to 10 mmol/L) produced an increase in [Ca2+]SR and lowered Ca2+ leak toward control values (Figure 3A, 3B, and 3E). Similar results in control and HF myocytes were obtained using the oxidizing and reducing reagents thimerosal and MPG instead of DTDP and DTT, respectively (online-only Data Supplement Figure III).
Figure 3.

Effects of oxidizing and reducing agents on intra-SR Ca2+ in saponin-permeabilized myocytes from normal and failing hearts. A, Representative traces of time-dependent changes of cell-averaged luminal Fluo-5N signals in control (left) and HF permeabilized (right) myocytes. The experimental protocol involved consecutive applications of DTDP (50 μmol/L), DTT (1 mmol/L) and RR (30 μmol/L) in control cells and consecutive applications of DTT (1 mmol/L) and RR (30 μmol/L) in HF cells, as indicated. The values of Fmin and Fmax in both control and HF myocytes after removal of RR were obtained on application of 10 mmol/L caffeine with low Ca2+ and high Ca2+ (10 mmol/L), respectively. B, Summary graph of averaged Fluo-5N signals. The values of normalized Fluo-5N signal were 0.812±0.016, 0.456±0.012, 0.688±0.019, and 0.895±0.028 for baseline and in the presence of DTDP, DTT, and RR, respectively, in myocytes from normal hearts. In myocytes from failing hearts, the values for baseline and in the presence of DTT and RR were 0.610±0.012, 0.712±0.021, and 0.860±0.029, respectively. *P<0.05 vs baseline (BL), **P<0.05 vs control (n=10 to 12 for control and HF, respectively). C, SR Ca2+ leak determined as the difference in [Ca2+]SR measured at the baseline conditions and in the presence of the 30 μmol/L RR in myocytes from healthy (0.083±0.026) and diseased hearts (0.25±0.017). D and E, Dose dependencies of DTDP and DTT effects on intra-SR [Ca2+] in myocytes from healthy (control) and failing hearts, respectively (n was from 7 to 14 for each data point). Data sets were fitted by logistic functions (red lines) with IC50 values of 26 μmol/L for DTDP and 28 μmol/L for DTT.
Consistent with the lack of effects of MPG in intact control cells, DTT did not affect intra-SR [Ca2+] in permeabilized cells from the control group (supplemental Figure II, C and D), again suggesting that RyR2s are not substantially modified by reversible oxidation in myocytes from normal hearts. At the same time, DTDP decreased [Ca2+]SR in HF myocytes in a manner similar to that in control cells (supplemental Figure II, E and F), indicating that augmenting oxidative stress can further elevate SR Ca2+ leak in HF myocytes. Interestingly, the extent of recovery of SR Ca2+ leak by DTT in HF myocytes treated with DTDP was significantly less than in untreated HF myocytes, suggesting that excessive oxidation, resulting from both HF and DTDP, can result in irreversible modification of RyR2s or that DTDP additionally modifies RyR2 at sites inaccessible to DTT.
In a separate series of experiments, we directly measured the RyR2-mediated SR Ca2+ leak following inhibition of SR Ca2+ uptake by thapsigargin (Tg) (Figure 4A and 4B). On application of the SERCA inhibitor Tg, luminal Fluo-5N fluorescence decreases with time, providing a measure of the rate of SR Ca2+ leak. Consistent with the results of steadystate [Ca2+]SR measurements, the SR Ca2+ leak rate was markedly enhanced in HF myocytes compared to controls. We were able to mimic the HF phenotype by treating the control cells with the oxidizing agent DTDP. As shown in Figure 4A, incubating control myocytes with DTDP lowered the basal level of intra-SR [Ca2+] and accelerated the SR Ca2+ leak, which was unmasked by Tg. Conversely, application of DTT normalized SR Ca2+ leak in HF myocytes. Pooled data for time constants of decay in these experiments are presented in Figure 4B. Taken together, these results suggest that the HF-related changes in myocyte Ca2+ handling could be attributed, in part, to redox modifications of components of the SR Ca2+ release machinery including the RyR2 channel.
Figure 4.
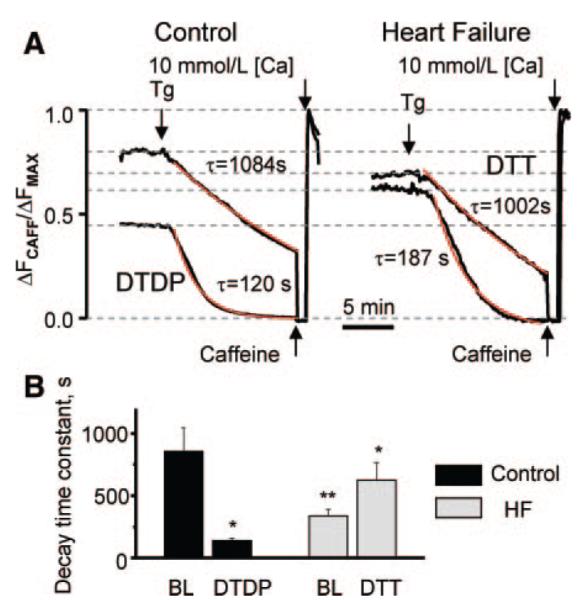
Effects of oxidizing and reducing reagents on SR Ca2+ leak in control and HF permeabilized myocytes. A, Representative recordings of intra-SR Fluo-5N fluorescence as measures of SR Ca2+ leak unmasked by inhibition of the SERCA pump with Tg (10 μmol/L) in control (left) and HF permeabilized myocytes (right) under baseline conditions and after treatment of the control and HF cells with DTDP (50 μmol/L) or DTT (1 mmol/L), respectively. The decline of Fluo-5N signal in the presence of Tg was fitted by exponential functions (red lines). B, Average time constants (from exponential fits) of SR Ca2+ leak for control cells at baseline (BL) conditions (856±191 s, n=12) or in the presence of DTDP (139±20 seconds; n=8) (black bars) and for myocytes from failing hearts at baseline conditions (337±52 seconds; n=10) or in the presence of DTT (625±140 seconds; n=6) (light bars). *P<0.05 vs baseline (BL), **P<0.05 vs control.
Changes in Single RyR2s
Consistent with our previous results,9 RyR2s from failing hearts exhibited higher functional activity than RyR2s from normal hearts (Figure 5). This difference was especially large at low luminal [Ca2+] because of the apparent HF-induced loss of ability of RyR2s to deactivate at reduced [Ca2+]SR. Oxidation by DTDP (10 μmol/L) increased the activity of RyR2 from the control group (Figure 5A, top, and 5B). Importantly, oxidation of control RyR2s resulted in a significant reduction in the ability to increase activity in response to elevation of luminal Ca2+, reminiscent of the behavior of HF channels. On the other hand, treatment of HF channels with the reducing agent DTT (5 mmol/L) decreased channel activity and at the same time restored their ability to respond to luminal Ca2+ (Figure 5A, bottom, and 5C). These results are summarized in Figure 5D, which shows relative values of open probabilities (Po) at low and high luminal Ca2+ for control and HF RyR2s at baseline and after exposure to DTDP or DTT, respectively. As the graph demonstrates, we were able to “convert” the control and HF RyR2 phenotypes from one to another using oxidizing and reducing reagents, respectively.
Figure 5.
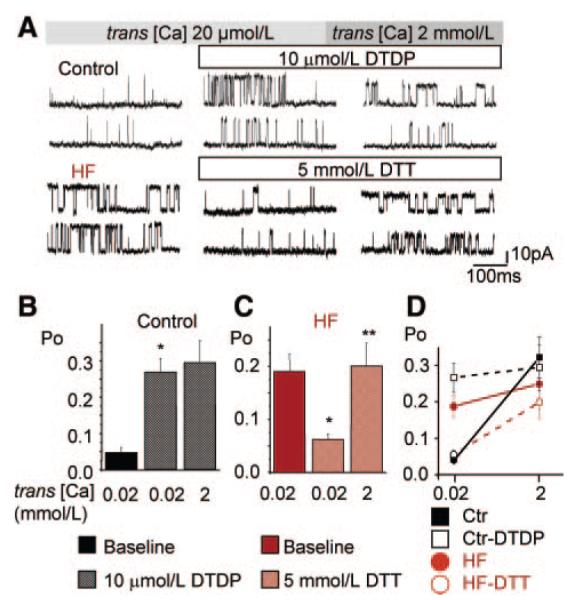
Effects of oxidizing and reducing reagents on activity of RyRs from control and failing hearts. A, Representative recordings of single RyR2 channels from control and HF samples (top and bottom recordings) at low and high trans [Ca2+] and in the presence of either DTDP (10 μmol/L) or DTT (5 mmol/L) as indicated. Single-channel activities were recorded in 350 mmol/L symmetrical CsCH3SO3 solutions at 40 mV. Channel openings are shown as upward deflections from the closed level. B, Summary data of relative values of Po in control RyR2s before and after the addition (to the cis side) of 10 μmol/L DTDP at 20 μmol/L Ca2+ trans [Ca2+] and following elevation of trans [Ca2+] to 2 mmol/L. *P<0.05 vs baseline (n=6 channels). C, Summary data of relative values of Po in HF RyR2s before and after the addition (to the cis side) of 5 mmol/L DTT with 20 μmol/L [Ca2+] trans and following elevation of trans [Ca2+]to 2 mmol/L. *P<0.05 vs baseline, **P<0.05 vs 20 μmol/L Ca2+ trans [Ca2+](n=7 channels). D, Summary data of relative values of Po at low and high luminal Ca2+ as in B and C.
It has been previously shown that RyRs contain different groups of thiols, the oxidation of which exerts different effects on channel function, ie, activation or inactivation.28–30 In agreement with the previous studies, high doses of the oxidant DTDP (200 μmol/L) caused a rapid and nearly complete activation of RyR2s, followed by reduction of Po back toward baseline levels in samples from both control and failing hearts (supplemental Figure IV, A and B). At the same time, low doses of DTDP (10 μmol/L) resulted in a less initially pronounced but sustained stimulation of RyR2s (supplemental Figure IV, B). Additionally, DTT caused a concentration-dependent reduction of Po in hyperactive HF channels toward normal (control) Po levels (supplemental Figure IV, C). These results support the notion that multiple functionally relevant thiols exist in individual RyR2s that become progressively modified with increasing levels of oxidative stress. Considering the concentration dependence of the effects of DTDP and the ability of high doses of DTDP to further increase the activity of HF RyR2s in our experiments, it appears that oxidative modifications of RyR2 in HF do not necessarily involve all the different classes of thiols and are mostly limited to those promoting channel openings at moderate oxidation. Collectively, our findings suggest that redox modifications are involved in acquisition of the HF phenotype by RyR2s.
Role of Different Types of Redox Modifications
Three types of reversible redox modifications have been described for the RyR2 channel, including S-nitrosylation, S-glutathionylation, and disulfide oxidation. To probe the involvement of these specific reactions in HF-related changes in SR Ca2+ leak, we used modification-specific reducing agents. Whereas DTT reverses all 3 types of modification, glutaredoxin reverses S-nitrosylation and S-glutathionylation without affecting oxidized disulfides, and ascorbate reverses only S-nitrosylation.31,32 Both glutaredoxin (1 units/mL+0.5 mmol/L GSH) and ascorbate (1 mmol/L) failed to produce measurable effects on [Ca2+]SR in failing myocytes as Fluo-5N ΔFCAF/ΔFmax on application of glutaredoxin was 0.624±0.027 (n=7) and in the presence of ascorbate was 0.627±0.031 (n=8) compared to this parameter under basal conditions 0.630±0.011 (n=12). At the same time, DTT significantly increased [Ca2+]SR (Figure 3A and 3B). Therefore, it appears that disulfide oxidation is the predominant type of modification involved in elevated RyR2-mediated SR Ca2+ leak in HF.
Discussion
Recently, we demonstrated using a chronic canine model of HF that increased leakage of Ca2+ from the SR via hyperactive RyR2s is a primary factor in determining the reduced SR Ca2+ content and decreased amplitude of cytosolic Ca2+ transients in HF myocytes.11 Our present study in the same model provides evidence that abnormal RyR2 activity in HF is at least partly attributable to modification of RyR2 by reactive oxygen species generated in the failing heart.
Multiple studies in human HF and various animal models of HF have demonstrated that the level of oxidative stress is elevated in HF because of increased production of ROS.20–23,33 In accordance with those previous studies, ROS were increased in failing hearts (Figure 1). Moreover, the number of oxidized thiols on RyR2s increased in HF as compared to control. These changes in redox balance were paralleled by alterations in myocyte Ca2+ cycling, including reduced cytosolic Ca2+ transients (Figure 2), the elevated SR Ca2+ leak and reduced [Ca2+]SR (Figures 3 and 4), abnormalities characteristic of HF. Treatment of HF myocytes with reducing reagents (DTT and MPG) normalized these parameters of intracellular Ca2+ cycling, by restoring normal redox balance. In contrast, treatment of normal myocytes with oxidizing agents (DTDP and thimerosal; Figure 3 and supplemental Figure III) produced alterations in SR Ca2+ leak and [Ca2+]SR similar to those observed in HF cells. Likewise, at the single channel level, RyR2s from failing hearts exhibited abnormally high open probability that could be partially normalized by DTT, whereas oxidation of normal RyR2 channels enhanced their activity, reproducing the HF phenotype (Figure 5).
Previously, we demonstrated that alterations in RyR2 activity in HF are associated with altered modulation of the channel by luminal Ca2+.9 In normal RyR2 channels, lowering luminal Ca2+ causes a decrease in open probability.34,35 This deactivation process is believed to be responsible for termination of systolic SR Ca2+ release and for the transient diastolic refractoriness of Ca2+ signaling that follows Ca2+ release.10 In HF, the ability of RyR2 to deactivate by reduced luminal [Ca2+] becomes compromised, accounting for the elevated diastolic SR Ca2+ leak characteristic of HF myocytes.9 If the HF-related changes in RyR2s were attributable to redox modification of the RyR2 channels, restoring normal redox balance would be expected to normalize RyR2 responsiveness to luminal Ca2+. Consistent with this expectation, application of reducing agents to restore redox balance normalized luminal [Ca2+] dependence of the RyR2s. Moreover, oxidizing normal RyR2s compromised their luminal Ca2+ modulation in a manner similar to that observed in HF RyR2s. Thus, by oxidizing and reducing normal and HF RyR2s, we were able to convert the channels between HF and normal phenotypes, respectively, providing strong evidence for the role of oxidative modifications in HF-related changes in RyR2s.
Because DTT is membrane-impermeant (at pH 7.2), the effects of this compound on RyR2 channels in HF, includingimprovement of luminal Ca2+ dependence, most likely occurred at the cytosolic side rather than the luminal side of RyR2s. The ability of cytosolic ligands to influence luminal Ca2+ sensitivity of the channel is consistent with the notion that RyR2 is an allosteric protein.30 Other examples of such allosteric effects include modulation of RyR2 responsiveness to cytosolic ligands, such as Ca2+,Mg2+, and tetracaine, by luminal Ca2+ and calsequestrin.34 Additionally, there is evidence that protein kinase A phosphorylation at Ser2030 alters the sensitivity of RyR2 to luminal Ca2+.36 Thus, it appears that cytosolic signals other than redox modulation are capable of influencing RyR2 luminal Ca2+ regulation, pre-sumably through allosteric mechanisms that traverse the channel protein. At the same time, given the partial restorative effects of DTT on SR Ca2+ leak and RyR2, it is also possible that some of the redox-mediated alterations of RyR2s in HF involve luminal sites. Consistent with this possibility, the activity of ryanodine receptors has been shown to be influenced by oxidation of the channel from the luminal side of the SR.37
RyR2 contains 89 cysteines, of which only approximately 21 are normally free,38 with even fewer remaining unmodified in HF.33 The redox modification of reactive cysteines is thought to involve disulfide crosslinking, S-nitrosylation, and/or S-glutathionylation.31 The failure of the S-nitrosylation– and S-glutathionylation–specific reducing reagents glutaredoxin and ascorbate to influence the RyR2-mediated leak in a manner consistent with that of DTT suggests that disulfide oxidation is the most likely form of redox-sensitive modulation in our experimental settings. Oxidation of reactive cysteines results in disulfide bonds between neighboring subunits, which are thought to drive a conformational change, promoting RyR2 opening by sensitizing the channel to Ca2+.38,39 Being the most stable of the 3 types of modification, disulfide oxidation may competitively replace40 S-nitrosylation and S-glutathionylation of susceptible reactive thiols during chronic HF, where there is likely to be long-term elevated oxidative stress.
Although treatment with antioxidants resulted in significant improvement of RyR2-mediated leak and single RyR2 function, the normalization was only partial in our experiments. The residual leak could be attributable to inaccessibility of modified sites to DTT, as discussed above; irreversible oxidative modification(s) of the RyR2 channel complex2;29,41 or to other types of posttranslational modifications such as phosphorylation at either protein kinase A7 or CaMKII sites.17 That is, although oxidative modification(s) appear(s) to be a major causal factor in abnormal RyR2 function in our particular HF model, our results do not exclude the possibility that changes in RyR2 phosphorylation status play a role in the alterations in cellular Ca2+ cycling and RyR2 activity in other models, stages, or durations of HF. Interestingly, elevated circulating catecholamines (which occur in the canine tachypacing model of HF42) while increasing phosphorylation, can also result in oxidative stress43,44 through autooxidation or other mechanisms. Previously, it has been proposed33 that altered interdomain interactions coupled with dissociation of FKBP12.6 caused by redox modification of the RyR2 contributes to the enhanced RyR2-mediated leak in HF. Although our data support the role of oxidative modification of RyR2s in HF, the role of FKBP12.6 is not supported by our experiments. Indeed, in our experiments normalization of SR Ca2+ leak in permeabilized cells and normalization of open probability of reconstituted RyR2s by reducing agents occurred in the absence of available FKBP12.6 to reassociate with RyR2s.
Limitations
Our studies performed in isolated myocytes and lipid bilayer experiments are limited by the fact that they may not fully represent redox modulation occurring in an intact organism. Our experiments were conducted at ambient (20%) oxygen concentrations. Increased ROS production at elevated oxygen levels could lead to more extensive redox-mediated alterations of Ca2+ handling in HF myocytes because of their reduced resistance to oxidative stress. Similarly, laser illumination and/or loading of myocytes with acetoxymethyl ester (AM) dyes could influence results by causing production of ROS. In separate experiments with physiological (2%) or ambient oxygen concentrations, there were no appreciable alterations in Ca2+ transients, SR Ca2+ leak, or ROS production when comparing the two oxygen concentrations in either HF or control myocytes (supplemental Figure V). Additionally, ROS levels were not affected by up to 2 hours of incubation with 10 μmol/L DCFDA or by incubation with 10 μmol/L DCFDA and 10 μmol/L Rhod-2AM (supplemental Figure VI). Nevertheless, because of limitations inherent to all experiments in ex vivo systems, our findings need to be interpreted cautiously. Although our results show that redox effects can account in part for the HF phenotype of abnormal Ca2+ handling in isolated myocytes and RyR2s, the applicability of these findings to in vivo settings will require further verification. Additionally, although our study focuses on redox-mediated changes in RyR2s, redox signaling has multiple targets, and additional mechanisms (eg, modification of other ion channels and transporters and of contractile filaments) are likely to contribute to alterations of cardiac contractility in HF.
Summary and Conclusions
Our study shows that abnormal oxidative modification of RyR2s by ROS contributes to the enhanced RyR2 activity and elevated SR Ca2+ leak that underlie the characteristic alterations of cytosolic Ca2+ transients and [Ca2+]SR in chronic HF. Importantly, enhanced oxidation of reactive cysteines results in altered modulation of RyR2s by luminal Ca2+. Luminal control is thought to contribute to Ca2+-release termination and Ca2+-signaling refractoriness following systolic release and thus plays an important role in preventing abnormal diastolic Ca2+ release. By linking redox effects to abnormal luminal regulation, our study provides new insights into understanding abnormal RyR2 behavior in HF. Targeted improvement of RyR2 redox-balance may present a rational therapeutic strategy for treating HF.
Supplementary Material
Online Figure I. Determination of free thiols in RyR2s from normal and failing hearts under baseline conditions and after treatment with the oxidizing or reducing reagents DTDP or DTT. A. Representative Coomassie Blue-stained gels and mBB fluorescence intensity of RyR2 from normal (left) and failing hearts (right) under baseline conditions and after 30 min incubation with of DTDP (0.2 mmol/L) or DTT (5 mmol/L). B. Pooled data for RyR2 free thiol content for the different experimental conditions relative to control, baseline conditions (BL). mBB fluorescence was normalized to RyR2 amount determined using Coomassie Blue staining of the gels run in parallel. RyR2 thiol content is significantly lower in HF than in control preparations (n=10 for control and n=7 for HF). The oxidizer DTDP reduces RyR2 free thiol content to a much larger extent in control than in HF preparations. The reducing reagent DTT increases the level of free RyR2 thiols to a larger extent in HF than in control preparations.
Online Figure II. Effects of reducing and oxidizing reagents on Ca2+ transients or [Ca2+]SR in control and HF myocytes, respectively. A. Representative Ca2+ transients recorded at baseline and after 30 minutes incubation with 800 μmol/L MPG. B. Ca2+ transient averages recorded under basal conditions (n=8) and after MPG incubation (n=6, p=NS). MPG or DTT did not have significant effects on Ca2+ transients or [Ca2+]SR recorded in control cells. C. Representative recordings of time-dependent changes of cell-averaged [Ca2+]SR with Fluo-5N in a saponin-permeabilized myocyte from a normal heart. Note that application of 1 mmol/L DTT did not affect [Ca2+]SR. D. Pooled data on [Ca2+]SR before and after application of DTT (n=8, p=NS). E, F Representative and pooled data illustrating that application of DTDP, increased SR Ca2+ leak in HF myocytes in a manner similar to that in control cells (Fig. 3, main text).
Online Figure III. Effects of oxidizing and reducing reagents, thimerosal and MPG on [Ca2+]SR in control and HF myocytes. A. Representative traces of time-dependent changes in cell-averaged luminal Fluo-5N before and after application of thimerosal (TL) or MPG in control and HF saponin-permeabilized myocytes as indicated. In control cells (left), application of the oxidant thimerosal (TL, 500 μmol/L) evoked a rapid drop in [Ca2+]SR similar to the effect of DTDP (Fig.3, main text). Treatment of HF myocytes (right) with 1 mmol/L MPG resulted in significant elevations of [Ca2+]SR similar to the effect of DTT (Fig.3, main text). B. Averaged [Ca2+]SR in control and HF myocytes under baseline conditions and after application of TL or MPG, as indicated. Control [Ca2+]SR was 0.79±0.02 and 0.48±0.01, under baseline conditions (BL) and in the presence of TL, respectively. In HF myocytes, [Ca2+]SR was 0.62±0.02 and 0.73±0.03 under baseline and in the presence of MPG, respectively. *Significantly different vs. baseline (BL, p<0.05, n=6 - 10) or **significantly different vs. control (p<0.05, n = 6 - 10 for HF and control respectively).
Online Figure IV. Effects of reducing and oxidizing reagents on RyR2 channels from control and HF samples. A, Representative recordings of single RyR2s from control and failing hearts (upper and lower panels, respectively) obtained under baseline conditions, and 1 or 5 min after addition of 200 μmol/L DTDP to the cis side. Single-channel activities were recorded in 350 mmol/L symmetrical CsCH3SO3 solutions at 40 mV. Channel openings are shown as upward deflections from the closed level. B. Summary data of RyR2 open probabilities (Po) before and after addition of DTDP at early (1 to 3 min) and later times (5 to 8 min) in control (grey squares and open squares , 10 and 200 μmol/L DTDP, respectively) and HF RyR2s (red circles, 200 μmol/L DTDP). *Significantly different vs. baseline (p<0.05, n= 3-6 channels). C, Summary data of RyR2 Po in HF RyR2s recorded under baseline conditions and after addition to the cis side of 0.5 or 5 mmol/L DTT. *Significantly different vs. baseline (p<0.05, n= 3 channels).
Online Figure V. Ca2+ handling or ROS production are similar at ambient and physiological oxygen levels in control and HF myocytes. A, Representative time-dependent profiles of cytosolic Ca2+ transients recorded during field-stimulation (0.3 Hz) of control (left) and HF (right) myocytes kept at a physiologic (low) oxygen concentration of 2% (LO) and 30 min after switching to ambient oxygen (20%, HO). The amplitude of Ca2+ transients was not significantly affected by O2, either in control or HF cells. B, Averaged amplitudes of Ca2+ transients (Rhod-2 ΔF/F0) were 0.51±0.05 for control (LO), 0.56±0.04 for control (HO), n=9, 0.35±0.05 for HF (LO) and 0.37±0.05 for HF (HO), n=7. *HF differed significantly from the corresponding controls at both oxygen concentrations (p<0.05). C, Representative traces of timedependent profiles of SR[Ca2+] leak measured with Fluo-5N in permeabilized myocytes from normal and failing hearts kept at low (2%) oxygen. Data were obtained at baseline (LO, black) and after switching to ambient oxygen (HO, red). The decline of Fluo-5N signal in the presence of Tg was fit by an exponential function. The rate of SR Ca2+ leak was not significantly affected by O2, either in control or HF cells. D. Averaged time constants (from exponential fit) of [Ca2+]SR leak in controls during LO (1165±300 s, n=12) and HO (985±213 s, n=14) and in HF myocytes during LO (521±69 s, n=14) and HO (446±58 s, n=11). * HF differed significantly from the corresponding controls at both oxygen concentrations (p<0.05). E. Representative images from DCFDA-loaded myocytes from normal and failing hearts under the same conditions as in A and C. ROS production was markedly increased in HF myocytes at both oxygen concentrations, compared to controls; however there was no significant difference in ROS levels at 20% oxygen relative to 2% oxygen within these groups. F, Summary graph for averaged ROS concentrations (DCFDA fluorescence) normalized to the signal in the presence of supraphysiologic H2O2 (10 mmol/L, FMAX) measured in control LO (2.25±0.38) and HO myocytes (2.33±0.15) and heart failure myocytes at LO (4.94±0.67) and HO (5.52±1.02). *HF differed significantly from the corresponding controls at both oxygen concentrations (p<0.05, n = 13 – 17 per group). After isolation cells were placed into gas-tight containers8 filled with a physiologically reduced (low) oxygen (2%O2, 5% CO2 and 93% N2) gas mixture and incubated in solution contained (mmol/L): 115 NaCl, 5.4 KCl, 2.0 CaCl2, 0.5 MgCl2, 10 HEPES, 5.6 glucose and 25 NaHCO3 (pH 7.4). Cells were loaded with ROS (DCFDA-AM) or Ca2+ (Rhod-2-AM) indicators and were perfused with solution bubbled with low oxygen (2%) gas mixture and Ca2+ transients or ROS levels were recorded. Cells were then switched to a solution with ambient pO2 and after 15 min of perfusion recordings were repeated. Paired Student’s t-tests were used to determine significant differences, with a criterion for significance of p<0.05.
Online Figure VI. Incubation of myocytes with AM forms of the dyes does not appreciably affect ROS production in control and HF myocytes. Our studies utilized the AM forms of fluorescent indicators (ROS and Ca2+ sensitive). Formaldehyde is a byproduct of the deesterification of these indicators, and that by itself can produce reactive oxidative species, although we expect that the concentrations would be quite low. To address this possibility, we measured ROS as a function of incubation time and concentration of AM dyes. Panel A shows representative images of myocytes from normal and failing hearts loaded with the ROS indicator DCFDA-AM (10 μmol/L) for 30 min and 2 hrs and in combination with Ca2+ indicator Rhod-2AM (10 μmol/L, 30 min) in control. Panel B shows averaged normalized DCFDA fluorescence. The values of normalized DFDA fluorescence in control cells were 2.83±0.24, 2.97±0.59, 2.40±0.39 for DCFDA loaded for 30 min and 2 hrs and DCFDA+Rhod-2 respectively. The values of normalized DCFDA fluorescence in HF myocytes were 8.30±0.91 and 9.29±1.12 for loaded 30min and 2 hrs. respectively. *Significantly different vs. control at p<0.05 (n=14 - 20). ROS levels were similar following 30 min and 2 hrs of incubation with 10 μmol/L DCFDA in both control and HF myocytes. Additionally, ROS levels were similar between cells incubated with 10 μmol/L DCFDA and 10 μmol/L DCFDA + 10 μmol/L Rhod-2AM for 30 min for control. Considering that most of our experiments were done with 10 μmol/L of AM dyes and 30 min incubation time, these results suggest that the differences in results between control and HF are not likely to be attributable to AM dye effects.
Acknowledgments
Sources of Funding This work was supported by the American Heart Association (to A.B., D.T., Y.N.), NIH grants HL074045 and HL063043 (to S.G.), HL089836 (to C.A.C.), and HL073087 (to C.K.S.).
Footnotes
Disclosures None.
References
- 1.Lindner M, Erdmann E, Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol. 1998;30:743–749. doi: 10.1006/jmcc.1997.0626. [DOI] [PubMed] [Google Scholar]
- 2.Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. doi: 10.1161/01.cir.103.11.1577. [DOI] [PubMed] [Google Scholar]
- 3.Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34:951–969. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- 4.Houser SR, Margulies KB. Is depressed myocyte contractility centrally involved in heart failure? Circ Res. 2003;92:350–358. doi: 10.1161/01.RES.0000060027.40275.A6. [DOI] [PubMed] [Google Scholar]
- 5.Sipido KR, Eisner D. Something old, something new: changing views on the cellular mechanisms of heart failure. Cardiovasc Res. 2005;68:167–174. doi: 10.1016/j.cardiores.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 6.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic Publishers; Dordrecht; Boston: 2001. [Google Scholar]
- 7.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 8.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 9.Kubalova Z, Terentyev D, Viatchenko-Karpinski S, Nishijima Y, Gyorke I, Terentyeva R, da Cunha DN, Sridhar A, Feldman DS, Hamlin RL, Carnes CA, Györke S. Abnormal intrastore calcium signaling in chronic heart failure. Proc Natl Acad Sci U S A. 2005;102:14104–14109. doi: 10.1073/pnas.0504298102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Györke S, Terentyev D. Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovasc Res. 2008;77:245–255. doi: 10.1093/cvr/cvm038. [DOI] [PubMed] [Google Scholar]
- 11.Belevych A, Kubalova Z, Terentyev D, Hamlin RL, Carnes CA, Györke S. Enhanced ryanodine receptor-mediated calcium leak determines reduced sarcoplasmic reticulum calcium content in chronic canine heart failure. Biophys J. 2007;93:4083–4092. doi: 10.1529/biophysj.107.114546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bridge JH, Savio E. Revealing the cellular basis of heart failure. Biophys J. 2007;93:3731–3732. doi: 10.1529/biophysj.107.116541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 14.Benkusky NA, Weber CS, Scherman JA, Farrell EF, Hacker TA, John MC, Powers PA, Valdivia HH. Intact beta-adrenergic response and unmodified progression toward heart failure in mice with genetic ablation of a major protein kinase A phosphorylation site in the cardiac ryanodine receptor. Circ Res. 2007;101:819–829. doi: 10.1161/CIRCRESAHA.107.153007. [DOI] [PubMed] [Google Scholar]
- 15.Xiao B, Sutherland C, Walsh MP, Chen SR. Protein kinase A phosphorylation at serine-2808 of the cardiac Ca2+-release channel (ryanodine receptor) does not dissociate 12.6-kDa FK506-binding protein (FKBP12.6) Circ Res. 2004;94:487–495. doi: 10.1161/01.RES.0000115945.89741.22. [DOI] [PubMed] [Google Scholar]
- 16.Stange M, Xu L, Balshaw D, Yamaguchi N, Meissner G. Characterization of recombinant skeletal muscle (Ser-2843) and cardiac muscle (Ser-2809) ryanodine receptor phosphorylation mutants. J Biol Chem. 2003;278:51693–51702. doi: 10.1074/jbc.M310406200. [DOI] [PubMed] [Google Scholar]
- 17.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 18.Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007;73:631–640. doi: 10.1016/j.cardiores.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 19.Yang D, Zhu WZ, Xiao B, Brochet DX, Chen SR, Lakatta EG, Xiao RP, Cheng H. Ca2+/calmodulin kinase II-dependent phosphorylation of ryanodine receptors suppresses Ca2+ sparks and Ca2+ waves in cardiac myocytes. Circ Res. 2007;100:399–407. doi: 10.1161/01.RES.0000258022.13090.55. [DOI] [PubMed] [Google Scholar]
- 20.Belch JF, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J. 1991;65:245–248. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS. Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol. 2002;34:379–388. doi: 10.1006/jmcc.2002.1526. [DOI] [PubMed] [Google Scholar]
- 22.Cesselli D, Jakoniuk I, Barlucchi L, Beltrami AP, Hintze TH, Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ Res. 2001;89:279–286. doi: 10.1161/hh1501.094115. [DOI] [PubMed] [Google Scholar]
- 23.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meissner G. Regulation of mammalian ryanodine receptors. Front Biosci. 2002;7:d2072–d2080. doi: 10.2741/A899. [DOI] [PubMed] [Google Scholar]
- 25.Hidalgo C, Aracena P, Sanchez G, Donoso P. Redox regulation of calcium release in skeletal and cardiac muscle. Biol Res. 2002;35:183–193. doi: 10.4067/s0716-97602002000200009. [DOI] [PubMed] [Google Scholar]
- 26.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 27.Nishijima Y, Feldman DS, Bonagura JD, Ozkanlar Y, Jenkins PJ, Lacombe VA, Abraham WT, Hamlin RL, Carnes CA. Canine nonischemic left ventricular dysfunction: a model of chronic human cardiomyopathy. J Card Fail. 2005;11:638–644. doi: 10.1016/j.cardfail.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 28.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 29.Marengo JJ, Hidalgo C, Bull R. Sulfhydryl oxidation modifies the calcium dependence of ryanodine-sensitive calcium channels of excitable cells. Biophys J. 1998;74:1263–1277. doi: 10.1016/S0006-3495(98)77840-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu Rev Physiol. 1994;56:485–508. doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- 31.Aracena-Parks P, Goonasekera SA, Gilman CP, Dirksen RT, Hidalgo C, Hamilton SL. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J Biol Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- 32.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci STKE. 2001:PL1. doi: 10.1126/stke.2001.86.pl1. [DOI] [PubMed] [Google Scholar]
- 33.Yano M, Okuda S, Oda T, Tokuhisa T, Tateishi H, Mochizuki M, Noma T, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation. 2005;112:3633–3643. doi: 10.1161/CIRCULATIONAHA.105.555623. [DOI] [PubMed] [Google Scholar]
- 34.Györke I, Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophys J. 1998;75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ching LL, Williams AJ, Sitsapesan R. Evidence for Ca(2+) activation and inactivation sites on the luminal side of the cardiac ryanodine receptor complex. Circ Res. 2000;87:201–206. doi: 10.1161/01.res.87.3.201. [DOI] [PubMed] [Google Scholar]
- 36.Xiao B, Tian X, Xie W, Jones PP, Cai S, Wang X, Jiang D, Kong H, Zhang L, Chen K, Walsh MP, Cheng H, Chen SR. Functional consequence of protein kinase A-dependent phosphorylation of the cardiac ryanodine receptor: sensitization of store overload-induced Ca2+ release. J Biol Chem. 2007;282:30256–30264. doi: 10.1074/jbc.M703510200. [DOI] [PubMed] [Google Scholar]
- 37.Feng W, Liu G, Allen PD, Pessah IN. Transmembrane redox sensor of ryanodine receptor complex. J Biol Chem. 2000;275:35902–35907. doi: 10.1074/jbc.C000523200. [DOI] [PubMed] [Google Scholar]
- 38.Abramson JJ, Salama G. Critical sulfhydryls regulate calcium release from sarcoplasmic reticulum. J Bioenerg Biomembr. 1989;21:283–294. doi: 10.1007/BF00812073. [DOI] [PubMed] [Google Scholar]
- 39.Pessah IN, Feng W. Functional role of hyperreactive sulfhydryl moieties within the ryanodine receptor complex. Antioxid Redox Signal. 2000;2:17–25. doi: 10.1089/ars.2000.2.1-17. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez DR, Beigi F, Treuer AV, Hare JM. Deficient ryanodine receptor S-nitrosylation increases sarcoplasmic reticulum calcium leak and arrhythmogenesis in cardiomyocytes. Proc Natl Acad Sci U S A. 2007;104:20612–20617. doi: 10.1073/pnas.0706796104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eager KR, Dulhunty AF. Activation of the cardiac ryanodine receptor by sulfhydryl oxidation is modified by Mg2+ and ATP. J Membr Biol. 1998;163:9–18. doi: 10.1007/s002329900365. [DOI] [PubMed] [Google Scholar]
- 42.Roche BM, Schwartz D, Lehnhard RA, McKeever KH, Nakayama T, Kirby TE, Robitaille PM, Hamlin RL. Changes in concentrations of neuroendocrine hormones and catecholamines in dogs with myocardial failure induced by rapid ventricular pacing. Am J Vet Res. 2002;63:1413–1417. doi: 10.2460/ajvr.2002.63.1413. [DOI] [PubMed] [Google Scholar]
- 43.Neri M, Cerretani D, Fiaschi AI, Laghi PF, Lazzerini PE, Maffione AB, Micheli L, Bruni G, Nencini C, Giorgi G, D’Errico S, Fiore C, Pomara C, Riezzo I, Turillazzi E, Fineschi V. Correlation between cardiac oxidative stress and myocardial pathology due to acute and chronic norepinephrine administration in rats. J Cell Mol Med. 2007;11:156–170. doi: 10.1111/j.1582-4934.2007.00009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Osadchii OW. Cardiac hypertrophy induced by sustained beta-adrenergic activation: pathophysiological aspects. Heart Fail Rev. 2007;12:66–86. doi: 10.1007/s10741-007-9007-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Online Figure I. Determination of free thiols in RyR2s from normal and failing hearts under baseline conditions and after treatment with the oxidizing or reducing reagents DTDP or DTT. A. Representative Coomassie Blue-stained gels and mBB fluorescence intensity of RyR2 from normal (left) and failing hearts (right) under baseline conditions and after 30 min incubation with of DTDP (0.2 mmol/L) or DTT (5 mmol/L). B. Pooled data for RyR2 free thiol content for the different experimental conditions relative to control, baseline conditions (BL). mBB fluorescence was normalized to RyR2 amount determined using Coomassie Blue staining of the gels run in parallel. RyR2 thiol content is significantly lower in HF than in control preparations (n=10 for control and n=7 for HF). The oxidizer DTDP reduces RyR2 free thiol content to a much larger extent in control than in HF preparations. The reducing reagent DTT increases the level of free RyR2 thiols to a larger extent in HF than in control preparations.
Online Figure II. Effects of reducing and oxidizing reagents on Ca2+ transients or [Ca2+]SR in control and HF myocytes, respectively. A. Representative Ca2+ transients recorded at baseline and after 30 minutes incubation with 800 μmol/L MPG. B. Ca2+ transient averages recorded under basal conditions (n=8) and after MPG incubation (n=6, p=NS). MPG or DTT did not have significant effects on Ca2+ transients or [Ca2+]SR recorded in control cells. C. Representative recordings of time-dependent changes of cell-averaged [Ca2+]SR with Fluo-5N in a saponin-permeabilized myocyte from a normal heart. Note that application of 1 mmol/L DTT did not affect [Ca2+]SR. D. Pooled data on [Ca2+]SR before and after application of DTT (n=8, p=NS). E, F Representative and pooled data illustrating that application of DTDP, increased SR Ca2+ leak in HF myocytes in a manner similar to that in control cells (Fig. 3, main text).
Online Figure III. Effects of oxidizing and reducing reagents, thimerosal and MPG on [Ca2+]SR in control and HF myocytes. A. Representative traces of time-dependent changes in cell-averaged luminal Fluo-5N before and after application of thimerosal (TL) or MPG in control and HF saponin-permeabilized myocytes as indicated. In control cells (left), application of the oxidant thimerosal (TL, 500 μmol/L) evoked a rapid drop in [Ca2+]SR similar to the effect of DTDP (Fig.3, main text). Treatment of HF myocytes (right) with 1 mmol/L MPG resulted in significant elevations of [Ca2+]SR similar to the effect of DTT (Fig.3, main text). B. Averaged [Ca2+]SR in control and HF myocytes under baseline conditions and after application of TL or MPG, as indicated. Control [Ca2+]SR was 0.79±0.02 and 0.48±0.01, under baseline conditions (BL) and in the presence of TL, respectively. In HF myocytes, [Ca2+]SR was 0.62±0.02 and 0.73±0.03 under baseline and in the presence of MPG, respectively. *Significantly different vs. baseline (BL, p<0.05, n=6 - 10) or **significantly different vs. control (p<0.05, n = 6 - 10 for HF and control respectively).
Online Figure IV. Effects of reducing and oxidizing reagents on RyR2 channels from control and HF samples. A, Representative recordings of single RyR2s from control and failing hearts (upper and lower panels, respectively) obtained under baseline conditions, and 1 or 5 min after addition of 200 μmol/L DTDP to the cis side. Single-channel activities were recorded in 350 mmol/L symmetrical CsCH3SO3 solutions at 40 mV. Channel openings are shown as upward deflections from the closed level. B. Summary data of RyR2 open probabilities (Po) before and after addition of DTDP at early (1 to 3 min) and later times (5 to 8 min) in control (grey squares and open squares , 10 and 200 μmol/L DTDP, respectively) and HF RyR2s (red circles, 200 μmol/L DTDP). *Significantly different vs. baseline (p<0.05, n= 3-6 channels). C, Summary data of RyR2 Po in HF RyR2s recorded under baseline conditions and after addition to the cis side of 0.5 or 5 mmol/L DTT. *Significantly different vs. baseline (p<0.05, n= 3 channels).
Online Figure V. Ca2+ handling or ROS production are similar at ambient and physiological oxygen levels in control and HF myocytes. A, Representative time-dependent profiles of cytosolic Ca2+ transients recorded during field-stimulation (0.3 Hz) of control (left) and HF (right) myocytes kept at a physiologic (low) oxygen concentration of 2% (LO) and 30 min after switching to ambient oxygen (20%, HO). The amplitude of Ca2+ transients was not significantly affected by O2, either in control or HF cells. B, Averaged amplitudes of Ca2+ transients (Rhod-2 ΔF/F0) were 0.51±0.05 for control (LO), 0.56±0.04 for control (HO), n=9, 0.35±0.05 for HF (LO) and 0.37±0.05 for HF (HO), n=7. *HF differed significantly from the corresponding controls at both oxygen concentrations (p<0.05). C, Representative traces of timedependent profiles of SR[Ca2+] leak measured with Fluo-5N in permeabilized myocytes from normal and failing hearts kept at low (2%) oxygen. Data were obtained at baseline (LO, black) and after switching to ambient oxygen (HO, red). The decline of Fluo-5N signal in the presence of Tg was fit by an exponential function. The rate of SR Ca2+ leak was not significantly affected by O2, either in control or HF cells. D. Averaged time constants (from exponential fit) of [Ca2+]SR leak in controls during LO (1165±300 s, n=12) and HO (985±213 s, n=14) and in HF myocytes during LO (521±69 s, n=14) and HO (446±58 s, n=11). * HF differed significantly from the corresponding controls at both oxygen concentrations (p<0.05). E. Representative images from DCFDA-loaded myocytes from normal and failing hearts under the same conditions as in A and C. ROS production was markedly increased in HF myocytes at both oxygen concentrations, compared to controls; however there was no significant difference in ROS levels at 20% oxygen relative to 2% oxygen within these groups. F, Summary graph for averaged ROS concentrations (DCFDA fluorescence) normalized to the signal in the presence of supraphysiologic H2O2 (10 mmol/L, FMAX) measured in control LO (2.25±0.38) and HO myocytes (2.33±0.15) and heart failure myocytes at LO (4.94±0.67) and HO (5.52±1.02). *HF differed significantly from the corresponding controls at both oxygen concentrations (p<0.05, n = 13 – 17 per group). After isolation cells were placed into gas-tight containers8 filled with a physiologically reduced (low) oxygen (2%O2, 5% CO2 and 93% N2) gas mixture and incubated in solution contained (mmol/L): 115 NaCl, 5.4 KCl, 2.0 CaCl2, 0.5 MgCl2, 10 HEPES, 5.6 glucose and 25 NaHCO3 (pH 7.4). Cells were loaded with ROS (DCFDA-AM) or Ca2+ (Rhod-2-AM) indicators and were perfused with solution bubbled with low oxygen (2%) gas mixture and Ca2+ transients or ROS levels were recorded. Cells were then switched to a solution with ambient pO2 and after 15 min of perfusion recordings were repeated. Paired Student’s t-tests were used to determine significant differences, with a criterion for significance of p<0.05.
Online Figure VI. Incubation of myocytes with AM forms of the dyes does not appreciably affect ROS production in control and HF myocytes. Our studies utilized the AM forms of fluorescent indicators (ROS and Ca2+ sensitive). Formaldehyde is a byproduct of the deesterification of these indicators, and that by itself can produce reactive oxidative species, although we expect that the concentrations would be quite low. To address this possibility, we measured ROS as a function of incubation time and concentration of AM dyes. Panel A shows representative images of myocytes from normal and failing hearts loaded with the ROS indicator DCFDA-AM (10 μmol/L) for 30 min and 2 hrs and in combination with Ca2+ indicator Rhod-2AM (10 μmol/L, 30 min) in control. Panel B shows averaged normalized DCFDA fluorescence. The values of normalized DFDA fluorescence in control cells were 2.83±0.24, 2.97±0.59, 2.40±0.39 for DCFDA loaded for 30 min and 2 hrs and DCFDA+Rhod-2 respectively. The values of normalized DCFDA fluorescence in HF myocytes were 8.30±0.91 and 9.29±1.12 for loaded 30min and 2 hrs. respectively. *Significantly different vs. control at p<0.05 (n=14 - 20). ROS levels were similar following 30 min and 2 hrs of incubation with 10 μmol/L DCFDA in both control and HF myocytes. Additionally, ROS levels were similar between cells incubated with 10 μmol/L DCFDA and 10 μmol/L DCFDA + 10 μmol/L Rhod-2AM for 30 min for control. Considering that most of our experiments were done with 10 μmol/L of AM dyes and 30 min incubation time, these results suggest that the differences in results between control and HF are not likely to be attributable to AM dye effects.