

Abstract
Inhibitors of neuronal nitric oxide synthase, based on a chiral pyrrolidine scaffold, show promise for the treatment of certain neurodegenerative diseases. We recently reported the synthesis of a series of selective inhibitors, but the method was limited at a key step of formingan allyl ether intermediate. Yields for this step were very inconsistent, and the presence of base sensitive functional groups limited the range of available methods for forming this ether bond. This work describes a novel application of palladium catalyzed decarboxylativeallylation, consistently resulting in 90% isolated yields, which is crucial for the synthesis of the critical allyl ether late stage intermediate. We also report a new quantitative yielding and straightforward synthesis of the allyl-t-butylcarbonate precursor.
Keywords: decarboxylativeallylation, tetrakis(triphenylphosphine)palladium(0), neutral O-allylation
1. Introduction
In our ongoing research to develop potential chemotherapeutic agents to prevent neurodegeneration, we identified a series of compounds, based on a chiral pyrrolidine core, that selectively inhibited neuronal nitric oxide synthase (nNOS) with respect to the other two enzyme isoforms, inducible (iNOS) and endothelial nitric oxide synthase (eNOS).1 A key intermediate in the synthesis of these compounds from (3R,4R)-1 is (3R,4R)-allyloxypyrrolidine2. Earlier work showed that the 2-amino nitrogen on the pyridine requires bulky
 protecting groups to prevent a side reaction, and for various reasons, we decided to employ a diBoc strategy.2 We discovered that an unexpected rearrangement takes place when Williamson conditions are used to convert precursor alcohol 1 into allyl ether 2. Formally, the mechanism is N- to O transfer of a tert-butylcarbonyl (Boc) group and allylation of the 2-amino group at the pyridine ring, forming N-allyl3 in a 95% yield. Recently, we reported an improved synthesis of intermediate 2.3By using a method for allylation that proceeds under neutral conditions, we were successful in avoiding the formation of 3 via the aforementioned rearrangement; however, the yields were very inconsistent, often as low as 20%. There is considerable effort associated with obtaining this late-stage intermediate in enantiomerically pure form, so we were interested in the identification of a more reliable and high yielding methodology for O-allylation of 1.
protecting groups to prevent a side reaction, and for various reasons, we decided to employ a diBoc strategy.2 We discovered that an unexpected rearrangement takes place when Williamson conditions are used to convert precursor alcohol 1 into allyl ether 2. Formally, the mechanism is N- to O transfer of a tert-butylcarbonyl (Boc) group and allylation of the 2-amino group at the pyridine ring, forming N-allyl3 in a 95% yield. Recently, we reported an improved synthesis of intermediate 2.3By using a method for allylation that proceeds under neutral conditions, we were successful in avoiding the formation of 3 via the aforementioned rearrangement; however, the yields were very inconsistent, often as low as 20%. There is considerable effort associated with obtaining this late-stage intermediate in enantiomerically pure form, so we were interested in the identification of a more reliable and high yielding methodology for O-allylation of 1.
2. Allylation strategies and their outcomes
Several general methods were explored to identify a method for forming 2. Based on the observation that the second Boc group is labile under basic and acidic conditions, we explored ether formation reactions that take place under neutral, or nearly neutral, conditions.
2.1. Des-Boc alcohol precursor
As mentioned above, we require the second Boc group on the 2-aminopyridine to prevent a side reaction at an earlier step in the synthesis, and having already served its purpose, we speculated that it might be possible to remove it at this stage, generating 4, which might be relatively easier to allylate, compared to 1. We screened various different conditions to find optimum parameters for quantitative removal of just one of the three Boc groups of 1. When 1 was treated with 1 M TFA in methylene chloride, the Boc group was removed selectively in a near quantitative yield in only 15 minutes, yielding 4 (Scheme 1)
Scheme 1.

Generation of 4 by mono deprotection of precursor 1
We predicted a priori that the alkoxide would be a better nucleophile than the carbamate nitrogen anion and hypothesized that 4 could be subjected to >2 equivalents of sodium hydride in the presence of a stoichiometric amount of allyl bromide to afford the O-allyl product under Williamson conditions. Subsequent aqueous workup would neutralize the carbamate nitrogen anion. Surprisingly, the only major product that was isolated was the N, O-diallyl species in a 20% yield. There is precedence for using trichloroalkylimidates along with an acid catalyst for the allylation of hindered alcohols, such as carbohydrate secondary hydroxyl groups.4We tried the trichloroacetimidate methodology on des-Boc4, but only obtained a trace amount of product along with loss of starting material. This seems to indicate that the substrate is not stable under the conditions of the reaction. Interestingly, treating 4 with palladium and allyl carbonate gave the N-allyl product in high yield, but did not lead to any significant O-allylation, confirming the difficulty in forming the allyl ether of this substrate (Scheme 2).
Scheme 2.

Attempted O-allylation of 4 using various conditions
2.2. Sterically hindered carbonate precursor
Next we revisited palladium0 catalyzed decarboxylativeallylation; an advantage of this reaction is that it takes place under nearly neutral conditions, since the amount of alkoxide that is present is never greater than the amount of catalyst. A disadvantage of Pd-catalyzed decarboxylativeallylations, in general, is that water competes as a nucleophile twice,5 first to attack the Pd π-allyl complex, then again, when the newly formed alcohol attacks another Pd π-allyl complex. We had limited success with allyl methyl carbonate, but inspection of the mechanism suggests that the reason may be because the pyrrolidine hydroxyl group is a secondary alcohol. The overall reaction can be viewed as proceeding in three steps (Scheme 3): In step 1, methoxide is generated by decarboxylation. In step 2, the pyrrolidinealkoxide is generated from an acid-base reaction with methoxide, but the equilibrium is expected to lie to the left side (favoring the secondary alcohol), which should have a slightly higher pKa. In step 3, both alkoxide species compete for the allylcation; however, the nucleophilicity of the secondary pyrrolidine oxide is not expected to exceed that of the methoxide ion.
Scheme 3.

Three-step mechanism for palladium catalyzed decarboxylative allylation reaction
Therefore, when allyl methyl carbonate is used, we are penalized twice, first in the acid-base reaction, then again in the nucleophilic attack on the π-allyl complex. In general, this hypothesis is attributed to Sinou and coworkers, who used allylethyl carbonate with Pd0 to form the allyl ether of carbohydrate substrates,6 but there are also other variations. In one example, an allyl carbonate precursor was formed from the alcohol substrate,7which was then treated with the palladium catalyst. Under strictly anhydrous conditions, this implies only one nucleophile (the alkoxide of the substrate) would be present to trap the allylcation, and the rate limiting step in the reaction would be the attack of the π-allyl complex by the alkoxide, since the acid-base equilibrium shown in step 2 of Scheme 2 would not be present in the mechanism. Similarly, forming allyl carbonate 8 from alcohol 1, then subjecting it to palladium catalyzed decarboxylation conditions seemed promising. The requisite allylchloroformate is commercially available, but we were unsuccessful in forming 8. These results suggest that formation of the carbonate precursor might be of comparable difficulty to forming the ether product (2) from this sterically hindered alcohol, so we did not pursue this method further (Scheme 4).
Scheme 4.

Attempted formation of 2by direct decarboxylative allylation of 8
It has been reported that using a less nucleophilicalkoxide, such as tert-butoxide (generated from decarboxylation of allyl-t-butylcarbonate) should minimize the formation of side products.8 In that work, they were able to obtain selective allylation of a hindered tertiary hydroxyl group on a carbohydrate substrate, in the presence of two unmasked secondary alcohols, while using only a slight excess (1.2 equiv) of allyl-t-butylcarbonate (9). The required allyltert-butylcarbonate precursor is not commercially available. It has previously been synthesized from allyl alcohol and di-tert-butyl-dicarbonate (Boc2O) in dichloromethane, using phase transfer catalysis and sodium hydroxide as base.8,8 In our hands, the reaction is very sluggish and, even after 48 hours, it does not go to completion. It is difficult to separate the carbonate product from the unreacted Boc2O using silica gel chromatography because the difference in the Rf values is very small. With these factors in mind, we set out to optimize the reaction conditions. It seems likely that the reaction is sluggish because the generation of the alkoxide is not very efficient. Enhancing the activity of the tert-butylcarbonyl group would be expected to facilitate the reaction with neutral allyl alcohol. 4-(N,N-Dimethyl)aminopyridine, DMAP, seemed promising based on the known mechanism involved in DMAP catalysis of acylations using acetic anhydride. It was speculated that running the reaction neat (Boc2O in allyl alcohol) would allow kinetics that are limited by diffusion rather than phase transfer. The hypothesis turned out to be correct; using DMAP catalysis enabled facile production of the required carbonate. The optimized conditions are as follows: Di-tert-butyldicarbonate (10 g)wasdissolved in 20 mL of anhydrous allyl alcohol. DMAP (275 mg, 5 mol%) was added all at once, and a reflux condenser was attached. Immediate evolution of CO2 occurred,which proceeded at a constant rate for about 1 h, at which time the reaction was complete, and Boc2O was undetectable in the crude mixture by TLC. The crude product was a solution in allyl alcohol, which was used in excess. It would be desirable to remove the excess unreacted allylalcohol before attempting the chromatographic purification of the carbonate, but when the crude reaction mixture was placed on the rotary evaporator, both the carbonate product and allyl alcohol were removed. Fractional distillation was not practical because the boiling points of the two compounds are very similar. Ultimately, the purification of the product was achieved by loading the crude reaction mixture directly onto silica gel. A mobile phase consisting of 5% ethyl acetate in hexanes eluted the product with an Rf of 0.5, while the DMAP catalyst and allyl alcohol were not elutedfrom the silica gel under these conditions. Potassium permanganate was used to visualize the olefin product by TLC. In this way, 7.3 g of 9wasproduced from 10 g of di-tert-butyl dicarbonate (Scheme 5).
Scheme 5.
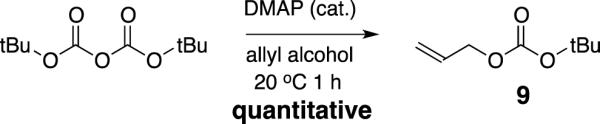
Synthesis of allyl-t-butylcarbonate (9)
On the basis ofour previous work, we expected the second Boc group to be very labile under basic conditions. We predicted that we could use a very low catalyst loading to minimize the concentration of alkoxide, and utilize tert-butyl carbonate precursor to prevent side reactions. Using 2.5% palladium catalyst and 5 equiv of 9 led to the formation of the desired product in 90% isolated yield in 6 hours (Scheme 6). Further optimization could include using a lower stoichiometric ratio of carbonate, but will also require more rigorous exclusion of water, and 9is easily synthesized in one step from inexpensive starting materials.
Scheme 6.

Decarboxylative allylation of 1 to form 2 in high yields
3. Conclusions
A simple protocol for the allylation of a complex substrate alcohol is reported. This robust method should be a useful addition to the synthetic toolkit for researchers interested in allylation of sterically-hindered alcohols in key pharmacophoric scaffolds containing base-sensitive functional groups. By using commercially available tetrakis(triphenylphosphine) palladium catalyst and a new simplified method for the synthesis of allyl-t-butylcarbonate (9), selective allylation of hindered alcohols proceeds in greater than 90% isolated yield.
4. Experimental
Allyltert-butyl carbonate (9)
A flame-dried flask containing a stir bar was charged with di-tert-butyl dicarbonate (10.0 g, 45.8 mmols), and anhydrous allyl alcohol (10.0 mL,147mmols) was added. A water-cooled condenser was attached and fitted with a calcium chloride drying tube. When the solids had dissolved, 4-(dimethylamino)-pyridine (275 mg, 2.25 mmols, 5%) was added all at once. Gas was evolved immediately, and continued at a steady rate for approximately 1 h. At this time, TLC indicated that the di-tert-butyl dicarbonate starting material had been completely consumed, as visualized with I2. The product was purified by flash chromatography using a 5.5 cm (od) column. The crude mixture was loaded directly onto the silica gel and eluted using a mobile phase consisting of 5% ethyl acetate / 95% hexanes, producing 7.3 g (quantitative) of 9. The product was visualized with potassium permanganate staining, Rf 0.4. 1H NMR (500 MHz, CDCl3, δ): 5.94 (m, 1H), 5.35 (ddd, J = 1.5, 3, 17.5 Hz, 1H), 5.26 (dd, J = 1.5, 10.5 Hz, 1H), 4.56 (m, 2H), 1.50 (s, 9H); 13C NMR (125 MHz, CDCl3, δ):153.31, 131.96, 118.60, 82.21, 67.62, 27.77.
(3R,4R)-tert-Butyl-3-(allyloxy)-4-((6-(bis(tert-butoxycarbonyl)amino)-4-methylpyridin-2-yl)methyl)pyrrolidine-1-carboxylate (2)
A flame-dried flask containing a stir bar was fitted with a water-cooled condenser under an argon atmosphere. (3R,4R)-tert-Butyl 3-((6-(bis(tert-butoxycarbonyl)amino)-4-methylpyridin-2-yl)methyl)-4-hydroxypyrrolidine-1-carboxylate (1, 175mg, 0.35mmol) was dissolved in 20 mL of anhydrous THF and added to the flask, followed by Pd(PPh3)4 (12 mg, 2.5%) as a solution in anhydrous THF (5 mL). The mixture was degassed by bubbling in argon for 20 min (the volume of solvent was reduced slightly). A shift in the color of the solution was observed from bright yellow to dark yellow. The mixture was heated to reflux for several minutes. A solution of 9 (320 mg, 1.74mmols) in 5 mL anhydrous THF was injected into the preheated, refluxing THF solution over approximately 1 min. The reaction was allowed to stir under reflux for 6 h, at which time, complete consumption of starting material was indicated by TLC, and a new spot was observed Rf = 0.8, 25% ethyl acetate / 75% hexanes. Silica gel chromatography gave 2 as a white solid (172 mg, 90%) 1H NMR (500 MHz, CDCl3, δ): 6.91 (d, J = 8.5 Hz, 1H), 6.90 (s, 1H), 5.88 (m, 1H), 5.27 (m, 1H), 5.16 (d, J = 10.5 Hz, 1H), 4.03 (dt, J = 5.5, 13 Hz, 1H), 3.78 (m, 2H), 3.52 (m, 2H), 3.27 (dd, J = 3.5, 12.5 Hz, 1H), 3.16 (m, 1H), 3.01 (m, 1H), 2.81 (m, 1H), 2.69 (m, 1H), 2.33 (m, 3H), 1.44 (s, 18H), 1.43 (s, 9H); 13C NMR (125 MHz, CDCl3, δ): 159.25, 159.19, 154.78, 154.48, 151.81, 151.48, 151.43, 149.50, 134.67, 134.62, 122.94, 119.55, 116.87, 116.67, 82.82, 79.21, 79.13, 78.61, 77.78, 70.26, 70.18, 51.00, 50.43, 49.20, 48.87, 43.32, 42.67, 34.76, 34.66, 28.51, 27.92, 20.90. HR ESI-MS: m/z = 548.3325 (mono-isotopic mass = 547.3258).
Acknowledgments
The authors are grateful for financial support from the National Institutes of Health (GM049725). JMK thanks the Center for Molecular Innovation and Drug Discovery at Northwestern University for a postdoctoral fellowship from the NIH training grant, Drug Discovery in Age Related Disorders (T32 AG000260).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References
- 1.Ji H, Delker SL, Li H, Martasek P, Roman LJ, Poulos TL, Silverman RB. J. Med. Chem. 2010;53:7804. doi: 10.1021/jm100947x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xue F, Silverman RB. Tetrahedron Lett. 2010;51:2536. doi: 10.1016/j.tetlet.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue F, Kraus JM, Labby KJ, Ji H, Mataka J, Xia G, Li H, Delker SL, Roman LJ, Martasek P, Poulos TL, Silverman RB. J. Med. Chem. 2011;54:6399. doi: 10.1021/jm200411j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wessel H-P, Iverson T, Bundle D. J. Chem. Soc., Perkin Trans. 1. 1985:2247. [Google Scholar]
- 5.Stoner EJ, Peterson MJ, Allen MS, DeMattei JA, Haight AR, Leanna MR, Patel SR, Plata DJ, Premchandran RH, Rasmussen M. J. Org. Chem. 2003;68:8847. doi: 10.1021/jo034883z. [DOI] [PubMed] [Google Scholar]
- 6.Lakhmiri R, Lhoste P, Dinou D. Tetrahedron Lett. 1989;30:4669. [Google Scholar]
- 7.Oltvoort JJ, Kloosterman M, van Boom JH. Recl. Trav. Chim. Pays-Bas. 1983;102:501. [Google Scholar]
- 8.Houlihan F, Bouchard F, Frechet JMJ. Can. J. Chem. 1985;63:153. [Google Scholar]