

Abstract
Haddad syndrome (congenital central hypoventilation syndrome and Hirschsprung's disease) is a rare disorder for which in-depth neuropathologic analysis is lacking. We report the brain findings in a full-term male infant with Haddad syndrome who died at 27 days of life. Bilateral hypoplasia of the superior temporal lobe and gyral anomalies in the frontal cortex were present. Immunohistochemistry with an antibody to tyrosine hydroxylase (noradrenaline synthesis) demonstrated hypoplasia of the locus coeruleus (implicated in chemoreception) and A5 region. Other findings included delayed maturation of the arcuate nucleus (putative human homologue of ventral medullary neurons in animals critical for chemoreception) and aberrant fascicles in the nucleus of the solitary tract. Efforts to determine the putative gene mutation were unsuccessful. This study implicates novel brain findings in Haddad syndrome mimicking those in murine Phox2b null mutants. This case suggests that abnormalities occur in CCHS in a network of sites critical to chemoreception.
Keywords: A5, Arcuate nucleus, Congenital central hypoventilation syndrome, Locus coeruleus, Nucleus of the solitary tract, Phox2b
Introduction
First reported by Haddad in 1978 [17], the combination of congenital central hypoventilation syndrome (CCHS) and Hirschsprung's disease (HCSR) (variable aganglionosis of the gastrointestinal tract) is extremely rare with approximately 57 cases reported in the worldwide literature [1, 8, 17]. In CCHS, patients breathe with inadequate depth due to impaired sensitivity to hypercapnia and hypoxia; they also experience autonomic and affective abnormalities [1, 8, 14, 17, 37, 43, 45]. CCHS remains a diagnosis of exclusion in patients who clinically lack neuromuscular, pulmonary, cardiac, or currently identifiable brainstem disease as a clinical explanation for abnormal chemoreception [1]. Haddad syndrome (MIM 209880) is interpreted not so much as a combination of two diseases but rather as a complicated form fruste of CCHS in which severe enteric nervous system aberration is superimposed on the core phenotype of ventilatory disease; almost 20% of patients with CCHS have concomitant HCSR [8]. Autopsy studies of patients with Haddad syndrome or CCHS are few [4, 5, 13, 26], in large part due to the rarity of clinically well-characterized cases that come to autopsy. In some cases, a brainstem lesion has been reported, i.e., neuronal depopulation and gliosis in the medullary tegmentum [26] and severe hypoplasia of the arcuate nucleus [13]. The latter lesion may contribute to impaired central chemoreception as the arcuate nucleus is postulated to contain neurons and glia, which are the human homologue of the respiratory chemosensitive fields for carbon dioxide monitoring in cats and rodents [12, 13, 33]. The paucity of comprehensive neuropathologic studies of patients with Haddad syndrome and CCHS prompted us to report a detailed neuropathologic study of a neonate with Haddad syndrome and well-documented central insensitivity to carbon dioxide.
A major advance in our understanding of CCHS is the discovery that mutations in the PHOX2B homeobox gene cause the majority of the cases [3, 29] and is critical for autonomic development [9, 34–36, 45]. Many patients with CCHS are heterozygous for a polyalanine expansion mutation in the second polyalanine repeat residue of PHOX2B, with one allele having 20 repeats and the affected allele having 25–33 repeats of the polyalanine sequence; in a small subset of patients, unique mutations in PHOX2B have been described [3, 38, 43, 46]. The PHOX2B gene maps to chromosome 4p12 and encodes a highly conserved homeobox transcription factor [43]. Homozygous Phox2b deficient mice embryos fail to form the hindbrain noradrenergic system, including the locus coeruleus (LC) [34]. All autonomic ganglia and the three cranial sensory ganglia that are part of the autonomic reflex circuits (visceral sensory ganglia of the seventh, ninth, and tenth cranial nerves) also fail to form properly [9, 36]. Moreover, the Phox2b knock-out mouse embryos lack peripheral nervous system expression of dopamine-beta-hydroxylase and tyrosine hydroxylase (TH), two genes encoding enzymes crucial for noradrenergic biosynthesis, supporting Phox2b as master regulator of central and peripheral noradrenergic differentiation [9, 34, 36]. In addition, the nucleus of the solitary tract, the vagal input of the autonomic nervous system, fails to form in homozygous Phox2b mouse mutant embryos [9]. Recently, absence of the retrotrapezoid nucleus, critical for chemoreception at the ventral surface of rats, was reported in Phox2b mouse knockouts [10, 16, 41], although the human homologue of the retrotrapezoid nucleus has yet to be determined. In the present case, we sought to determine if the anomalies of the LC and nucleus of the solitary tract that have been reported in Phox2b mice are present in Haddad syndrome. In our case, autopsy permission was limited to the examination of the brain only and precluded examination of relevant sites in the peripheral nervous system.
Clinical history
This male infant was the 39 5/7 weeks product of a 23-year-old G2P2, Caucasian mother following an uneventful pregnancy. The patient was born by vaginal delivery under epidural anesthesia. APGAR scores were 8 and 8 at 1 and 5 min, respectively. Birth weight was 3,668 grams (50–75th percentile), length was 52 cm (75th percentile), and head circumference was 36 cm (50th percentile). Soon after delivery, the infant developed grunting and respiratory retractions and was admitted to the neonatal intensive care unit for respiratory distress. Initial oxygen saturation was 96% but decreased to the 60s when the infant cried or was placed on his back. The infant was transferred to another hospital for the placement of a tracheotomy tube and mechanical ventilation. Several attempts to discontinue mechanical ventilation were unsuccessful: whenever the ventilator rate was decreased, the infant's arterial carbon dioxide levels (PaCO2) increased without an accompanying increase in the respiratory rate. Neonatal polysomnography demonstrated definite hypoventilation with increased PaCO2 when asleep, as well as intermittent hypoventilation episodes when awake. The arterial PaCO2 occasionally increased even on spontaneous intermittent mechanical ventilation at a rate of 30 breaths per minute, so the infant was kept on rates of 45–60 breaths per minute.
The infant appeared to have slightly dysmorphic features with prominent forehead and hypertelorism; he did not have the facial features described in CCHS in older patients, e.g., box-like facies, inferior inflection of the lateral segment of the vermillion border on the upper lip [42]. He had a right ptosis; pupils were 3.5 mm nonreactive to light. Fundoscopy revealed small optic discs bilaterally. The infant lacked blink to visual threat and corneal reflexes. There was bilateral facial weakness and a trace gag reflex. The tongue was midline without fasciculations. There was no suck reflex; the jaw had increased muscle tone. Motor examination revealed minimal spontaneous movements and mildly decreased muscle tone in the extremities. Deep tendon reflexes were 1+ and symmetric in all extremities; Babinski sign was negative bilaterally. An electroencephalogram, head ultrasound, head and neck computerized tomogram were normal. A two-dimensional and Doppler echocardiogram revealed normal pulmonary and cardiac structure and function.
In addition to respiratory insufficiency with ventilator-dependence, gastrointestinal dysfunction developed. Attempts to feed the infant were unsuccessful, leading to abdominal distension and high bilious output via the nasogastric tube. Thus, the infant was placed on total parenteral nutrition. Upper gastrointestinal series and small bowel follow-through studies were unremarkable. Rectal biopsy, however, revealed aganglionosis. At exploratory laparotomy, multiple biopsies of the gastrointestinal tract revealed ganglion cells first identified beginning 5 cm distal to the ligament of Treitz consistent with severe aganglionosis. Due to the combined severe respiratory and GI dysfunction, the infant was felt to have a very poor prognosis. The infant died shortly after withdrawal of the ventilator at 27 days of life. The parents donated the brain for research but a general autopsy was not performed.
Neuropathologic findings
Materials and methods
Tissue blocks of the entire brainstem, as well as multiple sections of cerebral cortex, periventricular white matter, deep cerebral gray matter structures, and cerebellum were embedded in paraffin, sectioned, and stained with hematoxylin-and-eosin/Luxol-fast-blue, as well as selected sections for immunostains for glial fibrillary acidic protein (GFAP) for reactive astrocytes and CD68 for reactive microglia. In addition, immunohistochemical analysis was performed on the serially sectioned brainstem of the patient (44 postconceptional weeks) and an age-related (52 post-conceptional weeks) control infant in the rostral pons (region of the LC) and the caudal pons (region of A5) using a polyclonal rabbit TH antibody (AB#151, Chemicon Int. Temecula, CA, USA). Control sections were analyzed with omission of the primary antibody. Standard methods of immunocytochemistry were applied. The density of TH-immunostained neurons in the LC was quantified using Neurolucida software in a sample of sections through the nucleus (n = 5 sections/case and control) (MicroBright-Field Inc. Colchester, VT, USA). A boundary contour was drawn around the LC, and neurons defined by a nucleolus were counted. The volumetric density of neurons was calculated as the number of neurons per the area of the boundary contour of the LC multiplied by the number of sections. The same approach was applied to quantitation in A5 in which 5 sections were examined per case and control through the region.
Findings in the cerebral hemispheres
The unfixed brain weighed 508 g (expected for age: 380 g); the increased brain weight was attributed to agonal vascular congestion as macroscopic cerebral edema was not present. External examination of the brain revealed open opercula and bilaterally small superior temporal gyri (Fig. 1); the middle temporal gyrus was malformed as it was divided into two gyri posteriorly (Fig. 1). The inferior temporal gyrus was unremarkable (Fig. 1). The cerebellum structurally appeared of appropriate size relative to the cerebrum (Fig. 1). Giant sections of the cerebrum revealed hyper-convoluted, slightly broad gyri in the frontal cerebral cortex (Fig. 2). Upon histological examination, the cortex demonstrated focal areas of irregular thickness and lamination with fused and shallow gyri (Fig. 2). The cytoarchitecture, however, of the temporal lobe gyri was unremarkable. Interstitial white matter neurons did not appear excessive in number by qualitative assessment. In the right parieto-occipital region, there was periventricular leukomalacia (PVL) characterized by focal periventricular necrosis and diffuse white matter gliosis (Fig. 3); the necrotic focus was infiltrated by macrophages indicative of the subacute/organizing stage of PVL (Fig. 3). There was diffuse gliosis in the cerebellar white matter as well. Examination of the thalamus, hypothalamus, hippocampi, and cerebellar cortex and roof nuclei were unremarkable. The degree of myelination throughout the brain was age-appropriate in comparison to human autopsy standards [19].
Fig. 1.
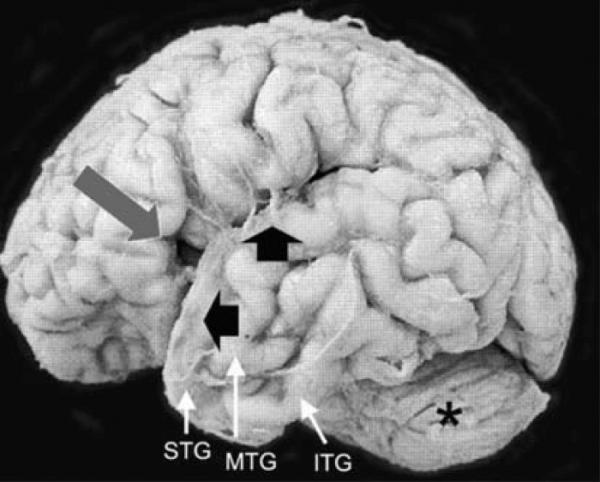
The external appearance of the brain of our neonatal case with Haddad syndrome was remarkable for a developmentally inappropriate opercula (long arrow) and small size of the superior temporal gyrus (STG) (short arrow). The middle temporal gyrus (MTG) appears split posteriorly; the inferior temporal gyrus (ITG) is unremarkable. The cerebellum (asterisk) is of appropriate size relative to the cerebrum
Fig. 2.

The cerebral cortex in our patient with Haddad syndrome is remarkable for focal anomalies characterized by hyperconvoluted gyri (arrows) in the frontal lobe (a) with microscopic evidence of short, fused sulci (arrows) (b, c) and focally anomalous lamination with a distinct Layer I but undifferentiated Layers II–VI (asterisk) (c). The deep gray nuclei are unremarkable (a). Hematoxylin-and-eosin
Fig. 3.
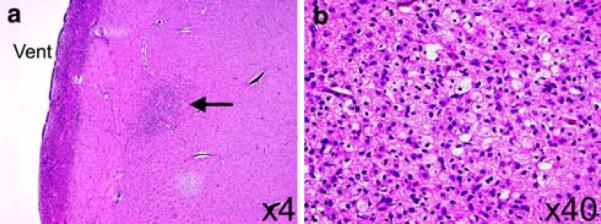
Periventricular leukomalacia was found in the parieto-occipital region of the patient's brain with Haddad syndrome (a, ×4). Focal necrosis (a) was noted in the white matter (arrow) close to the lateral ventricle (vent) which was characterized histologically by macrophages and reactive astrocytes (b, ×40). Hematoxylin-and-eosin
Brainstem findings
The brainstem and its vasculature appeared well formed upon macroscopic examination; several microscopic findings, however, were present. Transverse sections of the rostral pons of the case and age-related control stained with hematoxylin-and-eosin/Luxol-fast-blue demonstrated significant hypoplasia of the LC neuronal population in the case. Immunohistochemistry performed on serially sectioned brainstem with antibodies to TH confirmed substantial hypoplasia of the LC in the patient due to a marked deficit of TH-immunoreactive neurons compared to an age-related control (Fig. 4). The TH-neuronal cell density was 4.5 neurons/mm2 in the patient compared to 16.5 neurons/mm2 in the control in the LC; it was 1.5 neurons/mm2 in the patient compared to 3.4 neurons/mm3 in the control in A5. This reduction in cell density was not associated with gliosis. The external arcuate nucleus, which normally forms small clusters ventral to the medullary pyramids, appeared as a continuous cap over the ventral surface in sections throughout the medulla (Fig. 5). In addition, there was aberrant fasciculation in the nucleus of the solitary tract characterized by one large, one medium, and several smaller fascicles, the latter two were not as myelinated to the degree as the large fascicle (Fig. 5). Overall, the degree of myelination in the tractus solitarius was immature, as is age-appropriate according to human autopsy standards [19]. There was no gliosis in any brainstem site. The basis pontis, hypoglossal nucleus, trigeminal nucleus, facial nucleus, nucleus ambiguus, oculomotor nuclei, and rostral and caudal raphé neurons were unremarkable. The white matter tracts, including the decussation of the superior cerebellar peduncle, were also unremarkable. Periaqueductal gray sections were not available. Efforts to demonstrate immunocytochemical expression of PHOX2B protein in the brainstem in the case and control were technically unsuccessful with the available PHOX2B antibody (kindly supplied by Jean-Francois Brunet, Developmental Biology Institute of Marseille, France).
Fig. 4.
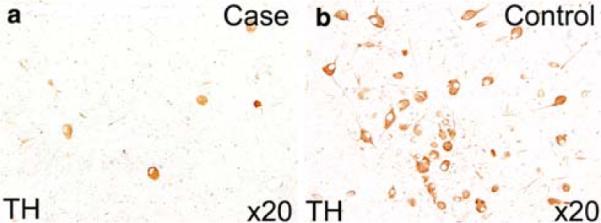
The locus coeruleus (LC) of the case with Haddad syndrome (a) is demonstrated at the same level of that of an age-matched control (b) and immunostained with an antibody to tyrosine hydroxylase (×20). There is a marked reduction of immunopositive LC neurons in the case (a) compared to the control (b)
Fig. 5.

Brainstem anomalies were detected in the Haddad case in two sites relevant to central chemoreception, i.e., the nucleus of the solitary tract (NTS) and ventral medullary surface (arcuate nucleus [ARC]) whose anatomic locations are indicated in a diagram of the infant medulla (e). The nucleus of the solitary tract contains an excessive number of fascicles of variable degrees of myelination (arrows) in the Haddad case (b) compared to the uniformly myelinated single fascicle (asterisk) in the control (a) (×20). In the Haddad case, the three fascicles are large, medium, and small (arrows). The arcuate nucleus forms a continuous cap of neurons and neuropil (arrows) overlying the pyramid (Pyr) in the Haddad case (d) compared to small clusters of neurons in the control (arrowhead) (×20). The degree of myelination in the pyramid is comparable between the case and control. Hematoxylin-and-eosin/Luxol-Fast-Blue (counter stain for myelin), ×40
Genetic analysis
A chromosome (karyotype) study in the infant was performed clinically and was normal. Microarray analysis to detect small deletions was not performed. At autopsy, DNA was extracted from paraffin-embedded brain tissue using the QIAamp DNA Mini Kit (QIAGEN Inc. Valencia, CA, USA) as frozen tissue was not available, and polymerase chain reaction (PCR) was performed using the specific primers to amplify the PHOX2B gene. Extensive efforts to isolate sufficient and technically optimal DNA were unsuccessful and the PHOX2B gene defect thus was not determined. Parental genetic testing was not performed.
Discussion
We report here the neuropathology of a neonatal case of Haddad syndrome whose clinical diagnosis was based upon the combined features of abnormal central chemoreception and severe aganglionosis of the gastrointestinal tract. Importantly, we observed brain findings which mimic in part those of the Phox2b knockout mouse [9, 34] and which have not been previously reported in Haddad syndrome or CCHS, i.e., developmental abnormalities in the LC, A5, and solitary tract. We also observed a developmental abnormality of the arcuate nucleus of the ventral surface of the medulla similar, albeit not identical, to that reported in CCHS previously [13]. Unfortunately, we were unable to prove a PHOX2B mutation in our case due to technical difficulties in DNA extraction from the available formalinfixed tissues; moreover, genetic testing upon the parents was not available. Nevertheless, the clinical phenotype of our case is identical to that of reports of PHOX2B mutations in Haddad syndrome and CCHS [1, 8, 14, 17, 43]; moreover, patients with Haddad syndrome who have undergone genetic testing have the PHOX2B mutation [38]. Irrespective of the determination of a specific PHOX2B mutation in our case, however, we found a spectrum of brainstem findings relevant to the regulation of chemoreception based upon extrapolation from animal studies, i.e., in the LC, A5, ventral surface (arcuate nucleus), and nucleus of the solitary tract, that ultimately may be found in subsequent neuropathologic studies of Haddad syndrome to be related to PHOX2B or other genes related to catecholaminergic and/or autonomic and respiratory control, e.g., RET, BDNF, MASH1. The finding of periventricular leukomalacia in our case further suggests that secondary hypoxic-ischemic insults likely compound neurological dysfunction in patients with Haddad syndrome and developmental brainstem abnormalities. Periventricular leukomalacia is a disorder of the immature cerebral white matter that is vulnerable to hypoxic-ischemic insult from midgestation through early infancy [20, 21]. While the peak occurrence of this white matter lesion is in the pre-term period, it is well-recognized to occur at term as well, particularly in the setting of cardiopulmonary disorders [20, 21]. The finding in our case of the organizing stage of PVL characterized by infiltrating macrophages in the periventricular necrotic focus indicates a subacute stage, i.e., after a week or more of the insult [20]. Below, we highlight the primary neuropathologic findings in our case in relationship to their clinical and biologic significance.
Abnormalities of brainstem regions implicated in central chemoreception
It has long been recognized that breathing is stimulated by brain acidification and the central chemoreflex [16, 31]. Historically, central chemoreception was thought to be exquisitely localized to the ventral surface of the medulla in the classic L, S, and M regions in cats and rats [16]. Subsequent research with different paradigms indicate, however, that many sites within the brainstem respond to focal acidification in vivo and many types of neurons respond to CO2/pH in vitro [6, 7, 10, 15, 16, 18, 24, 25, 28, 30–32, 39, 40]. These regions include the LC [7, 32], nucleus of the solitary tract [30, 32], caudal and rostral raphé [25, 39], retrotrapezoid nucleus [10, 15, 16, 25], preBötzinger complex [40], and fastigial nucleus of the cerebellum [28]. Central chemoreceptors provide the “tonic drive” to breathe, affect breath-to-breath variability, and modulate other control systems [31]. Yet, controversy exists whether there is a network of central chemoreceptors [31] relying on many types of pH-sensitive neurons, i.e., the so-called “distributed chemoreception theory”, or rather, a single specialized set of acid-sensitive neurons that drives the anatomically distinct central rhythm generator synaptically, i.e., the so-called “specialized chemoreceptor theory” [16]. In the latter theory, Guyenet et al. recently proposed that the retrotrapezoid nucleus at the ventral surface of the rostral medulla contains the most important central chemoreceptors [16], favoring the specialized chemoreceptor theory, based in part upon the findings in Phox2b knockout mice of a large decrease in the central chemoreceptor response [10], early mortality [10], abnormal breathing pattern [10], 85% reduction of neurons at this site [10], and the constitutive expression of PHOX2B protein by these neurons [41]. The human homologue of the retrotrapezoid nucleus has yet to be determined, although we suspect it is embedded in the arcuate nucleus of the rostral ventrolateral medulla (Kinney, unpublished observations). This idea is based upon the positional homology of arcuate neurons in the rostral ventrolateral medulla [12] and their glutamatergic phenotype in the majority of neurons [33]. Given the lack of a precisely defined homologue, however, the identification of the retrotrapezoid nucleus in our case was not feasible. Nevertheless, several sites known from animal data to be involved in chemoreception were affected in our case, i.e., LC, A5, ventral medullary surface, and nucleus of the solitary tract (see below), supporting the distributed chemoreception theory.
Perhaps the most profound lesion in a brainstem site involved in central chemoreception in our case was hypoplasia of the LC (A6). This nucleus, located in the rostral pons, is the major source of noradrenergic innervation to virtually the entire neuroaxis; this nucleus via its rostral projections is involved in arousal, cognition, attentive behavior, and sleep-wake cycles, and via caudal projections, in the control of the cardiovascular and respiratory systems [7, 18, 24]. Local tissue acidosis in the region of the LC produced by injections of acetazolamide significantly increases phrenic nerve activity; in addition, LC neurons in vitro are sensitive to carbon dioxide [7, 24, 32]. Given the functional anatomy of the LC, we speculate that the LC is a site of interface of chemoreception with arousal, attention, and cardiorespiratory control; experimental studies support this idea by the demonstration that brainstem catecholamine neurons promote wakefulness, participate in central chemoreception, stimulate breathing frequency, and minimize breathing variability in REM sleep [24]. The gene knockout studies in mice have shown that Phox2b is critical for the formation of the LC in fetal development [34]. In our case, the lack of proper formation of the LC likely contributes to impaired central chemoreception. An underpopulation of TH-neurons is also suggested in A5 as well as the LC in our case. This bilateral site in the ventrolateral tegmentum of the caudal pons is comprised of TH-neurons involved in sympathetic tone and the modulation of respiratory-related neurons, in part via projections to chemosensitive populations on the ventral medullary surface [6].
The nucleus of the solitary tract, like the LC, is implicated in central chemoreception [30, 32]. Its central neurons have also been shown to respond to both local changes in CO2 and acidification in vitro and in vivo [30, 31]. We observed aberrant fasciculation of the tract in the nucleus of the solitary tract in our case, which likely represents a developmental anomaly in path finding and fascicle formation. Of note, the degree of myelination appeared appropriate in these aberrant fascicles compared to human autopsy standards that indicate that the solitary tract is a “slow-myelinator” that has yet to attain mature myelination in infancy [19]. Like the LC, the nucleus of the solitary tract fails to develop properly in Phox2b knockout mice [9]. In the mutant, the anomaly of the NTS consists of lack of its development, compared to abnormal fasciculation in our case. The structural anomaly of fasciculation in our case suggests defective processing of oxygen and carbon dioxide levels as they interface with visceral sensory input and cardiorespiratory function.
A third site of putative chemoreception in the human brainstem is the arcuate nucleus, and aplasia or hypoplasia of this site has been reported in CCHS [13]. Embedded within the arcuate nucleus are serotonergic and glutamatergic neurons that in animals are specifically implicated in the cellular responses to carbon dioxide [33]. This nucleus forms from the germinal tissue of rhombic lip at the lateral pontomedullary junction; neurons migrate along the ventrolateral surface to form the arcuate nucleus at approximately 12 gestational weeks, with a second wave from the basis pontis over the ventral surface at approximately 20 weeks [11]. Initially, the arcuate nucleus is a continuous sheet of neurons overlying the entire pyramids at midgestation, but by the end of gestation, it forms clusters of 3–10 neurons that are “studded” along the pyramids. The defect in our case of a “sheet” of neurons at the ventral surface, potentially arrested at the midgestation stage, differs from aplasia or hypoplasia previously reported in CCHS but likely reflects a phenotypic difference based upon the specific gene mutation. Of note, the developmental nature of the lesions in the LC, solitary tract, and arcuate nucleus and the lack of gliosis suggest in our case that all of these lesions arise in the formation of the brainstem in utero and are not acquired, e.g., secondary to hypoxia-ischemia. A previous report of an 8-month-old with CCHS described mild generalized decrease in neuronal density and myelinated fibers in the “respiratory centers” of the medullary reticular formation that were attributed to a sublethal intrauterine lesion directly related to the functional abnormality in breathing [26]. A second report of a 2-year-old boy described slight to marked gliosis in the medullary reticular formation, nucleus of the solitary tract, and dorsal motor nucleus of the vagus in association with an intact arcuate nucleus that was attributed secondary to hypoxia [5]. It is likely that a spectrum of pathology exists that reflects the variations in the gene mutations and variability of secondary insults.
Cranial nerve nuclei pathophysiology
Despite the clinical finding of absent pupillary reflex and ptosis in this case, we found no obvious abnormalities of the cranial nerve three nuclei. Similar to the pathophysiology of Horner syndrome, the ptosis in this case may result from a functional sympathectomy to the eye secondary to atrophy or dysfunction of a PHOX2B-dependent superior cervical ganglion and ciliary ganglion. The restricted autopsy prevented the examination of the peripheral nervous system in our case. We did not observe morphological abnormalities in the trigeminal, facial, and ambiguous nuclei that could help explain the patient's clinical deficits in related cranial nerve dysfunction.
Discrepancies between the neuropathologic findings in our case of Haddad syndrome and neuroimaging studies
Conventional magnetic resonance studies have failed to define brainstem pathology in CCHS [44]. In a recent study of adolescent cases of CCHS, however, major structural abnormalities in the brainstem and cerebellum of the CCHS cases were reported based upon the superimposition of axial and radial diffusivity maps with diffusion tensor imaging [22]. Brainstem changes involved the lateral medulla, basis pontis, dorsal midbrain, oculomotor nuclei, periaqueductal gray, rostral raphé, and decussation of the superior cerebellar peduncle [22]. There was also injury in cerebellar structures, i.e., cerebellar cortex and deep (roof) nuclei and superior and inferior cerebellar peduncles [22]. This neuroimaging study did not demonstrate abnormalities in the LC or nucleus of the solitary tract which, given the microscopic nature of the findings in the Phox2b mutant and our case, is below the level of detection by these imaging techniques. In addition, cerebellar abnormalities as extensive as that demonstrated by diffusion tensor imaging in this series of CCHS patients neither have been reported in the Phox2b knockout mouse, nor did we observe cerebellar abnormalities in our case. We also did not find abnormalities in our case in the lateral medulla, basis pontis, dorsal midbrain, oculomotor nuclei, rostral and caudal raphé, or decussation of the superior cerebellar peduncle; sections of the periaqueductal gray were not available. The potential discrepancies between the neuroimaging and autopsy findings among CCHS cases likely reflect a heterogeneous group of disorders underlying CCHS and/or spectrum of abnormalities, which is not present in toto in every case. In addition, it is possible the neuroimaging studies detect functional abnormalities that are not apparent upon histopathologic examination at autopsy. Nevertheless, genotyping will be important in establishing clinical subtypes of CCHS and in deciphering the role of specific mutations in the pathogenesis of a particular cellular phenotype. In this regard, genotyping was performed or technically successful in only one-third of CCHS cases reported in the diffusion tensor imaging study [22], and was unsuccessful in our case.
Cerebral cortical anomalies
In our case, we found cortical anomalies in gyral development, notably in the frontal and temporal lobes. In the frontal cortex, we observed foci of abnormally thick and hyperconvoluted gyri. While these anomalies may be due to (unrecognized) intrauterine acquired insults, e.g., hypoxia-ischemia, or alternatively, due to the genetic defect itself, we speculate that they are secondary, at least in part, to the deficiency of the trophic influence of noradrenergic innervation from the LC during cortical development. Parenthetically, such cortical anomalies have not been described in the Phox2b mutant mice. Noradrenergic fibers from the LC innervate the developing cerebral cortex very early in embryonic development [23] and exert an important trophic influence upon the developing cortex [2, 23]; mice lacking vesicular monoamine transporters, for example, display cytoarchitectural anomalies within the somatosensory cortex [2]. Of note, was the finding of small superior temporal gyri, which has likewise been reported in a previous autopsy case of CCHS in infancy [26]. The clinical significance of this finding is unknown, particularly as chemosensory sites have not been reported in superior temporal gyri, i.e., temporal lobe components of the limbic system. The application of hypoxic challenges in CCHS, however, results in functional imaging changes within limbic sites, as well as the cerebellum and brainstem [27]. While the superior temporal lobe has not been specifically implicated to our knowledge in chemoreception, our case suggests that this site should be specifically sought as an abnormal limbic site in future neuroimaging studies of Haddad syndrome and CCHS. A developmental abnormality in overall brain growth is suggested by the increased brain weight at autopsy in our patient, but the effects of agonal changes (vascular congestion, cerebral edema) cannot be excluded.
Conclusion
The involvement in our case of neonatal Haddad syndrome of at least three brainstem regions known to regulate responses to carbon dioxide suggest the possibility that Haddad syndrome and CCHS involve a network of critically related chemosensitive sites rather than one specific site, and that these sites are likely to mediate different aspects of central chemoreception related to the function of the each individual nucleus within the network. Based upon extrapolation to the Phox2b knockout mouse, the brainstem anomalies reflect development, which is altered in some way by PHOX2B and its functions related to the cellular proliferation, migration, differentiation and/or pathfinding and fasciculation of the affected sites, which occurs or begins during gestation. In turn, the cortical anomalies likely reflect abnormal noradrenergic innervation from the LC and its trophic influence upon cortical development. This case underscores the important role of detailed neuropathologic examination to elucidate the pathogenesis of Haddad syndrome and CCHS.
Acknowledgments
The authors appreciate the help of Dr. Abbas R. Kingo M.D., Ms. Katrina Farwig, Ms. Lena Liu, and Mr. Richard A. Belliveau in the course of the study and the preparation of the manuscript. We appreciate the assistance of Dr. Elizabeth C. Engle and Ms. Wai-Man Chan in the analysis for PHOX2B mutations in the case. We are grateful for the careful reading of the manuscript by Dr. Eugene E. Nattie. This study was supported by the Christopher James Murphy Foundation (HCK).
References
- 1.Idiopathic congenital central hypoventilation syndrome: diagnosis and management. American Thoracic Society. Am J Respir Crit Care Med. 1999;160:368–373. doi: 10.1164/ajrccm.160.1.16010. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez C, Vitalis T, Fon EA, et al. Effects of genetic depletion of monoamines on somatosensory cortical development. Neuroscience. 2002;115:753–764. doi: 10.1016/s0306-4522(02)00484-0. [DOI] [PubMed] [Google Scholar]
- 3.Amiel J, Laudier B, Attie-Bitach T, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong D, Sachis P, Bryan C, Becker L. Pathological features of persistent infantile sleep apnea with reference to the pathology of sudden infant death syndrome. Ann Neurol. 1982;12:169–174. doi: 10.1002/ana.410120207. [DOI] [PubMed] [Google Scholar]
- 5.Becker LE, Takashima S. Chronic hypoventilation and development of brain stem gliosis. Neuropediatrics. 1985;16:19–23. doi: 10.1055/s-2008-1052538. [DOI] [PubMed] [Google Scholar]
- 6.Benarroch EE, Schmeichel AM, Low PA, Sandroni P, Parisi JE. Loss of A5 noradrenergic neurons in multiple system atrophy. Acta Neuropathol. 2008;115:629–634. doi: 10.1007/s00401-008-0351-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biancardi V, Bicego KC, Almeida MC, Gargaglioni LH. Locus coeruleus noradrenergic neurons and CO2 drive to breathing. Pflugers Arch. 2008;455:1119–1128. doi: 10.1007/s00424-007-0338-8. [DOI] [PubMed] [Google Scholar]
- 8.D'Souza S, Khubchandani RP. Haddad syndrome—congenital central hypoventilation associated with Hirschsprung's disease. Indian J Pediatr. 2003;70:597–599. doi: 10.1007/BF02723168. [DOI] [PubMed] [Google Scholar]
- 9.Dauger S, Pattyn A, Lofaso F, et al. Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development. 2003;130:6635–6642. doi: 10.1242/dev.00866. [DOI] [PubMed] [Google Scholar]
- 10.Dubreuil V, Ramanantsoa N, Trochet D, et al. A human mutation in Phox2b causes lack of CO2 chemosensitivity, fatal central apnea, and specific loss of parafacial neurons. Proc Natl Acad Sci USA. 2008;105:1067–1072. doi: 10.1073/pnas.0709115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Essick C. The development of the nucleus pontis and nucleus arcuatus in man. Am J Anat. 1912;13:25–54. [Google Scholar]
- 12.Filiano JJ, Choi JC, Kinney HC. Candidate cell populations for respiratory chemosensitive fields in the human infant medulla. J Comp Neurol. 1990;293:448–465. doi: 10.1002/cne.902930308. [DOI] [PubMed] [Google Scholar]
- 13.Folgering H, Kuyper F, Kille JF. Primary alveolar hypoventilation (Ondine's curse syndrome) in an infant without external arcuate nucleus. Case report. Bull Eur Physiopathol Respir. 1979;15:659–665. [PubMed] [Google Scholar]
- 14.Goldberg DS, Ludwig IH. Congenital central hypoventilation syndrome: ocular findings in 37 children. J Pediatr Ophthalmol Strabismus. 1996;33:175–180. doi: 10.3928/0191-3913-19960501-11. [DOI] [PubMed] [Google Scholar]
- 15.Guyenet PG, Mulkey DK, Stornetta RL, Bayliss DA. Regulation of ventral surface chemoreceptors by the central respiratory pattern generator. J Neurosci. 2005;25:8938–8947. doi: 10.1523/JNEUROSCI.2415-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guyenet PG, Stornetta RL, Bayliss DA. Retrotrapezoid nucleus and central chemoreception. J Physiol. 2008;586:2043–2048. doi: 10.1113/jphysiol.2008.150870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haddad GG, Mazza NM, Defendini R, et al. Congenital failure of automatic control of ventilation, gastrointestinal motility and heart rate. Medicine (Baltimore) 1978;57:517–526. doi: 10.1097/00005792-197811000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Kayama Y, Koyama Y. Control of sleep and wakefulness by brainstem monoaminergic and cholinergic neurons. Acta Neurochir Suppl. 2003;87:3–6. doi: 10.1007/978-3-7091-6081-7_1. [DOI] [PubMed] [Google Scholar]
- 19.Kinney HC, Brody BA, Kloman AS, Gilles FH. Sequence of CNS myelination in human infancy II. Patterns of myelination in autopsied infants. J Neuropathol Exp Neurol. 1988;47:217–234. doi: 10.1097/00005072-198805000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Kinney HC, Volpe JJ. Perinatal panencephalopathy in the premature infant: is it due to Hypoxia-Ischemia?”. In: Haddad GG, Ping YS, editors. Perinatal Brain Hypoxia and Ischemia. Contemporary Clinical Neuroscience. The Humana Press; Totowa: 2009. pp. 153–185. [Google Scholar]
- 21.Kinney HC, Panigrahy A, Newburger JW, Jonas RA, Sleeper LA. Hypoxic-ischemic brain injury in infants with congenital heart disease dying after cardiac surgery. Acta Neuropathol. 2005;110:563–578. doi: 10.1007/s00401-005-1077-6. [DOI] [PubMed] [Google Scholar]
- 22.Kumar R, Macey PM, Woo MA, Alger JR, Harper RM. Diffusion tensor imaging demonstrates brainstem and cerebellar abnormalities in congenital central hypoventilation syndrome. Pediatr Res. 2008;64:275–280. doi: 10.1203/PDR.0b013e31817da10a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levitt P, Moore RY. Development of the noradrenergic innervation of neocortex. Brain Res. 1979;162:243–259. doi: 10.1016/0006-8993(79)90287-7. [DOI] [PubMed] [Google Scholar]
- 24.Li A, Nattie E. Catecholamine neurones in rats modulate sleep, breathing, central chemoreception and breathing variability. J Physiol. 2006;570:385–396. doi: 10.1113/jphysiol.2005.099325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li A, Zhou S, Nattie E. Simultaneous inhibition of caudal medullary raphe and retrotrapezoid nucleus decreases breathing and the CO2 response in conscious rats. J Physiol. 2006;577:307–318. doi: 10.1113/jphysiol.2006.114504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu HM, Loew JM, Hunt CE. Congenital central hypoventilation syndrome: a pathologic study of the neuromuscular system. Neurology. 1978;28:1013–1019. doi: 10.1212/wnl.28.10.1013. [DOI] [PubMed] [Google Scholar]
- 27.Macey PM, Woo MA, Macey KE, et al. Hypoxia reveals posterior thalamic, cerebellar, midbrain, and limbic deficits in congenital central hypoventilation syndrome. J Appl Physiol. 2005;98:958–969. doi: 10.1152/japplphysiol.00969.2004. [DOI] [PubMed] [Google Scholar]
- 28.Martino PF, Davis S, Opansky C, et al. The cerebellar fastigial nucleus contributes to CO2–H+ ventilatory sensitivity in awake goats. Respir Physiol Neurobiol. 2007;157:242–251. doi: 10.1016/j.resp.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matera I, Bachetti T, Puppo F, et al. PHOX2B mutations and polyalanine expansions correlate with the severity of the respiratory phenotype and associated symptoms in both congenital and late onset Central Hypoventilation syndrome. J Med Genet. 2004;41:373–380. doi: 10.1136/jmg.2003.015412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nattie EE, Li A. CO2 dialysis in nucleus tractus solitarius region of rat increases ventilation in sleep and wakefulness. J Appl Physiol. 2002;92:2119–2130. doi: 10.1152/japplphysiol.01128.2001. [DOI] [PubMed] [Google Scholar]
- 31.Nattie EE, Li A. Central chemoreception is a complex system function that involves multiple brainstem sites. J Appl Physiol. 2008;106(4):1464–1466. doi: 10.1152/japplphysiol.00112.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nichols NL, Hartzler LK, Conrad SC, Dean JB, Putnam RW. Intrinsic chemosensitivity of individual nucleus tractus solitarius (NTS) and locus coeruleus (LC) neurons from neonatal rats. Adv Exp Med Biol. 2008;605:348–352. doi: 10.1007/978-0-387-73693-8_61. [DOI] [PubMed] [Google Scholar]
- 33.Paterson DS, Thompson EG, Kinney HC. Serotonergic and glutamatergic neurons at the ventral medullary surface of the human infant: observations relevant to central chemosensitivity in early human life. Auton Neurosci. 2006;124:112–124. doi: 10.1016/j.autneu.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 34.Pattyn A, Goridis C, Brunet JF. Specification of the central noradrenergic phenotype by the homeobox gene Phox2b. Mol Cell Neurosci. 2000;15:235–243. doi: 10.1006/mcne.1999.0826. [DOI] [PubMed] [Google Scholar]
- 35.Pattyn A, Hirsch M, Goridis C, Brunet JF. Control of hindbrain motor neuron differentiation by the homeobox gene Phox2b. Development. 2000;127:1349–1358. doi: 10.1242/dev.127.7.1349. [DOI] [PubMed] [Google Scholar]
- 36.Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of autonomic neural crest derivatives. Nature. 1999;399:366–370. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- 37.Pine DS, Weese-Mayer DE, Silvestri JM, Davies M, Whitaker AH, Klein DF. Anxiety and congenital central hypoventilation syndrome. Am J Psychiatry. 1994;151:864–870. doi: 10.1176/ajp.151.6.864. [DOI] [PubMed] [Google Scholar]
- 38.Sasaki A, Kanai M, Kijima K, et al. Molecular analysis of congenital central hypoventilation syndrome. Hum Genet. 2003;114:22–26. doi: 10.1007/s00439-003-1036-z. [DOI] [PubMed] [Google Scholar]
- 39.Severson CA, Wang W, Pieribone VA, Dohle CI, Richerson GB. Midbrain serotonergic neurons are central pH chemoreceptors. Nat Neurosci. 2003;6:1139–1140. doi: 10.1038/nn1130. [DOI] [PubMed] [Google Scholar]
- 40.Solomon IC, Edelman NH, O'Neal MH., 3rd CO(2)/H(+) chemoreception in the cat pre-Botzinger complex in vivo. J Appl Physiol. 2000;88:1996–2007. doi: 10.1152/jappl.2000.88.6.1996. [DOI] [PubMed] [Google Scholar]
- 41.Spengler CM, Gozal D, Shea SA. Chemoreceptive mechanisms elucidated by studies of congenital central hypoventilation syndrome. Respir Physiol. 2001;129:247–255. doi: 10.1016/s0034-5687(01)00294-8. [DOI] [PubMed] [Google Scholar]
- 42.Todd ES, Weinberg SM, Berry-Kravis EM, Silvestri JM, Kenny AS, Rand CM, Zhou L, Maher BS, Marazita ML, Weese-Mayer DE. Facial phenotype in children and young adults with PHOX2B-determined congenital central hypoventilation syndrome: quantitative pattern of dysmorphology. Pediatr Res. 2006;59:39–45. doi: 10.1203/01.pdr.0000191814.73340.1d. [DOI] [PubMed] [Google Scholar]
- 43.Weese-Mayer DE, Berry-Kravis EM, Zhou L, et al. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2b. Am J Med Genet A. 2003;123A:267–278. doi: 10.1002/ajmg.a.20527. [DOI] [PubMed] [Google Scholar]
- 44.Weese-Mayer DE, Brouillette RT, Naidich TP, McLone DG, Hunt CE. Magnetic resonance imaging and computerized tomography in central hypoventilation. Am Rev Respir Dis. 1988;137:393–398. doi: 10.1164/ajrccm/137.2.393. [DOI] [PubMed] [Google Scholar]
- 45.Woo MS, Woo MA, Gozal D, Jansen MT, Keens TG, Harper RM. Heart rate variability in congenital central hypoventilation syndrome. Pediatr Res. 1992;31:291–296. doi: 10.1203/00006450-199203000-00020. [DOI] [PubMed] [Google Scholar]
- 46.Yokoyama M, Watanabe H, Nakamura M. Genomic structure and functional characterization of NBPhox (PMX2B), a homeodomain protein specific to catecholaminergic cells that is involved in second messenger-mediated transcriptional activation. Genomics. 1999;59:40–50. doi: 10.1006/geno.1999.5845. [DOI] [PubMed] [Google Scholar]