

Abstract

Worldwide research efforts in drug discovery involving HIV integrase have produced only one compound, raltegravir, that has been approved for clinical use in HIV/AIDS. As resistance, toxicity, and drug–drug interactions are recurring issues with all classes of anti-HIV drugs, the discovery of novel integrase inhibitors remains a significant scientific challenge. We have designed a lead HIV-1 strand transfer (ST) inhibitor (IC50 70 nM), strategically assembled on a pyridinone scaffold. A focused structure–activity investigation of this parent compound led to a significantly more potent ST inhibitor, 2 (IC50 6 ± 3 nM). Compound 2 exhibits good stability in pooled human liver microsomes. It also displays a notably favorable profile with respect to key human cytochrome P450 (CYP) isozymes and human UDP glucuronosyl transferases (UGTs). The prodrug of inhibitor 2, i.e., compound 10, was found to possess remarkable anti-HIV-1 activity in cell culture (EC50 9 ± 4 nM, CC50 135 ± 7 μM, therapeutic index = 15 000).
Keywords: Integrase inhibitor, pyridinone, CYP/UGT profile, anti-HIV prodrug
The retroviral enzyme, HIV-1 integrase, which is encoded at the 3′-end of the pol gene of the human immunodeficiency virus (HIV), is essential for HIV replication and is a significant target for the discovery and development of anti-HIV therapeutic agents.1−11 However, research efforts in the area of anti-HIV integrase inhibitors for the treatment of acquired immunodeficiency syndrome (AIDS) have resulted in only one compound, raltegravir (Isentress), that has been approved by the FDA for the clinical treatment of HIV-AIDS.7,8 However, as resistance, toxicity, and drug–drug interactions are recurring issues with all classes of anti-HIV drugs, the discovery of new, anti-HIV active integrase inhibitors remains a significant scientific challenge. HIV-1 integrase is a 32 kDa protein,1,12,13 which catalyzes the incorporation of HIV DNA into host chromosomal DNA through a specifically defined sequence of reactions, which involves 3′-processing and a key strand transfer (ST) step.1,3,12−15 Initiation of integration occurs in the cytoplasm, where a complex is formed between viral cDNA, previously produced by reverse transcription, and HIV integrase. Following this is site-specific endonucleatic cleavage of two nucleotides from each 3′-end of double-stranded viral DNA, which produces truncated viral DNA with terminal CAOH-3′ (3′-processing). The next step, ST, occurs in the nucleus and involves staggered nicking of chromosomal DNA and joining of each 3′-end of the recessed viral DNA to the 5′-ends of the host DNA, followed by repair/ligation. The ST step is carried out after transport of the processed, preintegration complex from the cytoplasm into the nucleus. Both 3′-processing and ST steps require divalent metal ion cofactors.
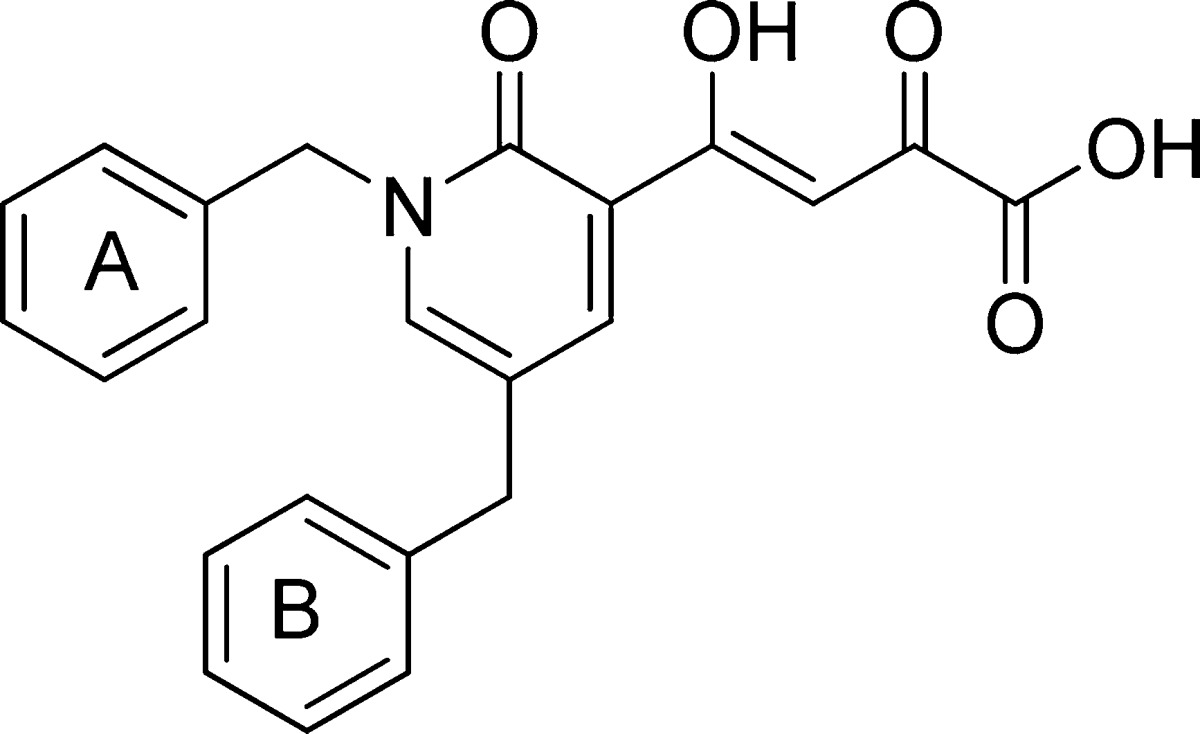
To explore whether a significantly anti-HIV active integrase inhibitor could be discovered that would also possess a favorable in vitro drug–drug interaction profile with respect to key cytochrome P450 (CYP) and UDP glucuronosyltransferase (UGT) isozymes, we carried out the design of such an inhibitor from a lead compound discovered in our laboratory. This lead compound was 4-(1,5-dibenzyl-1,2-dihydro-2-oxopyridin-3-yl)-2-hydroxy-4-oxobut-2-enoic acid (1, Figure 1), which was an inhibitor of the ST step of HIV-1 integrase (IC50 70 nM). Using compound 1 as a starting point, we undertook lead optimization studies on 1.16
Figure 1.

Structure of lead compound 1.
In the discovery of lead compound 1, it was established that the specific nature of the modified nucleobase scaffold (i.e., the pyridinone ring) and the nature of the substituents on the scaffold (the functional components as well as the hydrophobic benzyl groups) were critical for integrase inhibitory activity. For this reason, we focused our optimization studies on substituents on the hydrophobic phenyl groups of the pyridinone scaffold. In the subsequent study, we examined the effects of various substituents, e.g., methoxy, chloro, alkyls, and mixed halo/alkyl and others, on the phenyl rings and their effect on the enzymology involving ST step inhibition. There was considerable variation in the ST inhibitory activity for these compounds (IC50 <10 nM to >1500 nM). Fluoro substitution IC50 data, however, were more compelling. Among this entire group of fluorinated compounds, the difluoro, trifluoro, and tetrafluoro substituted compounds all had ST inhibitory IC50 values falling in the range of <10 nM, showing significant improvement over lead compound 1. Within this group of fluorinated compounds, the trifluoroaryl (o- and o,p) and tetrafluoroaryl (o,p and o,p) substituted analogues (involving both phenyl rings) were the most active in terms of the integrase IC50 and IC90 data (≤6 and <100 nM, respectively). While the detailed reason for the increase in inhibitory potency with appropriate fluorine substitution is not fully understood; hydrophobic and/or electrostatic interactions may contribute.17−19 In the next level of lead optimization, we investigated the antiviral cell culture data for these compounds. The results are summarized in Table 1 and show that the anti-HIV-1 EC50 values were largely in the 1–3 μM range. However, two compounds emerged from these studies that exhibited anti-HIV EC50 values of 500 nM or less. They were 4-(5-(2,4-difluorobenzyl)-1-(2-fluorobenzyl)-2-oxo-1,2-dihydropyridin-3-yl)-2-hydroxy-4-oxobut-2-enoic acid (2, entry 56, Table 1) and 4-(1,5-bis(2,4-difluorobenzyl)-2-oxo-1,2-dihydropyridin-3-yl)-4-hydroxy-2-oxobut-3-enoic acid (entry 11, Table 1). Their ST inhibition IC50 data were 6 ± 3 nM and 5.5 ± 1.5 nM, respectively. The eventual selection of compound 2 over entry 11 as the key compound to move forward is discussed in the prodrug section below.
Table 1. In Vitro Anti-HIV Data for Analogues of Compound 1.
| aryl ring A |
aryl ring B |
||||||
|---|---|---|---|---|---|---|---|
| entry | o | m | p | o | m | p | EC50 (μM)a,b |
| 1 | H | H | H | H | H | H | 2.1 |
| 2 | F | H | H | H | H | H | 0.9 |
| 3 | H | H | F | H | H | H | 1.0 |
| 4 | F | H | F | H | H | H | 0.6 |
| 5 | F | H | H | F | H | H | 0.7 |
| 6 | H | H | OMe | H | H | H | 1.8 |
| 7 | H | H | F | H | H | F | 0.8 |
| 8 | H | H | Me | H | H | F | 1.6 |
| 9 | H | Cl | F | H | H | H | 0.8 |
| 10 | H | H | OMe | H | H | OMe | >5.0 |
| 11 | F | H | F | F | H | F | 0.3 |
| 12 | H | F | H | H | H | H | 2.0 |
| 13 | H | Cl | H | H | Cl | H | 1.1 |
| 14 | H | Cl | F | H | Cl | H | 1.2 |
| 15 | F | H | H | H | H | F | 0.7 |
| 16 | H | Cl | F | H | Cl | F | 2.4 |
| 17 | H | H | Me | H | Cl | F | >5.0 |
| 18 | H | H | Me | H | Cl | H | 2.5 |
| 19 | H | H | Me | F | H | H | 0.7 |
| 20 | H | H | F | H | Cl | F | >5.0 |
| 21 | F | Cl | F | H | H | H | 0.6 |
| 22 | H | H | F | H | Cl | H | 1.4 |
| 23 | 2,6-di-F | H | H | H | H | F | 1.4 |
| 24 | H | H | F | F | H | H | 0.8 |
| 25 | F | Cl | H | F | Cl | H | 2.4 |
| 26 | H | H | Cl | H | Cl | H | 2.5 |
| 27 | H | Cl | H | H | Cl | H | 1.4 |
| 28 | F | Cl | H | H | Me | H | 1.4 |
| 29 | F | H | F | F | H | H | 0.6 |
| 30 | H | Me | H | H | Cl | F | 2.2 |
| 31 | F | Cl | H | H | H | H | 0.7 |
| 32 | F | 5-Cl | H | H | H | H | 0.9 |
| 33 | F | Cl | H | H | Cl | H | 1.4 |
| 34 | F | H | F | H | Cl | F | 1.4 |
| 35 | H | Cl | H | H | H | F | 1.2 |
| 36 | H | Cl | H | F | H | H | 0.7 |
| 37 | 2,6-di-F | H | H | H | Cl | F | 5.1 |
| 38 | 2,6-di-F | H | H | H | H | H | 1.5 |
| 39 | H | H | H | F | H | F | 0.8 |
| 40 | F | H | H | H | Cl | F | 2.2 |
| 41 | H | F | H | F | H | H | 1.9 |
| 42 | F | H | H | H | Cl | H | 1.9 |
| 43 | F | F | H | H | H | H | 0.9 |
| 44 | H | Me | H | F | H | H | 0.9 |
| 45 | F | F | H | H | H | F | 0.7 |
| 46 | H | H | CN | F | H | H | 1.1 |
| 47 | F | F | H | F | H | H | 0.8 |
| 48 | H | F | H | H | H | F | 1.0 |
| 49 | F | H | F | Cl | H | H | 0.8 |
| 50 | H | Cl | H | F | Cl | H | 1.5 |
| 51 | H | H | F | F | H | F | 0.6 |
| 52 | H | Me | H | F | H | F | 0.7 |
| 53 | H | H | H | H | COOH | H | >5.0 |
| 54 | H | H | H | Me | H | F | 1.3 |
| 55 | H | H | F | Me | H | F | 1.0 |
| 56 | F | H | H | F | H | F | 0.5 |
| 57 | F | H | H | Me | H | F | 1.3 |
EC50 values are the average of three determinations. Standard deviations for the EC50 are within 31% of the average.
EC50 = concentration for 50% inhibition of the replication of HIV-1.
A highly efficient synthesis of compound 2 (Scheme 1) was developed in our laboratory. Only seven steps (aromatic nucleophilic addition, demethylation/deoxygenation, radical bromination, benzylation, palladium-catalyzed cross-coupling, Claisen condensation, and acid-catalyzed hydrolysis)20−26 were required for the total synthesis of 2 from commercially available 5-bromo-2-methoxypyridine (3). The overall yield from 3 was 37%.
Scheme 1. Methodology Developed for the Synthesis of Integrase Inhibitor 2.

Abbreviations: (i) tert-butyl methyl ether (TBME); (ii) trimethylsilyl chloride (TMSCl), triethylsilane (TES), trifluoroacetic acid (TFA); (iii) N-bromosuccinimide (NBS); (iv–v) dimethylformamide (DMF); (vi) tetrahydrofuran (THF).
Stability studies of compound 2 in pooled human liver microsomes were carried out by preincubation, initiation with NADPH, incubation at 37 °C, quenching of samples at various time intervals with cold acetonitrile, centrifugation to remove precipitated proteins, and finally HPLC analysis that utilized UV detection.27−29 These studies revealed that integrase inhibitor 2 was relatively stable in human liver microsomes, exhibiting an in vitro half-life of >3 h, as evidenced from HPLC data, which showed that 80% of compound 2 remained after the 3 h incubation in microsomes (Figure 2). The key metabolite, which was slowly produced, was identified by HPLC and HRMS data to be the product of the retro-Claisen cleavage of the diketo group of 2 to produce the acetyl pyridinone 8.
Figure 2.
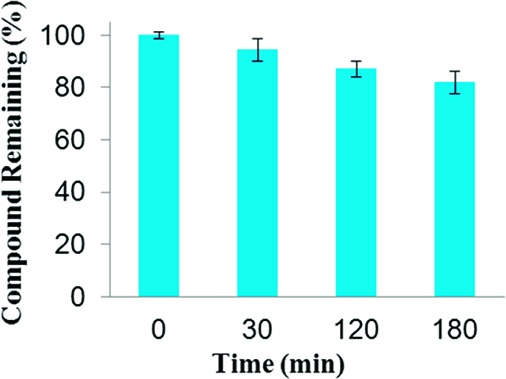
Stability of 2 in pooled human liver microsomes monitored by HPLC/UV.
The in vitro drug interaction profile involving key cytochrome P450 (CYP) isozymes28−30 and appropriate substrates in pooled human liver microsomes with varying concentrations of 2, followed by kinetic analysis (Table 2), showed that compound 2 was not an inhibitor of CYP3A4 and CYP2D6 isozymes and was a very weak inhibitor of the CYP2C8 isozyme (Table 1). These key isozymes account for a total of over 80% of drugs that are metabolized by different CYP isoforms. In addition, compound 2 did not exhibit any activation of these CYP isozymes. Thus, our studies suggest that this integrase-based, anti-HIV compound is anticipated to have a favorable drug interaction profile with respect to key CYP isozymes.
Table 2. IC50 Data for Inhibition of Key Cytochrome P450 Isozymes.
| CYP450 isozyme | substrate/stock solution | conc (μM) | protein (mg/mL) | incubation (min) | IC50 data (μM) |
|---|---|---|---|---|---|
| CYP3A4 | testosterone (50 mM) | 100 | 0.3 | 30 | >200 |
| CYP3A4 | triazolam (50 mM) | 200 | 0.4 | 30 | >200 |
| CYP2D6 | dextromethorphan (50 mM) | 200 | 2.0 | 60 | >200 |
| CYP2C8 | amodiaquine (5 mM) | 200 | 0.4 | 30 | >65 |
Because isozymes of UGT also play an important role in determining drug–drug interactions, we investigated the substrate activity of 2 toward key human UGTs.27,31 Compound 2 was not a substrate for the following key UGT isozymes: 1A1, 1A4, 1A6, 1A9, and 2B7. In comparison, the major mechanism for rapid clearance of raltegravir in humans is through UGT 1A1-mediated glucuronidation.27 The integrase inhibitor, S/GSK 1349572, is also a substrate for UGT 1A1, and its primary route of metabolism and subsequent clearance is glucuronidation.10
Antiviral data in cell culture of compound 2 revealed a significant disconnect of almost two or more orders of magnitude between the anti-HIV-1 activity (EC50 500 nM, MAGI cells) and the ST inhibition data for 2 (IC50 6 nM). For HIV-1 integrase inhibitors, there is normally a reasonably strong correlation between ST IC50 data and cell culture EC50 data.3,5 Because the disconnect between the IC50 and EC50 data for compound 2 and also other compounds, including entry 11 (Table 1), appeared to be a problem associated with the cellular permeability of the inhibitors, we examined prodrugs of these compounds. The isopropyl ester prodrug, 10 (Figure 3), was easily synthesized from compound 2 through acid-catalyzed esterification with 2-propanol. The cLog P values for compounds 2 and the isopropyl ester 10 are 2.38 and 4.24, respectively, suggesting that compound 10 is significantly less polar than compound 2 and thus would be expected to be more cellularly permeable. The antiviral data confirmed this, as prodrug 10 exhibited remarkable anti-HIV-1 activity (EC50 = 9 ± 4 nM, MAGI cells). This was the best activity achieved of all of the prodrugs studied in this work (EC50 values ranged from 9 to 46 nM for the isopropyl ester prodrugs). The isopropyl ester prodrug of entry 11 (Table 1) was significantly less active than compound 10, exhibiting an EC50 of 46 ± 18 nM (MAGI cells). The overall performance of the assay was validated by the MOI-sensitive positive control compound, raltegravir, which exhibited the expected level of antiviral activity (EC50 < 6 nM).7 Cell viability data for 10 showed only low toxicity at higher test concentrations (CC50 = 135 ± 7 μM, CC90 > 200 μM), although a CC90 was not reached at the highest test concentration (200 μM). It is of relevance to mention that the EC50 and EC90 data for 10 correlate exceptionally well with the ST inhibition IC50 and IC90 data for 2 (6 and 97 nM, respectively). The therapeutic (selectivity) index TI (CC50/EC50) for 10 was 15 000. Finally, isopropyl ester 10 was a poor inhibitor of HIV integrase in enzymatic studies (IC50 > 475 nM), suggesting that the anti-HIV activity of 10 was most likely the result of its hydrolysis in cell culture to produce the cellularly active anti-HIV compound 2. Consistent with this conclusion was our observation that CYP and UGT studies on compound 10 were precluded by its rapid hydrolysis in human liver microsomes to produce compound 2 (100% conversion in 15 min).
Figure 3.

Structures of prodrug 10 and raltegravir.
While a number of structurally diverse compounds have been reported to be inhibitors of HIV integrase, the data of two of these compounds (Figure 4), that have received considerable attention and that have a relationship, albeit peripheral, to the compounds described herein, are worthy of mention. Both compounds are ST inhibitors [IC50 20 nM (S-1360) and 170 nM (L-731,988)]. The in vitro anti-HIV data for the β-diketo triazole, S-1360,32 showed an EC50 of 140 nM and a CC50 of 110 μM (PBMC), resulting in a TI of 790. Compound L-731,988 is somewhat less active (EC50 1 μM in MT-4 cells).33 The CC50 value was not given.
Figure 4.

Two well-known β-diketo compounds.
In summary, our search for new integrase-based, anti-HIV compounds led to the discovery of a highly potent ST inhibitor of HIV-1 integrase, 2 (IC50 6 nM). This compound was relatively stable in pooled human liver microsomes (80% of compound remained after incubation for 3 h). It displayed a favorable interaction profile with respect to key human CYP isozymes, as it was not an inhibitor or activator of these isozymes. Also of significance was the observation that inhibitor 2 was not a substrate of important human UGTs. A prodrug of 2, i.e., compound 10, exhibited remarkable anti-HIV-1 activity in cell culture (EC50 = 9 nM, CC50 = 135 μM). The therapeutic or selectivity index of prodrug 10, which was 15 000, was also a notable finding. Further biological studies are in progress.
Acknowledgments
Some of the data cited here were determined at Inhibitex, Inc., Alpharetta, GA, and at the Southern Research Institute, Frederick, MD, and we express our thanks to them.
Supporting Information Available
Antiviral assays, microsome stability assays, cytochrome P450 inhibition assays, UDP-glucuronosyltransferase substrate assays, synthesis of integrase inhibitor and its prodrug, analytical data for all compounds, and HPLC purity trace of integrase inhibitor. This material is available free of charge via the Internet at http://pubs.acs.org.
This project was supported by research grant RO1 AI 43181 (NIAID) and shared equipment grant IS10RR016621 (NCRR) from the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. We also thank the Terry Chair Endowment, the Georgia Research Alliance, and the University of Georgia for additional research support.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Frankel A. D.; Young J. A. T. HIV-1: Fifteen Proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [DOI] [PubMed] [Google Scholar]
- Trono D.; Van Lint C.; Rouzioux C.; Verdin E.; Barre-Sinoussi F.; Chun T.-W.; Chomont N. HIV Persistence and the Prospect of Long-Term Drug-Free Remissions for HIV Infected Individuals. Science 2010, 329, 174–180. [DOI] [PubMed] [Google Scholar]
- Nair V.; Chi G. HIV Integrase Inhibitors as Therapeutic Agents in AIDS. Rev. Med. Virol. 2007, 17, 277–295. [DOI] [PubMed] [Google Scholar]
- Nair V.; Chi G.; Ptak R.; Neamati N. HIV Integrase Inhibitors with Nucleobase Scaffolds: Discovery of a Highly Potent Anti-HIV Agent. J. Med. Chem. 2006, 49, 445–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y.; Johnson A. A.; Marchand C. Integrase Inhibitors to Treat HIV/AIDS. Nat. Rev. Drug Discovery 2005, 4, 236–248. [DOI] [PubMed] [Google Scholar]
- Nair V. Novel Inhibitors of HIV Integrase: the Discovery of Potential Anti-HIV Therapeutic Agents. Frontiers Med. Chem. 2005, 2, 3–20. [DOI] [PubMed] [Google Scholar]
- Summa V.; Petrocchi A.; Bonelli F.; Crescenzi B.; Donghi M.; Ferrara M.; Fiore F.; Gardelli C.; Paz O. G.; Hazuda D. J.; Jones P.; Kinzel O.; Laufer R.; Monteagudo E.; Muraglia E.; Nizi E.; Orvieto F.; Pace P.; Pescatore G.; Scarpelli R.; Stillmock K.; Witmer M. V.; Rowley M. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855. [DOI] [PubMed] [Google Scholar]
- Laufer R.; Paz O. G.; DiMarco A.; Bonelli F.; Monteagudo E.; Summa V.; Rowley M. Quantitative Prediction of Human Clearance Guiding the Development of Raltegravir (MK-0518, Isentress) and Related HIV Integrase Inhibitors. Drug Metab. Dispos. 2009, 37, 873–883. [DOI] [PubMed] [Google Scholar]
- Garvey E. P.; Johns B. A.; Gartland M. J.; Foster S. A.; Miller W. H.; Ferris R. G.; Hazen R. J.; Underwood M. R.; Boros E. E.; Thompson J. B.; Weatherhead J. G.; Koble C. S.; Allen S. H.; Schaller L. T.; Sherrill R. G.; Yoshinaga T.; Kobayashi M.; Wakasa-Morimoto C.; Miki S.; Nakahara K.; Noshi T.; Sato A.; Fujiwara T. The Naphthyridinone GSK364735 is a Novel, Potent Human Immunodeficiency Virus Type I Integrase Inhibitor and Antiretroviral. Antimicrob. Agents Chemother. 2008, 52, 901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S.; Song I.; Borland J.; Chen S.; Lou Y.; Fujiwara T.; Piscitelli S. C. Pharmacokinetics and Safety of S/GSK1349572, a Next-Generation HIV Integrase Inhibitor, in Healthy Volunteers. Antimicrob. Agents Chemother. 2010, 54, 254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klibanov O. M. Elvitegravir, an Oral HIV Integrase Inhibitor, for the Potential Treatment of HIV Infection. Curr. Opin. Invest. Drugs 2009, 10, 190–200. [PubMed] [Google Scholar]
- Asante-Appiah E.; Skalka A. M. HIV-1 Integrase: Structural Organization, Conformational Changes, and Catalysis. Adv. Virus Res. 1999, 52, 351–369. [DOI] [PubMed] [Google Scholar]
- Esposito D.; Craigie R. HIV Integrase Structure and Function. Adv. Virus Res. 1999, 52, 319–333. [DOI] [PubMed] [Google Scholar]
- Liao C.; Marchand C.; Burke T. R.; Pommier Y.; Nicklaus M. C. Authentic HIV-1 Integrase Inhibitors. Future Med. Chem. 2010, 2, 1107–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S.; Gupta S. S.; Valkov E.; Engelman A.; Cherepanov P. Retroviral Intasome Assembly and Inhibition of DNA Strand Transfer. Nature 2010, 464, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox A. G.; Nair V. Novel HIV Integrase Inhibitors with Anti-HIV Activity: Insights into Integrase Inhibition from Docking Studies. Antiviral Chem. Chemother. 2006, 17, 343–353. [DOI] [PubMed] [Google Scholar]
- Urban J. J.; von Tersch R. L.; Famini G. R. Effect of Fluorine Substitution on Phenol Acidities in the Gas Phase and in Aqueous Solution. A Computational Study Using Continuum Solvation Models. J. Org. Chem. 1994, 59, 5239–5245. [Google Scholar]
- Resnati G. Synthesis of Chiral and Bioactive Fluoroorganic Compounds. Tetrahedron 1993, 49, 9385–9445. [Google Scholar]
- DiMagno S.; Sun H. The Strength of Weak Interactions: Aromatic Fluorine in Drug Design. Curr. Top. Med. Chem. 2006, 6, 1473–1482. [DOI] [PubMed] [Google Scholar]
- Boros E. E.; Burova S. A.; Erickson G. A.; Johns B. A.; Koble C. S.; Kurose N.; Sharp M. J.; Tabet E. A.; Thompson J. B.; Toczko M. A. A Scaleable Synthesis of Methyl 3-Amino-5-(4-fluorobenzyl)-2-pyridinecarboxylate. Org. Process Res. Dev. 2007, 11, 899–902. [Google Scholar]
- Singh B.; Bacon E. R.; Lesher G. Y.; Robinson S.; Pennock P. O.; Bode D. C.; Pagani E. D.; Bentley R. G.; Connell M. J.; Hamel L. T.; Silver P. J. Novel and Potent Adenosine 3′,5′-Cyclic Phosphate Phosphodiesterase-III Inhibitors: Thiazolo[4,5-b][1,6]naphthyridin-2-ones. J. Med. Chem. 1995, 38, 2546–2550. [DOI] [PubMed] [Google Scholar]
- Kowalski P. Reactions of 2-Aminopyridine with Benzyl-Chloride—Benzylation of Pyridine Ring. Pol. J. Chem. 1984, 58, 959–960. [Google Scholar]
- Nair V.; Turner G. A.; Chamberlain S. D. Novel Approaches to Functionalized Nucleosides via Palladium-Catalyzed Cross Coupling with Organostannanes. J. Am. Chem. Soc. 1987, 109, 7223–7224. [Google Scholar]
- Nair V.; Turner G. A.; Buenger G. S.; Chamberlain S. D. New Methodologies for the Synthesis of C-2 Functionalized Hypoxanthine Nucleosides. J. Org. Chem. 1988, 53, 3051–3057. [Google Scholar]
- Jiang X. H.; Song L. D.; Long Y. Q. Highly Efficient Preparation of Aryl Beta-Diketo Acids with Tert-butyl Methyl Oxalate. J. Org. Chem. 2003, 68, 7555–7558. [DOI] [PubMed] [Google Scholar]
- Uchil V.; Seo B.; Nair V. A Novel Strategy to Assemble the Beta-Diketo Acid Pharmacophore of HIV Integrase Inhibitors on Purine Nucleobase Scaffolds. J. Org. Chem. 2007, 72, 8577–8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassahun K.; McIntosh I.; Cui D.; Hreniuk D.; Merschman S.; Lasseter K.; Azrolan N.; Iwamoto M.; Wagner J. A.; Wenning L. A. Metabolism and Disposition in Humans of Raltegravir (MK-0518), an Anti-AIDS Drug Targeting the Human Immunodeficiency Virus 1 Integrase Enzyme. Drug Metab. Dispos. 2007, 35, 1657–1663. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R., Ed. Cytochrome P450: Structure, Mechanism, and Biochemistry, 3rd ed.; Kluwer Academic/Plenum: New York, 2005. [Google Scholar]
- Baranczewski P.; Stanczak A.; Sundberg K.; Svensson R.; Wallin A.; Jansson J.; Garberg P.; Postlind H. Introduction to In Vitro Estimation of Metabolic Stability and Drug Interactions of New Chemical Entities in Drug Discovery and Development. Pharmacol. Rep. 2006, 58, 453–472. [PubMed] [Google Scholar]
- Chauret N.; Gauthier A.; Martin J.; NicollGriffith D. A. Vitro Comparison of Cytochrome P450-Mediated Metabolic Activities in Human, Dog, Cat, and Horse. Drug Metab. Dispos. 1997, 25, 1130–1136. [PubMed] [Google Scholar]
- Williams J. A.; Hyland R.; Jones B. C.; Smith D. A.; Hurst S.; Goosen T. C.; Peterkin V.; Koup J. R.; Ball S. E. Drug-Drug Interactions for UDP-Glucuronosyltransferase Substrates: A Pharmacokinetics Explanation for Typically Observed Low Exposure (AUCi/AUC) Ratios. Drug Metab. Dispos. 2004, 32, 1201–1208. [DOI] [PubMed] [Google Scholar]
- Yoshinaga T.; Sato A.; Fujishita T.; Fujiwara T.. S-1360: In Vitro Activity of a New HIV-1 Integrase Inhibitor in Clinical Development. Presented at the 9th Conference on Retroviruses and Opportunistic Infections, Seattle, WA; February 24–28, 2002; Abstract 8, p 55.; Data given for compound S-1360 are those cited by Billich, A. S-1360 Shionogi-GlaxoSmithKline. Curr. Opin. Invest. Drugs 2003, 4, 206–209. [PubMed]
- Hazuda D. J.; Felock P.; Witmer M.; Wolfe A.; Stillmock K.; Grobler J. A.; Espeseth A.; Gabryelski L.; Schleif W.; Blau C.; Miller M. D. Inhibitors of Strand Transfer that Prevent Integration and Inhibit HIV-1 Replication in Cells. Science 2000, 287, 646–650. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.