

Abstract
Fluorescence resonance energy transfer (FRET) is an important source of long-range distance information in macromolecules. However, extracting maximum information requires knowledge of fluorophore, donor and acceptor, positions on the macromolecule. We previously determined the structure of the indocarbocyanine fluorophores Cy3 and Cy5 attached to DNA via three-carbon atom tethers, showing that they stacked onto the end of the helix in a manner similar to an additional basepair. Our recent FRET study has suggested that when they are attached via a longer 13-atom tether, these fluorophores are repositioned relative to the terminal basepair by a rotation of ∼30°, while remaining stacked. In this study, we have used NMR to extend our structural understanding to the commonly used fluorophore sulfoindocarbocyanine-3 (sCy3) attached to the 5′-terminus of the double-helical DNA via a 13-atom flexible tether (L13). We find that L13-sCy3 remains predominantly stacked onto the end of the duplex, but adopts a significantly different conformation, from that of either Cy3 or Cy5 attached by 3-atom tethers, with the long axes of the fluorophore and the terminal basepair approximately parallel. This result is in close agreement with our FRET data, supporting the contention that FRET data can be used to provide orientational information.
Introduction
Fluorescence can be used to provide distance information, on the molecular scale, by the measurement of fluorescence resonance energy transfer (FRET) between donor and acceptor fluorophores, providing important information on the structure and dynamics of macromolecules (1), particularly nucleic acids (2–5). The indocarbocyanine fluorophores are frequently used for this purpose, especially the cyanine 3 (donor) - cyanine 5 (acceptor) combination. In these experiments they will be tethered to specific points on the macromolecule. For nucleic acids this is frequently the 5′-terminus of a particular strand, and there are two main ways of achieving this objective. They may be coupled as phosphoramidites during synthesis, generally attaching the fluorophore via a three-carbon atom tether. Alternatively, they may be coupled postsynthetically via a 6-carbon-NHS linker, to a primary amine group that is itself covalently coupled to the 5′-terminus via a 6-carbon polymethylene chain, resulting in a much longer, potentially more flexible tether.
FRET is the transfer of excitation from the donor to the acceptor fluorophore due to dipolar coupling between the transition moment vectors of fluorophores. The efficiency of FRET (EFRET) as a function of fluorophore separation (R) is given by (6):
| (1) |
where R0 is the distance at which energy transfer is 50% efficient. R0 depends upon the spectroscopic properties of the fluorophores and the medium, given by
| (2) |
where the units of R0 and the wavelength λ are cm. ΦD is the quantum yield of the donor, N is the Avogadro number, n is the index of refraction of the medium, and J(λ) is the spectral overlap between donor emission and acceptor absorption. Although measurement of interfluorophore separation distance is the goal of most single-molecule FRET studies, EFRET is also a function of fluorophore orientation. κ2 in Eq. 2 describes the relative orientation of the fluorophores. It is given by
| (3) |
where the angles θT, θD, and θA describe the relative orientation of the donor and acceptor transition dipole moments. Ignorance of this term can potentially lead to incorrect estimations of distances (7–9). If the fluorophores undergo isotropic reorientation within the excited state lifetime of the donor then κ2 = 2/3 (8). In many studies this simplification is assumed to apply, but in some cases it may not be justifiable. For constrained fluorophores κ2 can take values between 0 and 4, or between 0 and 1 if the transition moments are coaxial and constrained to parallel planes.
When cyanine fluorophores are terminally attached to nucleic acids this becomes an important issue. FRET experiments had previously suggested that Cy3 attached to the 5′-terminus of double-stranded DNA via a three-carbon tether (termed L3-Cy3) was located close to the helical axis. Structure determination by NMR showed that L3-Cy3 was predominantly stacked onto the end of the helix in the manner similar to an additional basepair (10). We later used NMR to show that Cy5 equivalently tethered to DNA (termed L3-Cy5) was similarly stacked (11). These results strongly suggest that the donor-acceptor pair Cy3 and Cy5 tethered in this manner would exhibit a significant orientation dependence. This was demonstrated experimentally by measurement of FRET efficiency in a series of DNA and DNA-RNA duplexes of systematically varied length. EFRET was found to be modulated with length, with a periodicity that depended on the structure of the helix (11). The results could be fully explained on the basis of terminally stacked fluorophores, with a degree of lateral mobility about the mean positions determined by NMR.
This result indicates that the assumption that κ2 = 2/3 is not valid for the cyanine fluorophores tethered in this manner, and that any distances calculated on that basis will be suspect. However, it might have been thought that the observed orientation resulted from the short tether used, and that if a longer, more flexible, tether was used then perhaps orientation would no longer be significant. We therefore repeated the measurement of EFRET for a DNA duplex series with sulfoindocarbocyanine-3 and -5 fluorophores attached as N-hydroxysuccinimide esters to C6-linked amino groups generating a total tether length of 13 atoms (termed L13-sCy3 and L13-sCy5) (12). Surprisingly, we found that EFRET was strongly modulated as the length of the helix was varied, and the data could again be well explained by a model in which both fluorophores were terminally stacked, with a degree of lateral mobility. However, in contrast to the data for the L3-Cy3 - L3-Cy5 pair, those for the L13-sCy3 - L13-sCy5 pair exhibited a phase shift that required that the mean position for each fluorophore would be rotated by ∼30°, such that the long axes were now parallel to those of the terminal basepairs on which they were stacked.
In this work, we have set out to study the structure of a symmetrical DNA duplex with L13-sCy3 fluorophore attached at each end (Fig. 1). This would test the conclusion from the analysis of the FRET data for the duplex series, and also test our earlier claim that such FRET experiments could be used to provide information on orientation. Our results confirm that the L13-sCy3 is predominantly stacked onto the end of the DNA helix, and that its axis is indeed close to parallel with that of the terminal basepair, providing optimal stacking by positioning the cyanine methyl groups in the major groove.
Figure 1.
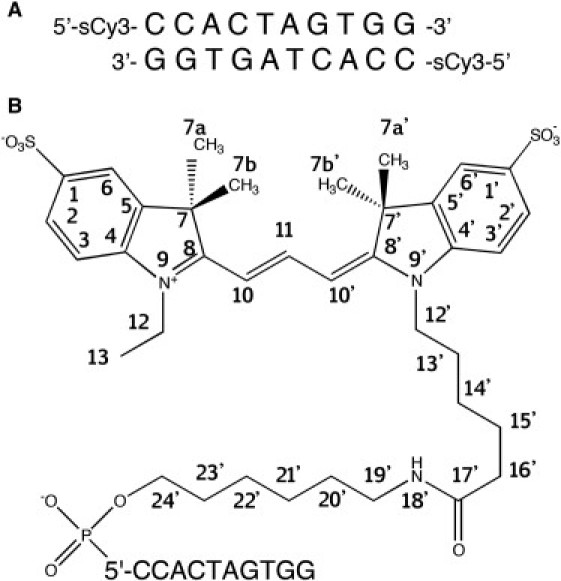
Chemical structure and attachment of L13-sCy3 used in this study. (A) The sequence and fluorophore position for the DNA used in this study. (B) Chemical structure and numbering scheme of sCy3 and tether.
Materials and Methods
Construction of DNA duplexes for NMR studies
DNA oligonucleotides were synthesized using cyanoethyl phosphoramidite chemistry (13) with a C6 amino modifier (Link Technologies, Bellshill, UK) coupled as a phosphoramidite in the final step of the synthesis with the 5′ monomethoxytrityl retained. The DNA was cleaved from the column and base deprotected using 35% ammonia in water. The partly deprotected product was then applied to a Glen-Pak cartridge (Glen Research, Sterling, VA) for partial purification and removal of monomethoxytrityl, eluted and then further purified by reversed-phase HPLC (Ace C18-300, Buffer A: 0.1 M TEAA pH 7.0, buffer B MeCN, 1 mL min−1, 0–12 min 0–20% B, 12–32 min 20–30% B, elution 15 min.) After ethanol precipitation the product was resuspended in 0.1 M sodium tetraborate pH 8.5 to give a stock solution of 1.37 mM and added to a 21 mM dimethyl sulfoxide solution of Cy3 N-hydroxysuccinimide ester (GE Healthcare, Amersham, UK) in a 1:3 molar ratio (DNA/Cy3-NHS ester) and allowed to react overnight. The reaction mixture was precipitated and then purified using reversed-phase HPLC (elution time 17 min) giving estimated labeling efficiency of 95%.
NMR spectroscopy and structure determination
Sample preparation
Labeled DNA was desalted using Nap-25 (Sepadex G-25 DNA grade from GE Healthcare) and ion exchanged into sodium ion form. Samples for NMR spectroscopy were dissolved in 0.6 mL of 0.1 M NaCl, 10 mM phosphate buffer, and 1 mM EDTA, in either 99.96% D2O or 90% H2O/10% D2O (v/v) at pH 7.0. Samples were either 46 OD A260 in D2O or 27 OD A260 units in H2O.
NMR experiments
All NMR data were acquired using a Bruker Avance 800 NMR (Bruker, Coventry, UK) spectrometer equipped with a cryoprobe. For the D2O sample, a series of NOESY spectra were acquired at 14°C, with mixing times of 50, 75, 100, 150, and 250 ms. The sweep width used was 10.00 ppm with o1 at 4.762 ppm. A relaxation delay of 1.4 s was used with 24 scans per experiment. Residual water suppression was achieved using presaturation. Time domain F2 was 4096 and F1 768. In addition, phase-sensitive COSY data were acquired. For the sample in H2O a NOESY experiment was performed at 2°C, with a mixing time of 250 ms.
Data processing and analysis
NMR data were processed using TopSpin (Bruker), and the spectra were analyzed and assigned using CCPN (14). NOE crosspeaks were quantified using Felix (Felix NMR, San Diego, CA) at 5 mixing times on the data acquired from the sample in D2O. The COSY experiments were used only for assignment purposes. Spectra taken in H2O buffer were assigned and analyzed qualitatively.
Structure determination
Structures were calculated using restrained molecular dynamics (MD) within the program X-PLOR-NIH version 2.28 (15). B-form DNA was constructed using Web 3DNA (16). L13-sCy3 coordinate, parameter and topology files were extrapolated from those used for L3-Cy3 (10). Initially, 50 starting coordinate files for double-stranded DNA with L13-sCy3 on both ends were generated within PyMol (17) by placing the dye in random orientations ∼12 Å away from the terminal basepair of the DNA. Starting structures were subjected to a short period of MD and energy minimization in the absence of experimental restraints, but with oligonucleotide conformation fixed, to generate a family of 50 starting structures. Structural refinement was carried out in several phases using direct application of full relaxation matrix refinement (18), utilizing the volume-integral measurements from all resolvable NOESY crosspeaks obtained at all mixing times. In the first stage, the L13-sCy3 was refined in the absence of the L13 tether, with the DNA restrained by a harmonic function to prevent significant fluctuation from standard B-form DNA conformation. The harmonic restraints were relaxed on the two terminal basepairs and removed from 5′-terminal sugar. Because of symmetry, at the end of this stage 100 unique structures were produced for the L13-sCy3-DNA interface, of which a family of 43 unique structures were selected based on the total and relaxation energy values (only energies arising from the Cy3 and the terminal basepairs were considered). These 43 structures were used to refine the full fluorophore structure, including the tether. Tether atoms were added using unrestrained MD, from random starting coordinates and full relaxation matrix refinement was repeated on these coordinates using harmonic restraint to hold the structures of the DNA and the refined portions of the fluorophore. An average structure was calculated and minimized, again using full relaxation restraints.
Simulation of FRET efficiency from NMR structure
The experimental dependence of FRET efficiency as a function of duplex length was simulated on the basis of the NMR-derived structure of L13-sCy3 attached to DNA. In the absence of structural data for L13-sCy5 we assume that this fluorophore will be structurally equivalent to L13-sCy3, and therefore use it for both ends of the modeled duplexes. We constructed computer models of a series of DNA duplexes in w3DNA (16), with the termini based on the NMR-derived fluorophore structure. The transition dipole moment vectors were approximated by the vector linking the indole N atoms, based on our earlier quantum mechanical calculations (11). Using a program written in MATLAB (The MathWorks, Natick, MA), we took the coordinates of the four N atoms (i.e., two in each of the two fluorophores) as input to calculate the value of EFRET, allowing the fluorophores to undergo lateral motion as previously deduced (12). From our fluorescence lifetime measurement of Cy3 attached to dsDNA by the 13-atom tether we know that a fraction of the fluorophores are unstacked, such that half the molecules should have one or both fluorophores unstacked at a given time (12). This fraction of the molecules was assigned a value of κ2 = 2/3. For the remaining fraction both fluorophores were placed according to the new NMR-derived structure, with lateral flexibility about the mean relative angle measured from the NMR-based model. For each duplex length κ2, R0, and thus EFRET were calculated for a distribution of angles either side of the mean. These values were then weighted, by a Gaussian distribution of given halfwidth. The weighted values of EFRET were then averaged to give the mean value for that duplex length. This was repeated for each length (between 10 and 24 basepairs), plotted as a function of duplex length, and compared to the experimental data. The only variable parameters in this analysis were the fraction of unstacked cyanine fluorophore, and the halfwidth for the lateral mobility.
Results
L13-sCy3-Conjugated DNA used for NMR studies
The self-complementary oligonucleotide 5′-CCACTAGTGG-3′ with sulfoindocarbocyanine-3 attached via a 13-atom tether at the 5′-terminus was chemically synthesized. The sequence was chosen to be directly comparable with that used in the previous determination of the structure of L3-Cy3 and L3-Cy5 on DNA (10,11). The numbering system for the DNA nucleotides and L13-sCy3 atoms is shown in Fig. 1.
Assignment of DNA proton resonances
The nonexchangeable protons in the L13-sCy3-conjugated DNA were assigned primarily by examination of the base to H1′ crosspeaks in the 250 ms NOESY spectra (Fig. 2 B). It was possible to assign the DNA proton resonances of the L13-sCy3-conjugated DNA, without assuming the identity of any peaks derived from the L13-sCy3 adduct. The spectra showed a typical base to H1′ NOE pattern, expected for a B-form double helix. Exchangeable protons were assigned from a NOESY spectrum acquired in H2O at 2°C. Assignments were made by comparison with the nonexchangeable resonances and by imino proton NOE connectivity.
Figure 2.
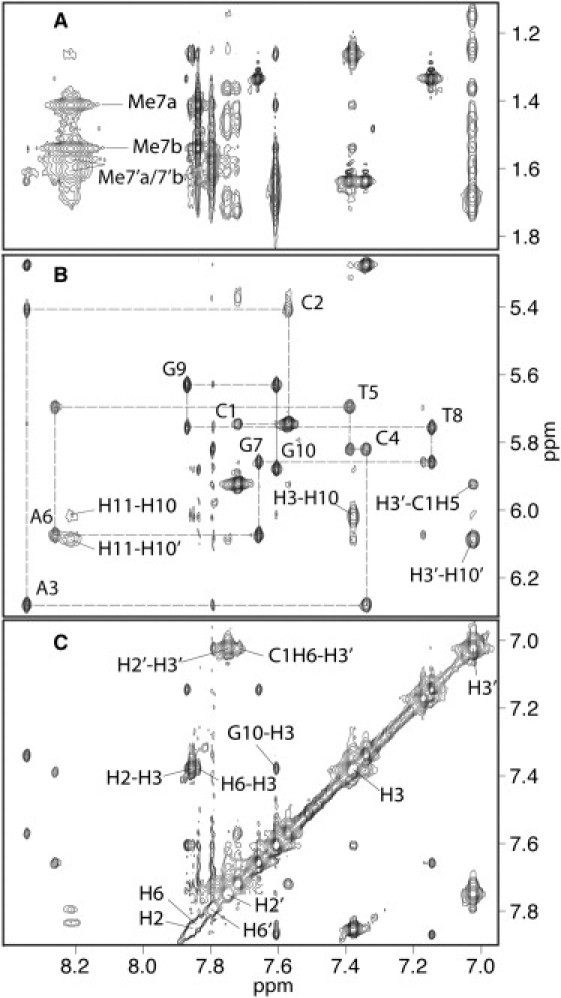
NMR NOESY spectrum of the L13-sCy3-labeled DNA duplex. Selected regions, of a 250 ms NOESY spectrum, recorded in D2O at 14°C. (A) The DNA base to methyl region. (B) The DNA base to H1′ region. The base to H1′ connectivities are represented by broken lines and assignments indicated on the intraresidue NOE crosspeaks. (C) The aromatic region of the two-dimensional NOESY spectrum, showing resonance positions of the two sCy3 indole ring systems and assignments on selected crosspeaks.
A comparison of base proton chemical shifts, between the unconjugated oligonucleotide (10) (11) and oligonucleotide, derivatized with either L3-Cy3 or L13-sCy3 showed a significant difference in the terminal basepair (most prominent in G10) (Fig. 3) indicative of a change in the stacking of the fluorophore compared to that observed with L3-Cy3. The chemical shifts of assigned protons from L13-sCy3-conjugated oligonucleotide are presented in Table 1.
Figure 3.

Changes in chemical shift between base proton resonances (H8 or H6) in unconjugated and fluorophore-conjugated DNA. Solid line: L3-Cy3-DNA. Broken line: L13-sCy3-DNA.
Table 1.
Assignments and chemical shifts (ppm) of the protons of the DNA and attached L13-sCy3
| L13-sCy3-conjugated DNA (5′-L13-sCy3-CCACTAGTGG-3′) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | 7.86 | H2′ | 7.75 | H14′ | 1.15/1.25 | H24′ | 3.96 | |||||
| H3 | 7.38 | H3′ | 7.02 | H15′ | 1.54/1.60 | H14′ | 1.15/1.25 | |||||
| H6 | 7.84 | H6′ | 7.80 | H16′ | 2.01/2.16 | H15′ | 1.54/1.60 | |||||
| Me7a | 1.41 | ∗Me7a′/Me7b′ | ∗1.57/1.61 | H18′ | 8.14 | H24′ | 3.96 | |||||
| Me7b | 1.54 | H19′ | 3.09/3.18 | |||||||||
| H10 | 6.02 | H10′ | 6.09 | H20′ | 1.48 | |||||||
| H11 | 8.21 | H21′ | 1.36 | |||||||||
| H12 | 3.99 | H12′ | 3.79/3.99 | H22′ | 1.45 | |||||||
| H13 | 1.26 | H13′ | 1.68 | H23′ | 1.72 | |||||||
| DNA | H8/H6 | H2/H5/Me | H1′ | H2′ | H2″ | H3′ | H4′ | H5′ | H5″ | H1/H3 | H4a | H4b |
| C1 | 7.72 | 5.92 | 5.75 | 2.00 | 2.37 | 4.75 | 4.07 | 4.05 | – | – | 8.23 | 7.19 |
| C2 | 7.57 | 5.75 | 5.41 | 2.17 | 2.42 | 4.88 | 4.13 | 4.06 | 3.88 | – | 8.61 | 7.09 |
| A3 | 8.35 | 7.79 | 6.28 | 2.83 | 2.95 | 5.09 | 4.48 | 4.08 | 4.20 | – | – | – |
| C4 | 7.34 | 5.28 | 5.82 | 1.98 | 2.49 | 4.71 | 4.24 | 4.21 | 4.35 | – | 8.11 | 6.85 |
| T5 | 7.39 | 1.64 | 5.69 | 2.15 | 2.52 | 4.91 | 4.16 | 4.11 | 4.18 | 13.75 | – | – |
| A6 | 8.26 | 7.17 | 6.07 | 2.78 | 2.92 | 5.08 | 4.46 | 4.11 | 4.19 | – | – | – |
| G7 | 7.66 | – | 5.86 | 2.48 | 2.71 | 4.94 | 4.43 | 4.24 | 4.29 | 12.72 | – | – |
| T8 | 7.15 | 1.33 | 5.76 | 1.93 | 2.34 | 4.87 | 4.19 | 4.13 | 4.22 | 13.84 | – | – |
| G9 | 7.87 | – | 5.63 | 2.67 | 2.72 | 5.00 | 4.35 | 4.04 | 4.12 | 13.01 | – | – |
| G10 | 7.61 | – | 5.87 | 1.64 | 1.68 | 4.42 | 4.18 | 4.01 | 4.16 | 13.20 | – | – |
No unique assignment.
Assignment of L13-sCy3 proton resonances
The Cy3 molecule comprises two indole ring systems connected by a planar trimethyne linker. The L13-sCy3 is covalently connected to the terminal 5′-phosphate of the DNA via a 13-atom tether joined at position N9′, containing an amide bond at its center. The pseudosymmetrical position, N9 of the distal indole ring, has a 2-carbon substituent attached, that we term the pseudotether. Unlike L3-Cy3, L13-sCy3 has sulfonate groups in the 1 and 1′, ring positions. Therefore, only two intense crosspeaks were observed in COSY and NOESY spectra in the region where indole proton resonances were expected, from reference to previous studies (10). Having tentatively assigned four resonance peaks to the indole ring protons (2, 3, 2′, 3′), based on COSY spectra, the remaining proton crosspeaks of the indole rings (6 and 6′) were identified based on NOE interactions with neighboring methyl groups, at ∼1.5 ppm, and the other indole protons. The assignment of the spin systems between the indole rings, the tether, and pseudotether was achieved by a combination of NOESY and COSY observations. The tether protons being largely in the form of CH2 were not stereospecifically assigned, although we accounted for all of them. Some of the tether resonances were split indicating the presence of alternative but relatively stable conformations on the NMR timescale.
Structure determination and analysis
Previous studies of FRET efficiencies between L13-sCy3 and L13-sCy5 in a series of DNA duplexes between 10 and 24 bp in length revealed a modulation of FRET efficiency with length consistent with an orientational dependence of EFRET. Modeling of these data using a model equivalent to that used in previous studies with L3-Cy3 and L3-Cy5 labeled systems (19) was indicative of a similar stacking of the fluorophores on the terminal basepairs, but to fit the observed phase of the modulation it was necessary to reorient the fluorophores by 30° so that the long axis of the fluorophores was now approximately parallel to that of the terminal basepairs on which they were stacked.
An altered stacked position of L13-sCy3 was also suggested by changes in the chemical shifts of the distal and proximal methyl groups (7 and 7′), in addition to those of the DNA protons. It was also noted from the NOESY spectrum acquired in H2O that the terminal imino proton, although broader than internal imino protons, exhibited a significant NOE to neighboring imino protons, implying considerably greater protection from exchange with solvent water compared to that observed in previous studies on the L3-Cy3-DNA species (Fig. 4).
Figure 4.

NOESY spectrum acquired in H2O at 2°C. The position of the diagonal imino proton peaks and the off-diagonal sequential imino-imino crosspeaks are indicated.
The structure of the L13-sCy3-DNA adduct was determined using MD, restrained against a full relaxation matrix populated in part by NOE buildup measurements derived from multiple mixing times. As in previous studies, the DNA was assumed to adopt a largely standard B-form conformation and only the terminal basepair and the cyanine dye components were freely refined. Refinement strategies were designed to determine the conformation of the L13-sCy3 relative to the B-DNA, only allowing for limited structural change in the terminal basepair.
DNA, fluorophore, and tether protons were almost completely assigned. A total of 331 NOE buildups were measured by volume integration of crosspeaks from spectra at five different mixing times. Because the aim of this work was to determine the structure of the fluorophore in relation to the terminal basepair of the DNA, the number of NOE buildups that could be reliably measured between the DNA and the fluorophore were critical. Although almost complete assignment of the spectra was possible, overlap of crosspeaks prevented many NOE buildups from being incorporated into the refinement. 12 NOE buildups between the fluorophore and the DNA could be accurately measured (Fig. 5 and Table 2). The long tether present in L13-sCy3 presented particular problems in the refinement. The nonstereospecific assignment of most of the tether protons limited the value of such restraints. After initial trials it was decided to refine the position of the fluorophore on the DNA in the absence of linker initially, and then to add in the tether under restraint before finally performing restrained minimization on the complete assembly. This approach facilitated the efficient discovery of a low energy ensemble of final structures. It should be noted that the positions of the fluorophores determined by NMR will represent only a single conformation of the stacked form of the fluorophore, while acknowledging that a number of studies have shown that cyanine fluorophores that are terminally attached to double-stranded nucleic acids can undergo both unstacking and lateral motions, and that these dynamic properties can be influenced by the nature of the terminal basepair (20–22). Structures refined from NMR data usually overestimate more compact forms and underestimate more dynamic, less compact forms. The structure studied here undoubtedly exhibits the same bias. Unstacked forms of the fluorophore will not contribute NOE interactions between itself and the underlying DNA. Lateral motion will, most likely, result in NOE interactions that cannot be wholly accounted for by a single tightly defined ensemble of structures. Despite this, the structure determined here makes stereochemical sense and also allows the successful back calculation of the orientationally sensitive FRET that incorporate estimates of unstacking and lateral motion (see below).
Figure 5.

Final minimized-average structure of L13-sCy3-DNA, with the tether not shown. Only the terminal basepair of the DNA is shown with lines to indicate the NOE interactions used in the refinement.
Table 2.
NOE buildup assignments used in the refinement of L13-sCy3-DNA
| DNA | L13-sCy3 |
|---|---|
| G10 H1′ | H13 |
| C1 H4′ | H3′ |
| G10 H1′ | H3 |
| C1 H4′ | H2′ |
| C1 H5′ | H2′ |
| G10 H8 | H2 |
| C1 H6 | H3′ |
| G10 H8 | H12 |
| C1 H5 | H3′ |
| G10 H8 | H7a |
| G10 H8 | H13 |
| G10 H8 | H3 |
Data illustrated in Fig. 5.
Refined structure of L13-sCy3
After refinement of the fluorophore position, followed by addition of the linker and further refinement against all NOE integrals, the final minimized average structure showed the fluorophore to be stacked on the terminal basepair with minimal rotation relative to the terminal basepair (analogous to the twist angle between successive basepairs) of 3° measured between G10N9- C1N1-N9′- N9). By comparison, the rotations previously estimated from NMR studies of L3-Cy3 and L3-Cy5 attached to DNA (10,11) are 27° and 34° respectively, values much closer to the standard 34° twist of DNA. The fluorophore is translated to the major groove side of the terminal basepair, allowing the methyl groups at positions 7a and 7b′ to fit at the side of the basepair thereby allowing the planar body of the fluorophore to make close contact to the terminal basepair. In this refined structure the planar body of the fluorophore is only 3.6 Å above the plane of the terminal basepair, a distance that is very close to the basepair rise observed in B-form DNA, and contrasts with the 5 Å rise found in the structures of L3-Cy3 and L3-Cy5 attached to DNA. The greater freedom, provided by the longer tether, has apparently allowed the fluorophore to adopt a stacked position that appears more stereochemically optimal, with the downward facing methyl groups accommodated in the major groove.
From the family of final structures (Fig. S1, a–e, in the Supporting Material), the coordinate root mean-square deviation for the sCy3 heavy atoms (not including the rotatable oxygen atoms of the SO3− groups) was 0.335 Å and the coordinate root mean-square deviation for the heavy atoms of the tether was 1.286 Å. Superimposition was made onto the average structure using only the central DNA basepairs. The coordinate deviation data indicate a system in which sCy3 heterocycle is well-defined relative to the DNA, but with the tether exhibiting much greater variability. How much of the greater variability of the tether is due to the lack of stereo-specific restraints, and how much reflects a real molecular flexibility is uncertain. However, the relatively broad crosspeaks observed, indicate that the tether is probably less well structured than the sCy3 heterocyclic region or the underlying DNA.
Comparison between short and long linker Cy3
The separation between the planes of L13-sCy3 and the terminal basepair of the DNA is ∼3.6 Å, which is almost the same as the basepair rise observed in B-form DNA. Both indole rings are partially stacked on the major groove edge of the terminal basepair. The two pendant methyl groups of the fluorophore are accommodated in the major groove, thereby improving van der Waals interaction between the DNA-proximal face of the fluorophore and the terminal basepair. NMR analysis of the structure of L3-Cy3 attached to DNA had previously revealed a significantly different stacking interaction (10), much more like an extra basepair, with a ∼30° angle between the long axes of the fluorophore and the terminal basepair making much less favorable stacking interactions due to the steric interference of the pendant methyl groups. The resulting difference between these two forms of Cy3 is a change in orientation of ∼30° with respect to the terminal basepair. The tethers in these two forms are very different in length and therefore must adopt completely different paths between the terminal phosphodiester and the attachment point on the indole ring. The L3 tether adopts a conformation very close to that of a ribose ring as it links the fluorophore to the DNA; the same number of carbon atoms being present in each case. The L13 tether might be expected to provide greater flexibility than the L3 tether. However, it appears to be more constrained in that it has to make two very tight turns to link to the indole ring while allowing the planar fluorophore to occupy a suitably low-energy stacked structure on the terminal basepair. The resultant L13 linker conformation is an elegantly close-packed structure.
The new structure is consistent with the previously acquired FRET data
With the refined structure of L13-sCy3 in hand, we can now model the structures of the 10–24 bp duplexes labeled with L13-sCy3 and L13-sCy5 and calculate the expected FRET efficiencies to compare with our earlier experimental data. For this purpose we make the assumption that L13-sCy5 will adopt the same position as L13-sCy3, and we calculate the mean angle subtended between the two transition dipole vectors for each member of the series, each approximated by the vector linking the indole N atoms. On the basis of our fluorescence lifetime measurements (12), we assume that half the molecules will have one or both fluorophores unstacked, and will have an orientation parameter κ2 = 2/3. For the remaining fraction where both fluorophores are stacked, both L13-sCy3 and L13-sCy5 were allowed lateral flexibility and κ2 was calculated for each point. An average for EFRET was then calculated that was weighted by a Gaussian distribution, corresponding to a Boltzmann population of angles where the angular displacement from the mean value obeys Hooke's law. The resulting values of EFRET as a function of duplex length were compared to the experimental data, shown in Fig. 6. The values calculated on the basis of the NMR structure fit well with those measured in the FRET experiments. In particular, the phases of the FRET modulation are in good agreement, and significantly different from that of the data obtained using L3-Cy3 and L3-Cy5, attached by the shorter tethers (the best simulation for the short-tether data is shown by the broken line in Fig. 6).
Figure 6.

Back calculation of FRET efficiencies using the NMR-derived structure of the L13-sCy3 fluorophore. Experimentally, EFRET was measured for a series of DNA duplex species of lengths between 10 and 24 bp, each labeled with L13-sCy3 and L13-sCy5. These data are plotted as a function of helix length (open circles) along with a simulation (solid line) based on B-form helical geometry, as described previously (12). The corresponding simulation for FRET data obtained with L3-Cy3 and L3-Cy5 is plotted (broken line) (19). The marked phase shift results from the angular reorientation of the fluorophores with the different length tethers. Values of EFRET were calculated from molecular models constructed on the basis of the NMR structure of the L13-sCy3 DNA with the inclusion of lateral mobility, as explained in the text and plotted (black circles).
Discussion
In this study, we have used NMR to determine the position of sulfoindocarbocyanine-3 attached via a potentially flexible 13-atom tether to the 5′-terminus of double stranded DNA. This structural analysis shows that the fluorophore is predominantly stacked onto the terminal basepair at the end of the helix, but that it is rotated by ∼30° compared to indocarbocyanine-3 and -5 attached via shorter, 3-carbon atom tethers (Fig. 7). The significance of this work lies entirely in its relevance to FRET studies of nucleic acids, where the cyanine fluorophores are extensively used as donor-acceptor pairs, especially in single-molecule studies.
Figure 7.

Comparison of the positions of L13-sCy3 (A and C) and L3-Cy3 (B and D) stacked onto the terminal basepair of DNA. The basepair shown using space-filling spheres, whereas the fluorophores and their tethers are depicted in stick representation. A and B are viewed down the DNA axis, showing the stacking arrangement and the angular relationships between the fluorophores and the DNA bases. C and D show a side view illustrating the closer stacking arrangement of L13-sCy3 compared to that of L3-Cy3.
Broadly speaking, there are two potential alternative strategies to the extraction of distance information in macromolecules using FRET, free of uncertainty in the orientation parameter κ2. The first is to attach both fluorophores flexibly and then assume that κ2 = 2/3. Using fluorescein, for example, is probably a good assumption. However, the very flexibility of the fluorophore means that the point of reference is not known precisely and must be estimated using a computational approach (23). The second approach is to use fluorophores that have fixed positions on the macromolecule. In this case the point of reference is now known, but any assumption of κ2 = 2/3 is unjustified. Progress with this approach requires a knowledge of the fluorophore position and orientation, and hence computation of κ2. The NMR study of L13-sCy3 now allows us to do just that.
In our initial studies, we chemically coupled the cyanine fluorophores as phosphoramidites, generating three-carbon atom tethers. NMR studies showed both Cy3 (10) and Cy5 (11) tethered in this manner to be terminally stacked, consistent with FRET studies in DNA and RNA duplex series (19). This might have been a result of a constraint imposed by the short tether, and we felt it possible that use of a longer tether would result in flexible attachment. However, FRET analysis of a DNA duplex series with L13-sCy3 and L13-sCy5 revealed a modulation of EFRET with distance indicative of terminally stacked fluorophores (12), and we have now confirmed this by direct structural study of L13-sCy3 attached to double-stranded DNA. It is clear that there is an intrinsic propensity of the indocarbocyanine fluorophores to stack onto the end of the helix, which is not a result of the manner of tethering. Thus, κ2 = 2/3 is not a safe assumption using these fluorophores. However, if the positions and dynamics of the fluorophores are known and the average orientation of the transition dipoles can be computed, the second approach to the analysis of FRET is possible. In light of this NMR study coupled with our FRET studies of DNA duplexes, this is now the case.
Given a good understanding of fluorophore position and dynamics, we have proposed that it is now possible to obtain angular information in FRET studies of nucleic acids. Analysis of the DNA duplex series with the L13-sCy fluorophores before the NMR study was initiated clearly showed a reorientation on the end of the helix so that the long axes of the fluorophores and the terminal basepairs were close to parallel. This has now been directly confirmed by the structural analysis using NMR. This is proof of principle that we can successfully extract information on orientation by FRET analysis. Moreover, if we can confidently analyze fluorophore orientation, this in turn provides great accuracy and confidence in distances calculated on the basis of FRET measurements. We are now extending this approach to the analysis of bent duplex species and helical junctions in DNA and RNA, where orientational information is essential for a full structural understanding.
Acknowledgments
The authors thank Juraj Bella and Lorna Murray of Edinburgh University, Chemistry Department, for gathering NMR spectra.
This work was financially supported by Cancer Research UK.
Supporting Material
References
- 1.Stryer L., Haugland R.P. Energy transfer: a spectroscopic ruler. Proc. Natl. Acad. Sci. USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clegg R.M. Fluorescence resonance energy transfer. In: Wang X.F., Herman B., editors. Fluorescence Imaging Spectroscopy and Microscopy. John Wiley & Sons; New York: 1996. pp. 179–252. [Google Scholar]
- 3.Lilley D.M., Wilson T.J. Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr. Opin. Chem. Biol. 2000;4:507–517. doi: 10.1016/s1367-5931(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 4.Clegg R.M. FRET tells us about proximities, distances, orientations and dynamic properties. J. Biotechnol. 2002;82:177–179. [PubMed] [Google Scholar]
- 5.Lilley D.M. The structure and folding of branched RNA analyzed by fluorescence resonance energy transfer. Methods Enzymol. 2009;469:159–187. doi: 10.1016/S0076-6879(09)69008-X. [DOI] [PubMed] [Google Scholar]
- 6.Förster T. Intermolecular energy transfer and fluorescence. Ann. Phys. 1948;2:55–75. [Google Scholar]
- 7.Haas E., Katchalski-Katzir E., Steinberg I.Z. Effect of the orientation of donor and acceptor on the probability of energy transfer involving electronic transitions of mixed polarization. Biochemistry. 1978;17:5064–5070. doi: 10.1021/bi00616a032. [DOI] [PubMed] [Google Scholar]
- 8.Dale R.E., Eisinger J., Blumberg W.E. The orientational freedom of molecular probes. The orientation factor in intramolecular energy transfer. Biophys. J. 1979;26:161–193. doi: 10.1016/S0006-3495(79)85243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu P., Brand L. Orientation factor in steady-state and time-resolved resonance energy transfer measurements. Biochemistry. 1992;31:7939–7947. doi: 10.1021/bi00149a027. [DOI] [PubMed] [Google Scholar]
- 10.Norman D.G., Grainger R.J., Lilley D.M. Location of cyanine-3 on double-stranded DNA: importance for fluorescence resonance energy transfer studies. Biochemistry. 2000;39:6317–6324. doi: 10.1021/bi992944a. [DOI] [PubMed] [Google Scholar]
- 11.Iqbal A., Wang L., Norman D.G. The structure of cyanine 5 terminally attached to double-stranded DNA: implications for FRET studies. Biochemistry. 2008;47:7857–7862. doi: 10.1021/bi800773f. [DOI] [PubMed] [Google Scholar]
- 12.Ouellet J., Schorr S., Lilley D.M. Orientation of cyanine fluorophores terminally attached to DNA via long, flexible tethers. Biophys. J. 2011;101:1148–1154. doi: 10.1016/j.bpj.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beaucage S.L., Caruthers M.H. Deoxynucleoside phosphoramidites—a new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett. 1981;22:1859–1862. [Google Scholar]
- 14.Vranken W.F., Boucher W., Laue E.D. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 15.Schwieters C.D., Kuszewski J.J., Clore G.M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 16.Zheng G.H., Lu X.J., Olson W.K. Web 3DNA—a web server for the analysis, reconstruction, and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res. 2009;37(Web Server issue):W240–W246. doi: 10.1093/nar/gkp358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeLano, W.L. The PyMOL Molecular Graphics System. Schrödinger, LLC, New York.
- 18.Yip P., Case D.A. A new method for refinement of macromolecular structures based on nuclear Overhauser effect spectra. J. Magn. Reson. 1989;83:643–648. [Google Scholar]
- 19.Iqbal A., Arslan S., Lilley D.M. Orientation dependence in fluorescent energy transfer between Cy3 and Cy5 terminally attached to double-stranded nucleic acids. Proc. Natl. Acad. Sci. USA. 2008;105:11176–11181. doi: 10.1073/pnas.0801707105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanborn M.E., Connolly B.K., Levitus M. Fluorescence properties and photophysics of the sulfoindocyanine Cy3 linked covalently to DNA. J. Phys. Chem. B. 2007;111:11064–11074. doi: 10.1021/jp072912u. [DOI] [PubMed] [Google Scholar]
- 21.Spiriti J., Binder J.K., van der Vaart A. Cy3-DNA stacking interactions strongly depend on the identity of the terminal basepair. Biophys. J. 2011;100:1049–1057. doi: 10.1016/j.bpj.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X., Yin Y., Xinxing Z., Zhi Z., Xin S. Temperature dependence of interaction between double stranded DNA and Cy3 or Cy5. Chem. Phys. Lett. 2011;513:271–275. [Google Scholar]
- 23.Sisamakis E., Valeri A., Seidel C.A. Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 2010;475:455–514. doi: 10.1016/S0076-6879(10)75018-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.