

Abstract
Nonantibody scaffolds such as designed ankyrin repeat proteins (DARPins) can be rapidly engineered to detect diverse target proteins with high specificity and offer an attractive alternative to antibodies. Using molecular simulations, we predicted that the binding interface between DARPin off7 and its ligand (maltose binding protein; MBP) is characterized by a hot-spot motif in which binding energy is largely concentrated on a few amino acids. To experimentally test this prediction, we fused MBP to a transmembrane domain to properly orient the protein into a polymer-cushioned lipid bilayer, and characterized its interaction with off7 using force spectroscopy. Using this, to our knowledge, novel technique along with surface plasmon resonance, we validated the simulation predictions and characterized the effects of select mutations on the kinetics of the off7-MBP interaction. Our integrated approach offers scientific insights on how the engineered protein interacts with the target molecule.
Introduction
Protein-based biosensors that detect molecules of biological interest hold promise for basic science and a variety of applications ranging from diagnostics to chem-bio threat detection. We have previously developed allosteric enzyme protein switches that utilize the interactions between a protein and its corresponding ligand to generate a detectable change in enzyme activity and thus function as protein-based molecular sensors (1–3). An understanding of such protein-ligand interactions and identification of protein domains that facilitate protein switch construction would guide the development of these protein-based biosensors. Allosteric protein switches developed to date have utilized natural protein domains. Synthetic antibody mimetic proteins have been evolved to bind a variety of protein targets (4,5) and have also been engineered to create fluorescent biosensors (6). Thus, antibody mimetics offer an excellent platform for developing biorecognition elements and modular biosensors.
Antibodies are the most widely used binding molecules in biomedical research with high specificity and affinity to the targets of interest; however, they are generally expensive to develop and produce, and have limited thermodynamic stability. To address these and other limitations, nonantibody scaffolds, including designed ankyrin repeat proteins (DARPins) (7,8), have been developed as binding proteins. The first example of an engineered DARPin was off7, a DARPin selected to bind maltose binding protein (MBP) with high affinity and specificity (5). The crystal structure of the off7-MBP complex has been solved (5). Since then, DARPins have been engineered to bind a diverse set of proteins including Her2 (9), aminoglycoside phosphotransferase (10), neurotensin receptor-1 (11), MAP kinase (12), and caspase-2 (13), and thus offer an attractive alternative to antibodies. DARPins have also been used to assist crystallographic studies (14).
Detailed analysis on the interaction between DARPins and their ligands would inform the design of protein switches for applications as biosensors. Here, we have studied the off7-MBP interface using simulations and biophysical tools. Protein-protein interactions are mediated by either a localized patch where the binding residues are clustered together, or by a large interaction zone where the binding residues are spread over the protein surface (15,16). For a localized patch, only a small subset of interfacial residues is believed to be critical for binding interactions. Referred to as “hot-spots”, modifying these crucial residues can disrupt protein-protein recognition (17,18). Hot-spots have been defined as sites at which an alanine mutation causes at least a 2.0 kcal/mol increase in the binding free energy (equivalent to a 30-fold increase in Kd) (19). Although the importance of hot-spots for natural protein interactions is well established, the occurrence and importance of hot-spots in artificial, engineered protein-protein interactions is less defined.
Membrane protein-ligand recognition is a dynamic process in which the association and dissociation kinetics are regulated by external forces (20,21). Single-molecule force spectroscopy has been applied to study receptor-ligand interactions between apposing live-cells as well as purified proteins at the single molecule level (22–24). The sensitivity of force spectroscopy enables us to study the tensile strength of receptor-ligand bonds with picoNewton resolution over a wide range of loading rates (25). To accurately characterize binding interactions, we need to ensure that the proper orientation of the testing molecules is preserved. However, current techniques to study nonmembrane, soluble proteins by force microscopy immobilize the protein in a random orientation via chemical attachment to a surface. Here, we addressed this limitation by orienting the MBP in a lipid bilayer using a trans-membrane domain (TMD) fused to the N-terminus of MBP at a location opposite from the off7-MBP binding interface. The hydrophobic interactions between TMD and lipid bilayer control the proper MBP orientation for experiments.
Combining molecular simulations, protein engineering, and biophysical tools, we characterized the affinity and hot-spot motif of the off7-MBP interaction. This integrated approach enables us to directly study how point mutation at the binding interface modulates the kinetics and micromechanical properties of the bonds. Detailed knowledge of DARPin-target interactions, including location of hot-spots, will facilitate the development of protein switch biosensors using DARPins as the recognition domain, because hot-spots for one ligand may be key sites for establishing interactions with other binding partners.
Methods
Construction, expression, and purification of TMD-MBP
The DNA segment encoding the CD44 trans-membrane domain (TMD) and a flexible GGSG linker (amino-acid sequence: LWLIILASLLALALILAVCIAVGGSG) was genetically linked to the 5′ end of the malE gene (see the Supporting Material) in plasmid pMAL-TMD-MBP, which is derived from pMAL-c5X (NEB, Ipswich, MA). The plasmid's DNA sequence was confirmed by DNA sequencing. Plasmid pMAL-TMD-MBP lacks the protease factor Xa recognition sequence present in pMAL-c5X. This plasmid allows cytoplasmic expression of TMD-linked wt-MBP under an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible T7 promoter.
The TMD-MBP protein was expressed in Escherichia coli DH5α after induction with IPTG and purified using an amylose affinity chromatography (see the Supporting Material). TMD-MBP concentration was determined using its extinction coefficient, and the yield was ∼3 mg/L culture volume.
Construction, expression, and purification of DARPins
The pQE30 (AmpR) vector backbone was employed to construct the plasmids that encode the DARPins employed in this study. The plasmids were constructed using commercial gene synthesis, gene assembly, restriction enzyme-based cloning, and site-directed mutagenesis (see the Supporting Material). The gene encoding for off7 protein was commercially synthesized and cloned into vector pQE30 (AmpR) to create pQE30-off7 (see the Supporting Material). Plasmids pQE30-off7-Y125H and pQE30-off7-W90A were constructed by swapping the relevant DNA from the pQE30-off7 plasmid with PCR-assembled DNA incorporating the desired mutation (see the Supporting Material). Site-directed mutagenesis of pQE30-off7 plasmid was employed to construct plasmids pQE30-off7-Y81A, pQE30-off7-D110A, and pQE30-off7-F79A (see the Supporting Material). E3_5 is a DARPin that lacks affinity for MBP and was used as a negative control for DARPin-MBP binding assays (5).
The gene encoding for E3_5 protein was commercially synthesized and cloned into vector pQE30 to create pQE30-E3_5 (see the Supporting Material). All pQE30-DARPin (AmpR) plasmids (pQE30-off7, pQE30-off7-Y125H, pQE30-off7-W90A, pQE30-off7-Y81A, pQE30-off7-D110A, pQE30-off7-F79A, and pQE30-E3_5) have an open reading frame that codes for the respective DARPin with a RGSHHHHHH amino-acid tag at the N-terminus. This open reading frame is under an IPTG-inducible T5 promoter. This allows cytoplasmic expression of His-tagged DARPin. Circular dichroism was used to confirm the mutations did not significantly alter the folded structure of off7 (see Fig. S1 in the Supporting Material).
E. coli XA90 cells harboring the pQE30 vectors were used for DARPin expression (see the Supporting Material). Protein expression was induced with IPTG. The expressed DARPins were purified using immobilized metal affinity chromatography via its N-terminal hexahistidine tag (see the Supporting Material). Invision His-tag in-gel staining (Invitrogen, Carlsbad, CA) confirmed the correct expression of all His-tagged DARPins. Purified DARPins were dialyzed against phosphate-buffered saline (PBS, pH 7.4). DARPin concentrations were determined using their extinction coefficients.
Molecular dynamics simulation
The crystal structure of the off7 in complex with the maltose binding protein (Protein Data Bank PDB ID, 1SVX) (5) was used as starting coordinates and fully solvated in a rectangular water box of 68 × 74 × 161 Å3 by using the Visual Molecular Dynamics program (26). Sodium and chloride ions were added to neutralize the system, which yielded 76,842 atoms in total. The molecular dynamics (MD) simulation was performed in the NAMD program using the CHARMM22 force field and the TIP3P water model (27–29). Periodical boundary conditions were applied to avoid finite size effects. The electrostatic interactions were simulated by using particle-mesh Ewald sum method (30) and van der Waals interactions were calculated with a cutoff distance of 12 Å.
The system was energy-minimized by a 20,000-steps-of-conjugate gradient with heavy atoms fixed followed by another 20,000 steps with all atoms free. Next, the system was gradually heated from 0 to 300 K in 60 ps and equilibrated for at least 1.5 ns with temperature at 300 K and pressure at 1 atm by using the Langevin dynamics method (31,32). A typical profile of RMSD plot for the backbone atoms of DARPin off7-MBP complex during the annealing and equilibrium in MD simulation is shown in Fig. S2. In this work, five pairs of off7-MBP complexes were simulated by using a computer equipped with six CPUs and 6 GB RAM. Each simulation took approximately five days to finish, which brought the protein complex to the experimental conditions before alanine-scanning mutagenesis.
Computational alanine-scanning mutagenesis
After annealing and equilibrium using MD simulation, five individual off7-MBP complexes were entered to the computational alanine-scanning in Robetta server (33) (http://robetta.bakerlab.org/). In this simulation, all interfacial amino acids of the protein-protein complex were identified and individually mutated to alanine. The variation of the change of free binding energy (ΔΔGbind) was calculated based on a free energy function that accounts for the shape complementarity of interface atoms, polar interactions involving ion pairs and hydrogen bonds, and solvation interactions (33).
Lipid bilayer preparation
The lipid solutions were prepared by dissolving 8 mg of DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) into 8 mL of lipid buffer B (20 mM Tris-HCl, 50 mM NaCl, 1 mM CaCl2, 0.1% (w/v) Triton X-100) (22,34). TMD-MBP (130 μL) at a concentration of 80 μg/mL was added to 380 μL of lipid solution and incubated at 37°C for 2 h. The lipid-protein solution was transferred to a 10 kDa MWCO dialysis cassette and dialyzed against 1 L of lipid buffer A (20 mM Tris-HCl, 50 mM NaCl, 1 mM CaCl2) three times for 12 h each (22). Lipid-protein solutions were stored at 4°C under nitrogen for up to one month. To prepare bilayer platforms, glass slides were plasma cleaned for 5 min and immediately submerged in a solution of 100 ppm polyethyleneimine (PEI) in 0.5 mM KNO3 for 20 min before rinsing with DI water and drying with nitrogen (22). The slides were further dried in a vacuum desiccator for at least three days before use. A 4 μL droplet of the lipid-protein solution was incubated on the PEI-coated glass slide for 2 h under a slightly dampened towel to prevent complete dehydration. The slides were later rinsed and immersed in Hank's balanced salt solution for force spectroscopy experiments. Blank lipid solutions (without TMD-MBP protein) were incubated on PEI-glass as a negative control to determine the binding specificity.
Cantilever functionalization
To prepare a surface for coating soluble proteins, molecular force probe cantilevers (Veeco, Plainview, NY) were silanized with 2% (v/v) 3-aminopropyltriethoxysilane in acetone (22,23). The cantilevers were incubated for 1 h in a 3 μg/mL solution of wild-type off7, mutated off7, or E3_5 protein in Dulbecco's PBS containing 50-fold molar excess of the cross-linker bis(sulfosuccinimidyl) suberate (BS3; Pierce, Rockford, IL) followed by quenching with Tris buffer. A higher concentration, 30 μg/mL, of D110A solution was used to obtain sufficient binding events for kinetic analysis. Cantilevers were subsequently incubated in 1% bovine-serum albumin in Dulbecco's PBS to block nonspecific interactions. The concentrations of protein solution were optimized to result in a low proportion of binding events during force-spectroscopy experiments (∼20 binding events per 100 contacts).
Single-molecule force spectroscopy
Force spectroscopy assays were conducted using a Molecular Force Probe (MFP-1D; Asylum Research, Santa Barbara, CA). A triangular-shaped cantilever with nominal spring constants of 10 pN/nm was calibrated using thermal noise amplitude, and its deflection was measured by laser reflection onto a split photodetector (35,36). The 50-mm petri dish containing the MBP-incorporated or blank lipid bilayer and Hanks' balanced salt solution buffer was placed on the MFP-1D stage and positioned so that the cantilever was directly above the testing region. The distance between the cantilever and the lipid bilayer was adjusted, such that each approach cycle generated a slight depression force (∼1–2 nN) on the lipid bilayer before reproach. The reproach velocity was varied from 5 to 25 μm/s, and the dwell time was set to 20 ms to minimize the occurrence of multiple events (35,36).
Rupture forces were derived from force-versus-distance traces using IgorPro 4.09 software (WaveMetrics, Lake Oswego, OR). The Bell model parameters were tabulated by a least-squares fit to the rupture force against the logarithm of loading rate (22,23,36). Force spectra for MBP binding to the wild-type, Y125H off7, and D110A off7 comprise at least four individual experiments and represent >1000 successful events in each sample. The other mutants and control samples were tested by three individual experiments. A new cantilever was freshly prepared, calibrated, and tested in each individual experiment.
Surface plasmon resonance
The interactions of wild-type or mutated off7 and MBP were studied by surface plasmon resonance using a BIAcore 3000. The running buffer was HBS-P, which contains10 mM HEPES, pH 7.4, 150 mM NaCl, and 0.005% (v/v) Tween 20 surfactant (BIAcore, Piscataway, NJ). Experiments were conducted on a streptavidin SA chip (BIAcore) coated with a low-density (∼200 RU) biotinylated MBP (Avidity, Aurora, CO) to avoid mass-transport limited binding. Binding assays were performed at a flow rate of 60 μL/min with 100 s injection of wild-type or mutated off7 proteins (2–1000 nM) followed by washing with buffer for 10 min. The measured signals were double-referenced from reference curves generated by an uncoated flow cell and several injections of running buffer. The kinetic constants kon and koff were calculated by the global fitting of a 1:1 bimolecular interaction model, and the dissociation equilibrium constant (Kd) was calculated by the dividing kon by koff. Experimental data were analyzed using BIAevaluation 3.0 software (BIAcore) supplied with the instrument and examined for self-consistency as described in the literature (37,38).
Results
Computational simulation predicts that the off7-MBP binding energy is concentrated on few residues at the interface
To characterize the off7-MBP interaction, we first performed computational alanine-scanning mutagenesis to analyze the binding interface using molecular dynamics (MD) and Robetta server simulations. In these simulations, all interface residues in the off7-MBP interface were identified. Each of these was individually mutated to an alanine residue, and the change in binding free energy (ΔΔGbind) was predicted. An increase in binding free energy is indicative of destabilization of the protein-protein complex upon alanine mutation (Fig. 1 A). A threshold value of ΔΔGbind >1.5 kcal/mol was used to identify the key residues that affect off7-MBP binding (19,33). Simulations predict that the off7-MBP binding energy is not equally distributed among all the interfacial residues. Three amino acids (F79, Y81, and W90) were predicted to play pivotal roles in biomolecular recognition. As revealed by the simulation scores, F79 and W90 are expected to have a stronger impact than Y81 on DARPin off7 binding to the target MBP. These three amino acids separate in two regions based on their locations at the interface (Fig. 1, B and C).
Figure 1.

Simulation data predict that the off7-MBP interaction is characterized by a hot-spot motif. (A) Simulation result on the changes in binding free energy (ΔΔGbind) upon alanine mutation at the off7 interfacial amino acids. Those amino acids with a ΔΔGbind value >1.5 kcal/mol (red dashed line) are considered to be critical at the interface, because the point mutations at these sites are expected to significantly destabilize the complex. Data represent the mean ± SE of five independent simulations. (B and C) Two perpendicular views of the crystal structure of off7 (blue, on the right) in complex with MBP (cyan, on the left). (Spheres) Interfacial residues. (Orange) Off7 interfacial residues with ΔΔGbind value >1.5 kcal/mol. Simulation predicted that binding energy between off7-MBP concentrates at two locations. The PDB ID is 1SVX. The figure was made with the software PyMOL (DeLano Scientific, San Francisco, CA).
The F79A, Y81A, and W90A mutations were selected for experimental testing. As examples of residues predicted to have a moderate effect on binding, we chose residues at the interface that were DARPin framework residues—residues that were not allowed to vary in the combinatorial library design. Framework residues D110A and Y125H mutations were selected for testing. Both were reported to be part of the off7-MBP interface (5) but scored low in our simulation. D110 is a designed DARPin framework residue whereas Y125 was designed to be histidine but mutated to tyrosine during library selection (5). The biomolecular interactions between wild-type/mutated off7 and MBP were characterized experimentally using two complementary biophysical tools: single-molecule force spectroscopy and surface plasmon resonance.
Insertion of TMD via a flexible linker directionally orients the MBP on lipid bilayer
To study the off7-MBP interface by single-molecule force microscopy, we chose to attach off7 to the cantilever and orient the MBP in a lipid bilayer via an attached TMD from the well-characterized human CD44 transmembrane protein (39–41). The CD44 TMD DNA was assembled using PCR and fused to the DNA encoding MBP such that the TMD was fused to the N-terminus of MBP through a flexible GGSG linker (Fig. 2 A). This genetic fusion was designed not only to incorporate MBP onto a lipid bilayer but also to provide sufficient flexibility for binding interactions. TMD-MBP was expressed in E. coli and purified using affinity chromatography. The purity of TMD-MBP was >95%. To ensure that insertion of the TMD segment did not alter MBP function, we compared the fluorescent spectra between the native MBP and TMD-MBP in the presence and absence of maltose (42). Both the native MBP and TMD-MBP spectra showed a similar red shift (∼1 nm) (42) when maltose was added to their solutions, indicating that TMD-MBP retained maltose-binding activity.
Figure 2.

CD44 trans-membrane domain (TMD)-linked maltose binding protein. (A) The TMD is linked to the N-terminus of the MBP via a flexible GGSG linker. (B) Incorporation of TMD-linked MBP onto a lipid-bilayer. HRP conjugated anti-MBP antibody was used to confirm the presence of TMD-linked MBP on the lipid bilayer. Both blank bilayer and MBP lacking the TMD served as negative controls.
The hydrophobic interactions between the lipid and the transmembrane domain ensure that the off7 binding site of MBP is directed outward for molecular recognitions. Incorporation of TMD-MBP into a lipid bilayer was demonstrated using HRP-conjugated anti-MBP antibody (Fig. 2 B). The even signal intensity suggests a uniform surface occupation of TMD-MBP across the lipid bilayer. In contrast, a very weak signal was detected when immobilization of native MBP on the lipid was attempted, proving that the insertion of TMD facilitates the incorporation of MBP into the bilayer, presumably in the desired orientation based on the geometry of the TMD-MBP fusion.
Characterization of wild-type/mutated off7-MBP bond by using single-molecule force spectroscopy confirms the hot-spot hypothesis
We studied the binding kinetics and micromechanical properties of the off7-MBP bond under forces using single molecule force microscopy (Fig. 3 A). The detailed configuration of the equipment can be found in our previous publications (22,24,35). A cantilever coated with dilute concentrations of either wild-type (WT) or mutated off7 was brought into contact for a prescribed period of time with TMD-MBP-incorporated lipid bilayer supported on a polymer-cushioned glass substrate, before being retracted at constant, predefined velocity. The unbinding of single WT/mutated off7-MBP pairs was detected by the emergence of predominantly single rather than multiple steps in the force-time traces, as indicated by the red arrows in the Fig. 3 B. The force required to dissociate the bond (i.e., the tensile strength) and the corresponding loading rate were systematically recorded over a wide range of retraction velocities to accurately determine the unstressed off-rate (k0off) and reactive compliance (xβ) of the WT/mutated off7-MBP pairs based on Bell model (22,43).
Figure 3.

Single-molecule force spectroscopy for measuring off7-MBP interactions. (A) TMD-MBP was oriented onto the lipid bilayer via the flexible GGSG-linked TMD. Purified off7 protein was covalently attached to the AFM tip and lowered onto the directionally oriented MBP on the lipid bilayer. (B) Typical force-versus-displacement traces acquired from force spectroscopy experiments in which an off7 protein functionalized cantilever tip was brought in contact hundreds of times with MBP incorporated into lipid vesicles on a PEI-coated glass slide. (Red arrows) Rupture events. Loading rates are estimated based on the linear increase in slope just before bond dissociations.
The concentrations of wild-type off7 protein immobilized on the cantilever tip and TMD-MBP immobilized into the lipid bilayer were chosen to yield a low percentage (∼20%) of successful adhesion events (Fig. 4 A), such that the majority of the binding events (>89%, as estimated from the Poisson distribution (44)) were mediated by single protein-protein interaction (22,23). Interestingly, the F79A, Y81A, and W90A off7 mutations drastically reduced the frequency of binding events to <5%, which indicates the critical role of these three interface residues in the off7-MBP intermolecular binding. In contrast, a ∼20% binding frequency was obtained for the Y125H mutant (Fig. 4 A), suggesting that the unexpected mutation from histidine to tyrosine at this framework residue, which was generated by the library selection process, does not play a pivotal role in off7-MBP molecular recognition. A higher concentration of D110A off7 compared to that of the wild-type off7 was required to functionalize the cantilever for mediating a similar binding frequency, which indicates this mutation lowered the binding affinity of off7 to the target MBP.
Figure 4.

Characterization of kinetic and micromechanical properties of wild-type or mutated off7 binding to MBP using single-molecule force spectroscopy. (A) Frequency of binding events between wild-type (WT) or mutated off7 and MBP-incorporated DMPC lipid bilayer. The last two columns represent control experiments using either a blank bilayer or the negative control protein E3_5. Data represent the mean ± SE of at least three independent experiments. (B) Rupture force as a function of loading rate for wild-type off7 (black circles), Y125H mutant (red triangles), and D110A mutant (blue squares) binding to MBP. At least four individual experiments with independent cantilevers were tested for each sample. (Superimposed solid lines) Nonlinear least-squares fitting of the Bell dissociation model for determining the unstressed off-rate k0off and reactive compliance xβ of a bond. (C–E) Representative distribution of adhesion forces for wild-type, Y125H-, and D110A-mutated off7-MBP bonds that were obtained experimentally (bars) or computed using Monte Carlo simulations (line) based on Bell model kinetic parameters listed in Table 1. ∗P < 0.002 with respect to wild-type off7-MBP binding.
The rupture force of wild-type off7-MBP bond was in the range of 50–80 pN over a wide range of loading rates (Fig. 4 B). Both Y125H and D110A mutations significantly weakened the bond strength by ∼10–15 pN, as quantified by reduced bond rupture forces over a wide range of loading rates as shown in the Fig. 4 B. Based on these force spectroscopy data, the Bell model kinetic constants were estimated and listed in the Table 1. The D110A mutation induced a 43-fold increase in the bond off-rate k0off, whereas Y125H mutation exhibited a relatively smaller (approximately eightfold) increase in the k0off constant. A higher k0off represents a shorter bond lifetime in the absence of force. It is noteworthy that neither mutation had any significant effect on the reactive compliance (xβ) of the bond.
Table 1.
Comparison of simulation results and experimental kinetic parameters for the wild-type or mutated off7 interacting with MBP
| Sample | Robetta simulation |
Single-molecule force microscopy |
Surface plasmon resonance |
|||
|---|---|---|---|---|---|---|
| ΔΔGbind (kcal/mol) | k0off (×10−3 s−1) | xβ (nm) | kon (×105 M−1 s−1) | koff (×10−3 s−1) | Kd (nM) | |
| Wild-type | — | 6.1 ± 2 | 0.59 ± 0.03 | 9.41 ± 0.4 | 7.3 ± 0.5 | 7.7 ± 0.6 |
| Y125H | 0.41 ± 0.03 | 47.3 ± 9 | 0.54 ± 0.02 | 6.14 ± 0.7 | 25.9 ± 0.8 | 42.2 ± 4.6 |
| D110A | 0.61 ± 0.10 | 262.8 ± 58 | 0.43 ± 0.03 | 2.45 ± 0.2 | 28.6 ± 1.2 | 116.7 ± 10.7 |
| Y81A | 1.88 ± 0.09 | N.B. | — | 0.64 ± 0.1 | 103.3 ± 7.8 | 1614 ± 280 |
| F79A | 2.79 ± 0.16 | N.B. | — | N.B. | — | — |
| W90A | 2.74 ± 0.19 | N.B. | — | N.B. | — | — |
| E3_5 | — | N.B. | — | N.B. | — | — |
N.B. represents no binding that is defined as <5 RU response was detected in SPR assay up to 1 μM concentration or <5% of successful binding events observed in single-molecule force microscopy.
Based on the Bell model (20), applied rupture force (f) generates mechanical energy f·xβ (force × displacement) on the stressed bond and alters the bond off-rate (koff) by following an exponential function: koff /k0off = exp(fxβ/kBT). The close values of xβ among wild-type, D110A, and Y125H mutants reflect their similar sensitivity of bond rupture under force. The distributions and means of rupture forces obtained in experiments are in accord with Monte Carlo simulations based on kinetic constants listed in the Table 1, which further validated the accuracy of our estimated Bell model parameters as shown in the Fig. 4, C–E. The binding specificity was demonstrated by the use of a blank lipid bilayer or a negative control E3_5 DARPin (5) functionalized cantilever, which reduced the binding frequency to the background level.
Measurement of the three-dimensional kinetics of off7-MBP interactions by surface plasmon resonance further substantiates the hot-spot motif
Surface plasmon resonance (SPR) assays were performed using a BIAcore 3000 (BIAcore). The wild-type and mutated off7 were perfused over the biotinylated MBP immobilized sensor chip at a wide range of concentrations (Fig. 5, A–C) to accurately determine the kinetic constants: kon, koff, and Kd. The binding specificity was demonstrated by perfusing the negative control E3_5 DARPin (5), which drastically reduced the binding signal to the background level.
Figure 5.
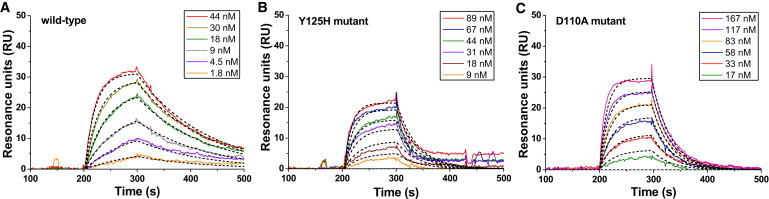
Binding interactions of wild-type/mutated off7 and MBP analyzed by SPR. Representative sensorgrams were recorded by perfusing (A) wild-type, (B) Y125H, or (C) D110A mutated off7 into a MBP-immobilized flow cell at different concentrations for 100 s followed by washing with buffer for 10 min. (Black dashed lines) Global fitting of a 1:1 bimolecular interaction model for the calculation of kinetic parameters kon, koff, and Kd. The values of χ2 are 0.3, 2.8, and 0.6 for wild-type, Y125H, and D110A off7, respectively. Data were analyzed by using BIAevaluation 3.0 (BIAcore).
In agreement with force spectroscopy, SPR experiments show that the F79A and W90A mutations abolished the off7-MBP binding interactions (<5 RU at concentrations up to 1 μM), which corroborates their pivotal roles in the off7-MBP intermolecular recognition. The Y81A mutation has a significant effect on both the on-rate and off-rate, thereby reducing the affinity 200-fold (Table 1). In agreement with the force microscopy data, D110 contributes more than Y125 to the off7-MBP interaction though both contributions are moderate compared to F79, W90, and Y81 (Table 1). The Y125H mutation only induced a modest decrease in the binding on-rate kon, whereas the D110A mutation reduced the kon value by four times. This result explains why a higher concentration of D110A was needed to obtain a similar percentage of binding events compared to the wild-type and Y125H mutant.
Discussion
Computational alanine scanning simulations, single-molecule force spectroscopy, and SPR were applied to characterize the off7-MBP interactions. Our simulation and experimental data suggest the existence of two classes of interfacial amino-acid residues: 1), hot-spot residues (including F79, Y81, and W90) at which the binding energy concentrates, because mutation at any of these sites will drastically (>100-fold increase in Kd) disrupt the ability of off7 to recognize MBP; and 2), auxiliary interfacial residues (including D110 and Y125) that have a rather moderate contribution to this biomolecular interaction. This establishes that the off7-MBP interface is characterized by a hot-spot motif in which binding energy is largely concentrated on a group of few amino acids.
Layton and Hellinga (45) recently developed a new method to measure Kd that exploits shifts in thermal stability upon protein binding, and used off7-MBP binding as an example. The resulting affinities were well determined at an elevated temperature (51°C), but the uncertainty at extrapolating to other temperatures resulted in a large uncertainty (2–3 orders of magnitude) for affinity values at 25°C. Nonetheless, their estimations show reasonable correlation with our simulations and experimental data (see Fig. S3). Two notable exceptions (D77 and Y89) colocalize with the hot-spot residues we identified (Fig. 6 A). The importance of these residues was further supported by a semiquantitative ELISA (46).
Figure 6.

Schematic illustrations show how the interfacial amino acids of off7 are in complex with the binding surface of MBP. (A) The open sandwich surface representation of off7 (in blue). (Stained in yellow) Recognition patch for the target MBP molecule (auxiliary residues); (orange) hot-spots F79, Y81, and W90; and (green) hot-spots D77 and Y89. (b and c) The cartoon (in cyan) and surface representation (in gray) of MBP are shown (left) with two interfacial residues, K137 and K200 (stick representation). (Ribbon representation, in blue) DARPin off7 (right). (B) Five hot-spot residues of off7 protein (stick representation in orange and green). F79, Y89, and W90 are extensively inserted into the valley of MBP binding pockets, whereas Y81 has a large contact area with MBP surface. (C) Two auxiliary interfacial residues (stick representation in yellow). The figure was prepared using the software PyMOL (DeLano Scientific).
The D77A mutation scored low in our simulation and D77 does not directly interact with MBP. The residue may help to position F79 or Y81 for optimal interaction—offering a possible explanation of why predicting the effects of the D77A mutation might be more challenging. Another possibility is that mutation of D77 to a smaller residue allows water access to F79 or Y81, reducing their ability to contribute to the interaction. Y89 colocalizes with W90 and its addition as a hot-spot residue results in a continuous strip of hot-spot residues from W90 to F79 (Fig. 6 A).
The off7-MBP binding interface has a specific topography that resembles a two-humped camel. Hot-spot residues F79, Y89, and W90, insert deep into the two hydrophobic valleys of MBP between the humps with a large surface area of interaction (Fig. 6 B). Such topology is typical of hot-spots, as 79% of hot-spot residues in natural protein interfaces are located as protruding or complemented pocket residues (47). Y81's area of interaction with the top of one hump, although considerable, is not as large as the residues inserted into the valleys—offering an explanation for why the Y81A mutation dramatically reduces, but does not eliminate, the ability to detect binding by SPR. The Y81A mutant's 15-fold decrease in the kon value along with 14-fold increase in the koff value explain the lack of binding events in single-molecule force spectroscopy experiments (Fig. 4 A). It is also possible that Y81A alters the structure of the DARPin off7 binding site such that a rate-limiting conformational change must take place before binding to MBP.
Each consensus ankyrin repeat module has eight nonframework residues that can be randomly changed for mediating specific binding to target molecules without disrupting the ankyrin repeat module. In addition to these designed binding sites, a previous study has shown the involvement of framework residues in this biomolecular interaction (5). The hydrogen-bond distances between two framework residues in off7 protein (D110 and Y125) and MBP are the two shortest distances among all hydrogen bonds (5), suggesting that they may play key roles in the off7-MBP molecular recognition. However, our data indicate that they are auxiliary interfacial residues at which mutation will have a rather modest effect on the binding interaction (Fig. 4 B and Table 1). Framework residues D110 and Y125 interact with two humps of the camel of the MBP-binding surface (Fig. 6 C).
Typically, hot-spot residues tend to cluster together near the center of the binding interface (17). The hot spots on off7 concentrate in a narrow, contiguous patch in the center of the binding interface that extends to the edge of the interface at W90 at one end (Fig. 6 A). Exposure to bulk solvent can significantly reduce a residue's contribution to the overall binding free energy (17). Solvent is excluded from contacting the F79, whereas W90 is partially exposed to the solvent. Hot-spot residues at the edge of an interface are uncommon, but our data indicate that the exclusion of bulk solvent is not a necessary condition for a hot-spot residue. Hot-spot residues in natural proteins have been found to have a nonrandom composition in which nearly half of them are tryptophan (21%), arginine (13.3%), and tyrosine (12.3%) (17). Our data support the hypothesis that hot-spot amino acids in synthetic binding proteins will share this bias, as three of the five off7 hot-spot residues are tryptophan or tyrosine.
Attachment of a TMD to the target molecule (e.g., MBP) enables the incorporation of the target molecule into the lipid bilayer platform in the proper orientation, thereby enabling the study of how point mutation(s) at the binding interface modulates the kinetics and micromechanical properties of synthetic binding proteins under force (48). This strategy can be applied not only to force spectroscopy but also to other binding assays that utilize microchannel and biosensing devices. Our proposed technique will be useful for the characterization of sensor proteins pertinent to detection of target molecules attached on surfaces, such as human skin, clothes, and shoes.
In summary, we delineated how an engineered DARPin recognizes its target at the single-molecule level using what we consider a novel approach that integrates protein engineering, in silico hot-spots prediction, and two complementary quantitative biophysical assays. This cross-disciplinary research provides knowledge-based designing cues for engineering the next generation of tunable synthetic binders.
Acknowledgments
We thank Dr. Ruben T. Almaraz for technical assistance with the SPR assays and Phrabha S. Raman for the single-molecule force spectroscopy training.
This work was supported by Defense Threat Reduction Agency grant HDTRA1-09-1-0016 (to K.K. and M.O.), National Institutes of Health/National Cancer Institute grant R01 CA101135 (to K.K.), National Institute of General Medicine at the National Institutes of Health grant R01-GM066972 (to M.O.), and a postdoctoral fellowship from the American Heart Association (to L.S.-L.C.).
Contributor Information
Marc Ostermeier, Email: oster@jhu.edu.
Konstantinos Konstantopoulos, Email: konstant@jhu.edu.
Supporting Material
References
- 1.Guntas G., Mitchell S.F., Ostermeier M. A molecular switch created by in vitro recombination of nonhomologous genes. Chem. Biol. 2004;11:1483–1487. doi: 10.1016/j.chembiol.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 2.Guntas G., Mansell T.J., Ostermeier M. Directed evolution of protein switches and their application to the creation of ligand-binding proteins. Proc. Natl. Acad. Sci. USA. 2005;102:11224–11229. doi: 10.1073/pnas.0502673102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tullman J., Guntas G., Ostermeier M. Protein switches identified from diverse insertion libraries created using S1 nuclease digestion of supercoiled-form plasmid DNA. Biotechnol. Bioeng. 2011;108:2535–2543. doi: 10.1002/bit.23224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koide A., Gilbreth R.N., Koide S. High-affinity single-domain binding proteins with a binary-code interface. Proc. Natl. Acad. Sci. USA. 2007;104:6632–6637. doi: 10.1073/pnas.0700149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binz H.K., Amstutz P., Plückthun A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 2004;22:575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- 6.Brient-Litzler E., Plückthun A., Bedouelle H. Knowledge-based design of reagentless fluorescent biosensors from a designed ankyrin repeat protein. Protein Eng. Des. Sel. 2010;23:229–241. doi: 10.1093/protein/gzp074. [DOI] [PubMed] [Google Scholar]
- 7.Kohl A., Binz H.K., Grütter M.G. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc. Natl. Acad. Sci. USA. 2003;100:1700–1705. doi: 10.1073/pnas.0337680100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boersma Y.L., Pluckthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr. Opin. Biotechnol. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Zahnd C., Pecorari F., Plückthun A. Selection and characterization of Her2 binding-designed ankyrin repeat proteins. J. Biol. Chem. 2006;281:35167–35175. doi: 10.1074/jbc.M602547200. [DOI] [PubMed] [Google Scholar]
- 10.Kohl A., Amstutz P., Grütter M.G. Allosteric inhibition of aminoglycoside phosphotransferase by a designed ankyrin repeat protein. Structure. 2005;13:1131–1141. doi: 10.1016/j.str.2005.04.020. [DOI] [PubMed] [Google Scholar]
- 11.Milovnik P., Ferrari D., Plückthun A. Selection and characterization of DARPins specific for the neurotensin receptor 1. Protein Eng. Des. Sel. 2009;22:357–366. doi: 10.1093/protein/gzp011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amstutz P., Koch H., Plückthun A. Rapid selection of specific MAP kinase-binders from designed ankyrin repeat protein libraries. Protein Eng. Des. Sel. 2006;19:219–229. doi: 10.1093/protein/gzl004. [DOI] [PubMed] [Google Scholar]
- 13.Schweizer A., Roschitzki-Voser H., Grütter M.G. Inhibition of caspase-2 by a designed ankyrin repeat protein: specificity, structure, and inhibition mechanism. Structure. 2007;15:625–636. doi: 10.1016/j.str.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 14.Sennhauser G., Grütter M.G. Chaperone-assisted crystallography with DARPins. Structure. 2008;16:1443–1453. doi: 10.1016/j.str.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Jones S., Thornton J.M. Principles of protein-protein interactions. Proc. Natl. Acad. Sci. USA. 1996;93:13–20. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakrabarti P., Janin J. Dissecting protein-protein recognition sites. Proteins. 2002;47:334–343. doi: 10.1002/prot.10085. [DOI] [PubMed] [Google Scholar]
- 17.Bogan A.A., Thorn K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 18.Moreira I.S., Fernandes P.A., Ramos M.J. Hot spots—a review of the protein-protein interface determinant amino-acid residues. Proteins. 2007;68:803–812. doi: 10.1002/prot.21396. [DOI] [PubMed] [Google Scholar]
- 19.Thorn K.S., Bogan A.A. ASEdb: a database of alanine mutations and their effects on the free energy of binding in protein interactions. Bioinformatics. 2001;17:284–285. doi: 10.1093/bioinformatics/17.3.284. [DOI] [PubMed] [Google Scholar]
- 20.Bell G.I. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 21.Evans E.A., Calderwood D.A. Forces and bond dynamics in cell adhesion. Science. 2007;316:1148–1153. doi: 10.1126/science.1137592. [DOI] [PubMed] [Google Scholar]
- 22.Raman P.S., Alves C.S., Konstantopoulos K. Single-molecule binding of CD44 to fibrin versus P-selectin predicts their distinct shear-dependent interactions in cancer. J. Cell Sci. 2011;124:1903–1910. doi: 10.1242/jcs.079814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hanley W.D., Wirtz D., Konstantopoulos K. Distinct kinetic and mechanical properties govern selectin-leukocyte interactions. J. Cell Sci. 2004;117:2503–2511. doi: 10.1242/jcs.01088. [DOI] [PubMed] [Google Scholar]
- 24.Panorchan P., Thompson M.S., Wirtz D. Single-molecule analysis of cadherin-mediated cell-cell adhesion. J. Cell Sci. 2006;119:66–74. doi: 10.1242/jcs.02719. [DOI] [PubMed] [Google Scholar]
- 25.Neuman K.C., Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat. Methods. 2008;5:491–505. doi: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
- 27.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 29.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 30.Darden T., York D., Pedersen L. Particle mesh Ewald—an N.log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 31.Schneider T., Stoll E. Molecular-dynamics study of a 3-dimensional one-component model for distortive phase-transitions. Phys. Rev. B. 1978;17:1302–1322. [Google Scholar]
- 32.Feller S.E., Zhang Y.H., Brooks B.R. Constant-pressure molecular-dynamics simulation—the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 33.Kortemme T., Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. USA. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshall B.T., Long M., Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 35.Dobrowsky T.M., Panorchan P., Wirtz D. Chapter 15: Live-cell single-molecule force spectroscopy. Methods Cell Biol. 2008;89:411–432. doi: 10.1016/S0091-679X(08)00615-8. [DOI] [PubMed] [Google Scholar]
- 36.Hanley W., McCarty O., Konstantopoulos K. Single molecule characterization of P-selectin/ligand binding. J. Biol. Chem. 2003;278:10556–10561. doi: 10.1074/jbc.M213233200. [DOI] [PubMed] [Google Scholar]
- 37.Schuck P., Minton A.P. Kinetic analysis of biosensor data: elementary tests for self-consistency. Trends Biochem. Sci. 1996;21:458–460. doi: 10.1016/s0968-0004(96)20025-8. [DOI] [PubMed] [Google Scholar]
- 38.Rich R.L., Myszka D.G. Grading the commercial optical biosensor literature—Class of 2008: ‘The Mighty Binders’. J. Mol. Recognit. 2010;23:1–64. doi: 10.1002/jmr.1004. [DOI] [PubMed] [Google Scholar]
- 39.Alves C.S., Yakovlev S., Konstantopoulos K. Biomolecular characterization of CD44-fibrin(ogen) binding: distinct molecular requirements mediate binding of standard and variant isoforms of CD44 to immobilized fibrin(ogen) J. Biol. Chem. 2009;284:1177–1189. doi: 10.1074/jbc.M805144200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomas S.N., Zhu F., Konstantopoulos K. Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E- and L-selectin in shear flow. J. Biol. Chem. 2008;283:15647–15655. doi: 10.1074/jbc.M800543200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thorne R.F., Legg J.W., Isacke C.M. The role of the CD44 transmembrane and cytoplasmic domains in co-ordinating adhesive and signaling events. J. Cell Sci. 2004;117:373–380. doi: 10.1242/jcs.00954. [DOI] [PubMed] [Google Scholar]
- 42.Hall J.A., Ganesan A.K., Nikaido H. Two modes of ligand binding in maltose-binding protein of Escherichia coli. Functional significance in active transport. J. Biol. Chem. 1997;272:17615–17622. doi: 10.1074/jbc.272.28.17615. [DOI] [PubMed] [Google Scholar]
- 43.Tees D.F., Waugh R.E., Hammer D.A. A microcantilever device to assess the effect of force on the lifetime of selectin-carbohydrate bonds. Biophys. J. 2001;80:668–682. doi: 10.1016/S0006-3495(01)76047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chesla S.E., Selvaraj P., Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys. J. 1998;75:1553–1572. doi: 10.1016/S0006-3495(98)74074-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Layton C.J., Hellinga H.W. Quantitation of protein-protein interactions by thermal stability shift analysis. Protein Sci. 2011;20:1439–1450. doi: 10.1002/pro.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Layton C.J., Hellinga H.W. Integration of cell-free protein coexpression with an enzyme-linked immunosorbent assay enables rapid analysis of protein-protein interactions directly from DNA. Protein Sci. 2011;20:1432–1438. doi: 10.1002/pro.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keskin O., Ma B., Nussinov R. Hot regions in protein-protein interactions: the organization and contribution of structurally conserved hot spot residues. J. Mol. Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 48.Orsello C.E., Lauffenburger D.A., Hammer D.A. Molecular properties in cell adhesion: a physical and engineering perspective. Trends Biotechnol. 2001;19:310–316. doi: 10.1016/s0167-7799(01)01692-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.