

Abstract
Nanomechanical analysis of proteins by single-molecule force spectroscopy based on atomic force microscopy is increasingly being used to investigate the inner workings of mechanical proteins and substrate proteins of unfoldase machines as well as to gain new insight into the process of protein folding. However, such studies are hindered by a number of technical problems, including the noise of the proximal region, ambiguous single-molecule identification, as well as difficulties in protein expression/folding and full-length purification. To overcome these major drawbacks in protein nanomechanics, we designed a family of cloning/expression vectors, termed pFS (plasmid for force spectroscopy), that essentially has an unstructured region to surmount the noisy proximal region, a homomeric polyprotein marker, a carrier to mechanically protect the protein of interest (only the pFS-2 version) that also acts as a reporter, and two purification tags. pFS-2 enables the unambiguous analysis of proteins with low mechanical stability or/and complex force spectra, such as the increasingly abundant class of intrinsically disordered proteins, which are hard to characterize by traditional bulk techniques and have important biological and clinical implications. The advantages, applications, and potential of this ready-to-go system are illustrated through the analysis of representative proteins.
Introduction
The study of protein nanomechanics typically relies on stretching the protein of interest using single-molecule force spectroscopy (SMFS) techniques to measure its mechanical resistance. A commonly used technique is atomic force microscopy (AFM). In this versatile technique, a protein that is attached to a substrate and the tip of a cantilever (the sensor) is stretched by a piezoelectric device and the forces of resistance are measured (1).
Investigators have developed several approaches to unambiguously identify and select single-molecule recordings. The first such approach was based on the use of homopolyproteins (identical repeats of the protein or protein region under study), which enabled researchers to identify single-molecule recordings as those carrying periodical patterns, and to discard aperiodic patterns (1,2). According to this strategy, the force-extension traces obtained with the length-clamp mode of the atomic force microscope should display a pattern of equally spaced peaks (the so-called sawtooth pattern) in which each peak typically originates from the unfolding of an individual (structured) protein. Thus, the process of unfolding can usually be modeled as a two-state (folded and unfolded) process (1). The height of each force peak is used to calculate the mechanical stability of the protein (Fu, defined as the average unfolding force), and the distance between peaks is used to measure the length of the protein region that is being unfolded (hidden to the force) by calculating the increase in contour length of the molecule (ΔLc, obtained after fitting the force-extension recordings to the worm-like chain (WLC) equation of polymer elasticity) (3). From this last parameter, we can easily calculate the number of amino acids contained in the force-hidden region of the protein (applying the ratio of 0.4 nm/amino acid (1,4)). These parameters depend on the pulling geometry, which in proteins is usually the N-C direction (the natural geometry of their peptide bonds).
In an alternative approach, the protein of interest is fused to a known AFM-SMFS homopolyprotein marker (i.e., a fusion heteropolyprotein) that acts as both a useful internal positive control of the technique and a single-molecule reporter (5–7).
To avoid the tedious process of synthesizing identical repeats of the protein of interest, a versatile cloning/expression vector was later developed in which the protein of interest is cloned as a fusion protein with a built-in polyprotein marker, a series of I27 modules (an immunoglobulin module from human cardiac titin, a model system in AFM-SMFS (1)), that acts as a single-molecule marker (8). This strategy allows the simple construction of heteropolyproteins such that the force peak(s) resulting from the unfolding of the protein under study can usually be identified unambiguously in the force curves (Fig. 1). However, although this system is very helpful, its use is limited by several drawbacks depending on the mechanical properties of the protein of interest.
Figure 1.
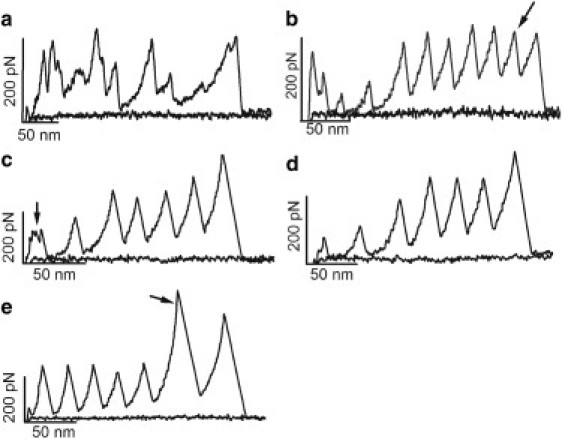
Examples of AFM-SMFS recordings obtained using a standard heteropolyprotein approach. (a) In AFM-SMFS, it is usual to obtain contaminated force-extension recordings. (b) In some cases, one may obtain a longer-than-expected recording that may originate from oligomers of the protein under study, and thus such recordings should be discarded from the analyses. In this example, the protein is (I27)3-EC1,2-(I27)2, where EC refers to C-cadherin extracellular domains (12). Although no more than seven unfolding peaks are expected if the full-length polyprotein is picked up, an extra I27 peak was obtained (arrow). (c and d) The proximal region of the recording can often appear contaminated, hiding the unfolding peaks of the protein of interest when its mechanical stability is lower than that of the marker. These recordings were obtained using the same polyprotein as in panel b, where the first unfolding peak that originated from an EC could be masked by the contamination of the proximal region of the recording (arrow in c). In such cases, the only way to increase the chance of getting clean recordings is to lower the protein concentration (d). (e) For proteins that are more mechanically stable than the marker (I27 in this figure), this standard heteropolyprotein approach is adequate for unambiguous single-molecule identification. The force peak of the protein of interest will always appear after the force peaks of the protein marker in the recording (arrow). In this case, a recording from (I27)3-c7A-(I27)2 is shown (c7A comes from the seventh cohesin module from CipA scaffoldin of Clostridium thermocellum) (13).
More recently, a new approach based on the use of single proteins flanked by long handles was reported (9). This strategy was shown to be extremely useful in mechanical folding studies (10), and it has the advantage of validating the suitability of the polyprotein approach. However, its utility for analyzing proteins whose mechanical properties have not been previously characterized remains to be demonstrated.
Variations on the polyprotein strategy, such as those described above, have been successfully applied to several proteins. The presence of a simple sawtooth pattern is taken as a fingerprint to identify the recordings that carry information about the unfolding of individual molecules (Fig. 1). In general, to maximize the number of good single-molecule recordings, one needs to ensure that the concentration of pure protein is not too high (0.2–0.5 mg/ml).
However, there are still several drawbacks that hamper this approach. Regardless of the strategy used to identify single-molecule recordings in AFM-SMFS, the proximal (to substrate) region of the recordings (typically the first 50–75 nm) is often contaminated with what are collectively called unspecific interactions (Fig. 1, a–c). These are undefined patterns of force peaks that originate from many sources, including protein desorption (from either the substrate or the cantilever tip), surface-denatured or aggregated proteins, and multiple molecules in parallel, among other unwanted events (1). Frequently, good recordings display these unspecific interactions followed by a typical sawtooth pattern that can be fitted to the WLC. However, if the protein of interest has a lower mechanical stability than the marker, its force resistance events may appear in this contaminated region and be masked by this undefined pattern, followed by the events of the marker (Fig. 1 c) (11,12). In this case, the single criterion of observing a simple sawtooth pattern carrying the flanking markers is not sufficiently stringent, and it becomes a necessary but insufficient condition that can lead to an erroneous selection of false-positive recordings as real data (11). The only solution in these cases is to lower the concentration of the sample until a clean (featureless) proximal region is obtained (Fig. 1 d). After examining many of these recordings, if the ΔLc is unique (i.e., not complex or polymorphic), one can take this value as an additional single-molecule fingerprint.
Conversely, when the mechanical stability of the protein of interest is higher than that of the marker (typically the I27 module (1,8)), its force peak will normally appear in the recordings after those of the marker, and hence it will not be affected by this problem (Fig. 1 e) (13).
Still, in any SMFS experiment that uses proteins, aperiodic force-extension recordings are often obtained even with very clean protein preparations (resulting from an optimal purification) as a result of nonspecific interactions. Furthermore, even if the polyprotein is kept in conditions that prevent oligomerization in solution (usually reducing conditions to avoid the formation of disulfide bonds between cysteine residues, typically placed at one end of the polyprotein for covalent attachment to gold substrates), longer-than-expected (supernumerary) recordings are occasionally obtained. These may result from either intermolecular disulfide-bonding or intermolecular entanglement. Moreover, as would be expected from sampling statistics, recordings that are shorter than the expected length of the full-length molecule (pulled from its termini) are commonly obtained. This problem is aggravated if degraded species of the polyprotein are present in the preparation.
To aid in the selection of data in AFM-SMFS, the following general guidelines have been proposed: 1), the proximal region should be clean to avoid masking the protein of interest; 2), the peak-to-peak distance (or distances, if there are unfolding intermediates) should be reproducible and close to the expected contour length of the force-hidden region of the protein, but it should not display other intercalated peaks (which may originate from more than one protein attached to the tip); 3), the protein should not show more unfolding peaks than those expected from the cloning strategy; and 4), the total length of the unfolded protein should not be longer than that expected from the length of the complete polypeptide (14,15).
These criteria work reasonably well for most structured proteins. However, if the protein of interest produces complex or polymorphic AFM-SMFS spectra that include a variety of low unfolding forces and variable ΔLc values, it is virtually impossible to distinguish the real data from the proximal noise. In this case, as with some intrinsically disordered proteins (IDPs; e.g., neurotoxic proteins (11)), all of the aforementioned conditions, although necessary, are insufficient.
To overcome these limitations, we developed a system we call pFS (plasmid for force spectroscopy), which has several advantages that make it an optimal strategy to produce polyproteins for general use in AFM-SMFS (Fig. 2 a). This system is based on the cassette (intercalary) strategy described elsewhere (8), although the single-molecule markers used are ubiquitin repeats instead of I27 modules. Ubiquitin's nanomechanical properties are well known (9,10,16) and it has the additional advantage of behaving like a chaperone, which in principle may prevent incorrect folding (and possibly its subsequent aggregation) of the protein of interest (17). The first member of this family of vectors, termed pFS-1, contains a fragment of an IDP, the N2B polypeptide of human cardiac titin, the featureless unfolding pattern of which will appear in the proximal region of the force-extension recording (18). This element acts as a spacer, such that the observable force peaks (from both the protein under study and the marker) will appear farther away from this noisy region of the recordings (Figs. 1 and 2). Moreover, it contains an affinity purification tag at each terminus of the protein (a His-tag and a Strep-tag), which, in addition to increasing the versatility of the purification process, allows the full-length form of the protein to be purified, thereby limiting the presence of degraded or truncated species in the purified sample (Fig. 2, b and c). Furthermore, as the N-terminus contains a His-tag, it can also be used for Ni2+-nitrilo triacetic acid (NTA) substrate attachment (19). The C-terminus, which contains two cysteine residues, can be used for covalent attachment to a gold substrate (20).
Figure 2.
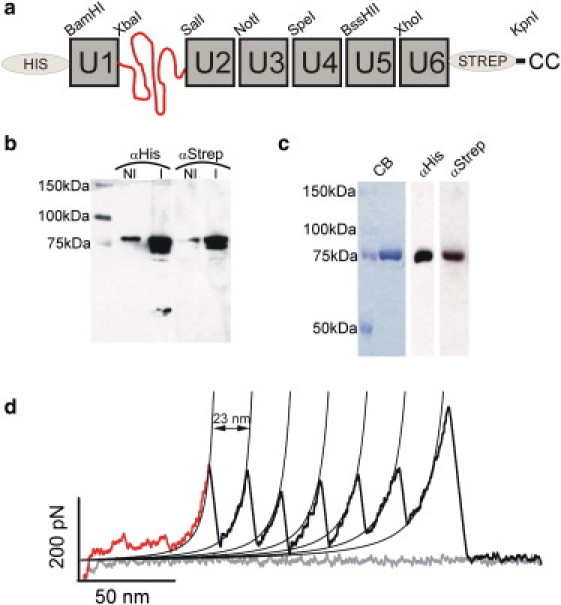
pFS-1 protein. (a) Schematic representation of the pFS-1 protein. The ubiquitin repeats are represented as gray boxes and termed U, followed by the number of the repeat in the construct. The N2B fragment is represented as a nonfolded polypeptide (curving line). This vector contains two purification tags to optimize the purification of the full-length form (His-tag at the N-terminus and Strep-tag at the C-terminus). At the C-terminus there are also two cysteine residues for attachment to gold-coated substrates (20). The vector contains eight restriction sites (BamHI, XbaI, SalI, NotI, SpeI, BssHII, XhoI, and KpnI), facilitating the cloning of the protein of interest (ideally performed with NotI, SpeI, BssHII, or XhoI to make full use of the advantages of this vector). (b) Western blot to monitor the induction of the expression of pFS-1 protein cloned into the pRSETA vector (Stratagene) using the C41(DE3) E. coli strain (29). The nominal molecular mass of the protein is 80 kDa according to ProtParam (http://www.expasy.org/tools/protparam.html). An anti-Histag antibody labels the N-terminus, and an anti-Strep-tag labels the C-terminus. NI, noninduced; I, induced. (c) After two-step affinity purification (Ni2+ affinity and Strep affinity), a single band is observed by CB staining and in Western blots probed with each antibody. (d) Typical force-extension recording of pFS-1. The extension gained by the stretching of the N2B fragment (aperiodical region of the retraction recording) serves as a spacer to avoid the usually contaminated proximal region of the force-extension recordings. The ubiquitin force peaks are shown in black.
A variant of this vector, termed pFS-2 (Fig. 3), was designed for the unequivocal single-molecule analysis of proteins with low (12) or/and complex/polymorphic mechanical stability (e.g., amyloidogenic IDPs (21) such as neurotoxic proteins (22)). The possibility of unambiguously analyzing the single-molecule mechanics of IDPs is of particular relevance given that ∼40% of eukaryotic proteins possess at least one long disordered region (>50 residues), and it is difficult to characterize these proteins by traditional bulk techniques (21). This vector is particularly useful for analyzing neurotoxic proteins, a subset of IDPs that cause neurodegenerative diseases such as Alzheimer's or Parkinson's (22). In this vector, we modified a ubiquitin repeat that is present in pFS-1 to introduce a multicloning site (MCS) within a tolerant loop of the protein to facilitate the cloning of the protein of interest in a mechanically protected way. In this manner, the carrier ubiquitin repeat unfolds mechanically before the protein of interest (which is force-hidden within) is stretched, announcing that the mechanical protection has been broken and the force has access to the protected region and can stretch the protein of interest (the mechanical features of which would appear afterward in the recording). Peng and Li (23) recently reported mechanical protection by domain insertion independently using different proteins. We call this kind of approach the carrier-guest strategy (Fig. 3 a). To effectively avoid the contamination of the noisy proximal region of the AFM recordings, pFS-2 combines the spacer (N2B fragment) and a carrier (e.g., ubiquitin or else) into which the protein of interest (the guest) can be inserted by means of regular DNA cloning procedures (Fig. 3, b and e). Thus, pFS-2 is particularly useful for mechanically polymorphic proteins. Here we show different applications of this strategy to demonstrate its potential (Fig. 4).
Figure 3.
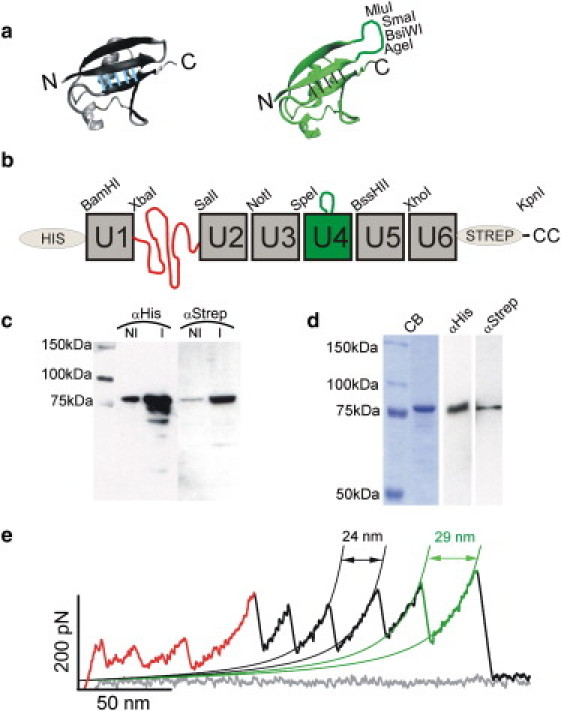
pFS-2 protein. (a) Representation of the structure of ubiquitin (left, PDB code 1D3Z) and the ubiquitin+MCS (right). The MCS is inserted into loop A-B of the ubiquitin structure (between residues T9 and G10). It is composed of 14 amino acids and contains restriction sites for the enzymes AgeI, BsiWI, SmaI, and MluI. The atomic structures were visualized by VMD 1.8.6 (48). (b) Schematic representation of the pFS-2 protein. (c) Western blot to monitor the induction of the expression of the pFS-2 protein cloned in the pRSETA vector (Stratagene) and using the C41(DE3) E. coli strain (29). The nominal molecular mass of the protein is 82 kDa according to ProtParam. The protein yield is very similar to that obtained with pFS-1 (Fig. 2b). (d) Purification of the recombinant pFS-2 protein. As with the pFS-1 protein (Fig. 2c), a single band is observed in CB staining and in Western blots probed with each antibody. (e) Typical force-extension recording of the pFS-2 containing (in this order) the N2B region, the ubiquitin unfolding peaks (in black), and the ubiquitin+MCS unfolding peak. This peak can readily be identified by its larger ΔLc due to the insertion of the MCS into the ubiquitin cDNA sequence.
Figure 4.
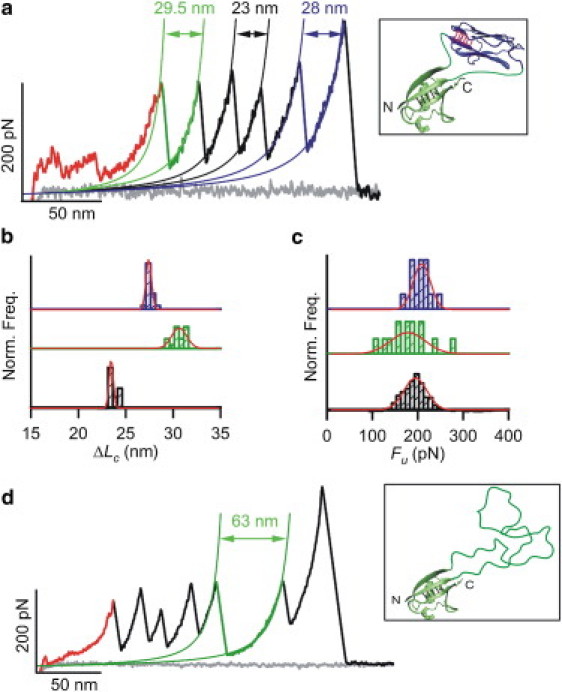
Use of the carrier-guest strategy. (a) Typical force-extension recording of the pFS-2+I27 protein carrying (in this order) the N2B region and unfolding peaks from the “carrier” ubiquitin, three ubiquitin repeats, and the I27. The unfolding peak of the carrier ubiquitin repeat must always precede the unfolding peak of the guest I27 module (23). This carrier ubiquitin shows a larger ΔLc due to the inclusion in its fold of the MCS and the folded I27. The inset shows a representation of the ubiquitin carrier with the I27 module grafted inside (top; PDB code 1TIT) as visualized by VMD 1.8.6 (48). (b) The ΔLc analysis of the elements in this construct shows that the folds of the ubiquitin repeats (bottom), ubiquitin carrier (middle), and I27 guest (top) are preserved. (c) Fu histograms of the construct modules (same order). Both histograms show that the carrier-guest strategy does not alter the mechanical properties of the modules involved. All distributions are normalized in b and c. The Gaussian fits to the histograms are shown. (d) Typical force-extension recording of the pFS-2+VAMP2 protein. VAMP2 shows no force peaks in SFMS (i.e., no mechanical stability), thus the unfolding of the ubiquitin carrier is followed by the peakless stretching of the guest VAMP2. The inset shows a representation of the ubiquitin carrier with the cytoplasmic region from VAMP2 (residues 1–94, top) hosted within it, visualized by VMD 1.8.6 (48).
Materials and Methods
Design and construction of the pFS-1 vector
The pFS-1 vector was originally designed on the basis of a previously described strategy (8). A human Ubi9 cDNA clone was used as a template to PCR-clone the ubiquitin repeats (16). We used a fragment of the N2B region from titin instead of its entire length for two reasons: first to reduce the size of the final fusion protein, because proteins > 100 kDa typically are poorly expressed in Escherichia coli (pET System Manual, Novagen, Darmstadt, Germany), and second to avoid the formation of internal disulfide bonds (24). The fragment of choice should not have any tertiary structure, as it occurs for the full-length N2B (18). Based on the predicted secondary structure of different candidate sequences of N2B run at PROF (http://www.aber.ac.uk/∼phiwww/prof/), we selected the fragment of N2B from D145 to L349 (with both residues included).
With the exception of NotI, all of the restriction enzymes chosen (BamHI, XbaI, SalI, SpeI, BssHII, XhoI, and KpnI, from the N- to C-terminus; Fig. 2 a) leave cohesive ends encoding two amino acids (GS, SR, VD, TS, AR, LE, and GT, respectively, in the one-letter code). NotI requires the introduction of an extra nucleotide to avoid altering the reading frame, and thus we introduced an extra adenosine (this restriction site with this extension will code for AAA).
The cloning procedure to generate this vector was as follows: 1) The first ubiquitin repeat was cloned in the plasmid pT7blue (Novagen) via BamHI-XbaI. 2) The N2B fragment was inserted into the same vector after the previous repeat via XbaI-SalI. 3) To clone the next ubiquitin repeat, the BamHI-Ubiq-N2B-SalI DNA fragment was cloned into pET24d (Novagen). Then, as SalI and NotI are very close in this vector and digestion with these enzymes could become problematic, we inserted synthetic oligonucleotides to separate these sites and facilitate digestion. These oligonucleotides contained the sequences 5′-GGCCGCCACTGAACCTGC-3′ and 5′- GGCCGCAGGTTCAGTGGC-3′ in such a way that upon hybridization, a double-stranded oligonucleotide is formed with NotI cohesive ends and phosphate groups at the 5′-termini. Thus, we digested the pET24d plasmid carrying BamHI-Ubiq-N2B-SalI with NotI and inserted the double-stranded oligonucleotide. Then, the next ubiquitin repeat was cloned via SalI-NotI. 4) The next ubiquitin repeat was first cloned into pT7blue via BamHI-NotI-Ubiq-SpeI, and the fragment was then released via NotI-SpeI. The BamHI-Ubiq-N2B-Ubiq-NotI fragment was also released from the pET24d plasmid via BamHI-NotI, and both fragments were simultaneously cloned into pT7blue via BamHI-SpeI. 5) The next cloning step involved the ubiquitin repeat at the 3′ position in the DNA sequence of the pFS-1 construct. The BamHI-XhoI-Ubiq-KpnI fragment was cloned into pT7blue via BamHI-KpnI. 6) The plasmid was then digested with HindIII-XhoI and the previous ubiquitin repeat in the pFS-1 vector was cloned here via HindIII-BssHII-Ubiq-XhoI, obtaining the BssHII-Ubiq-Ubiq-KpnI sequence cloned into pT7blue. 7) The resulting fragment was then released using BssHII-KpnI and cloned simultaneously with the SpeI-Ubiq-BssHII PCR fragment into pT7blue via SpeI-KpnI. 8) This sequence was then released using SpeI-KpnI. Then, the BamHI-Ubiq-N2B-Ubiq-Ubiq-SpeI sequence was also released from the pT7blue vector using BamHI-SpeI. Both fragments were simultaneously cloned into pRSETA (Stratagene, La Jolla, CA) or pQE80L (Qiagen, Duesseldorf, Germany) via BamHI-KpnI.
Design and construction of the pFS-2 vector
We next modified the pFS-1 vector by inserting an MCS into a ubiquitin repeat as a site for cloning the protein of interest according to the carrier-guest strategy. To avoid affecting the fold of the carrier ubiquitin repeat, the best candidate insertion sites were the loops that were naturally present. Based on previous studies describing the modification of ubiquitin stability by insertion of sequences in the different loops of its fold (25–27), we chose to use the loop A-B (between residues T9-G10) because it showed the highest tolerance to insertions in terms of global fold, thermodynamic stability, and loss of hydrogen bonds (Fig. 3 a). Also, we selected the ubiquitin repeat at position 4 of the pFS-1 construct because it is placed in a central position and therefore would allow us to gain the full benefit from the use of this vector (Fig. 3 b).
For the restriction enzymes, we chose to use AgeI, BsiWI, SmaI, and MluI, which code for the amino acids TG, RS, PG, and TR, respectively. We also inserted two glycine residues at each terminus of the MCS to make this region more flexible and to decrease the possibility of deleterious effects on ubiquitin folding. Furthermore, we inserted a spacer between the restriction sites to improve the efficiency of the enzymatic digestion in this MCS (TCATCA, which codes for SS). The complete sequence of the MCS was CTCACTGGTGGAACCGGTCGTACGTCATCACCCGGGACGCGTGGAGGAGGC (nucleotides that belong to the ubiquitin repeat are shown in italics, and those that form the sites of the restriction enzymes described above are shown in bold).
The cloning process was as follows: the SpeI-Ubiq-BssHII DNA fragment was first cloned into the pCR2.1 vector (Invitrogen, San Diego, CA). Then, using the QuickChange kit (Stratagene), we inserted the MCS sequence in two steps: first the GGTGGAACCGGTCGTACGTCA sequence and then the TCACCCGGGACGCGTGGAGGA sequence. Finally, we cloned the SpeI-Ubiq+MCS-BssHII DNA fragment into the pFS-1 via SpeI-BssHII.
Cloning guest proteins into pFS-2
To clone the I27 module inside the MCS (pFS-2+I27), we selected BsiWI and MluI sites. Therefore, the GGTGRS and TRGG amino acid sequences of the MCS remained in the N- and C-termini of the I27 module, respectively (Fig. 4 a). For PCR amplification we used as a template a plasmid containing 12 repeats of the I27 module (1). The cytoplasmic region (residues 1–94) of VAMP2 from Rattus norvegicus was cloned into AgeI-SmaI restriction sites (pFS-2+VAMP2), leaving the amino acids GGTG and PGTRGG of the MCS on either side of VAMP2. We used a plasmid containing the entire VAMP2 sequence (pGEX-KGVAMP2 (28)) as a template for PCR amplification.
In all of these cloning steps, we used the XL1-Blue E. coli strain (Stratagene). We verified all of the sequences by sequencing both strands of the DNA.
Expression and purification of the pFS-1 and pFS-2 proteins
To optimize the production of pFS-1 and pFS-2 recombinant proteins, we tested a battery of E. coli strains for induction and yield as well as for minimizing degradation of the recombinant protein. Specifically, we tested the C41 (DE3) (29), BL21star (DE3) (Invitrogen), and M15[pREP4] (Qiagen) strains with the pFS-1 and pFS-2 constructs inserted into either the pRSETA or pQE80L vectors. Cells were grown at 37°C shaking at 300 rpm until an OD595 of 0.5–0.8 was reached, and expression was induced by addition of 1 mM of isopropyl β-D-thiogalactopyranoside (IPTG) for 4 h. We found that the best combination was that of the pRSETA plasmid and the C41(DE3) strain (Figs. 2 b and 3 c) (29).
Bacterial cells were lysed by treatment with 1 mg/ml lysozyme and 1% Triton X-100 (30). We purified the recombinant proteins by Ni2+-affinity chromatography using Histrap HP FPLC columns (GE Healthcare, Uppsala, Sweden) on an FPLC apparatus (ÄKTA Purifier, GE Healthcare) using 50 mM sodium phosphate buffer/500 mM NaCl (pH 7.4) with 50 mM and 500 mM imidazole in the binding and elution buffers, respectively. The purified fractions were concentrated and the buffer exchanged to phosphate buffer saline (PBS)/5 mM DTT by ultrafiltration using Amicon 10K filters (Millipore; Billerica, MA). After a brief sonication pulse, we further purified the sample by Strep-tag affinity purification using Streptrap HP FPLC columns (GE Healthcare). The binding buffer was PBS and the elution buffer was PBS/2.5 mM desthiobiotin. For pFS-2+VAMP2, after the Ni2+-affinity chromatography purification step, the sample was further purified by size exclusion chromatography using a HiLoad 16/60 Superdex TM 200 column (GE Healthcare) with 100 mM TrisHCl buffer (pH 7.0) containing 1.25 M guanidinium chloride (this concentration does not denature the ubiquitin repeat and it allows the contaminants that coelute with the protein to be removed) (31). The purified fractions were concentrated again and the buffer was exchanged to PBS/5 mM DTT/0.2 mM EDTA (pH 7.4; for pFS-1 and pFS-2+I27) or TrisHCl 10 mM/5 mM DTT (pH 7.5; for pFS-2+VAMP2) by ultrafiltration using Amicon 10K filters (Millipore).
Although the yield of pure recombinant protein was not very high (∼1 mg pure protein per liter of bacterial culture), it was very pure (>95%). Thus, a single band was observed in Coomassie Blue (CB)-stained gels, as well as in Western blots probed with antibodies against the two purification tags present in the pFS-1/pFS-2 proteins (Figs. 2 c and 3 d).
AFM-SMFS of the pFS-1 and pFS-2 proteins
The AFM we used is a slightly modified version of a previously described in-house-made AFM (32) with added imaging capabilities (33). We determined the spring constant of each Si3N4 cantilever (MLCT-AUNM; Veeco Metrology Group, Santa Barbara, CA; and BioLever, Olympus, Tokyo, Japan) using the equipartition theorem (34), and found that it ranged from 35 to 70 pN/nm for MLCT-AUNM and ∼30 pN/nm for Biolever cantilevers. The noise calculated for the cantilevers was ∼6 pN (13). All experiments were performed in the so-called length-clamp mode of the AFM (1) at a constant pulling speed of 0.4 nm/ms. The elasticity of the stretched proteins was analyzed fitting the force curves to the WLC model (3,35):
where F is the force, p is the persistence length, x is the end-to-end length, and Lc is the contour length of the stretched protein. Lc and p are the adjustable parameters.
We used two different types of AFM substrates to test the efficiency of pulling. The pFS vectors can be covalently attached to gold-coated coverslips through the cysteine residues located at the C-terminus (20), or to Ni2+-NTA functionalized glass coverslips through the His-tag present at the N-terminus (19). A drop of pure protein preparation (∼2–8 μl, ∼0.2–0.5 mg/ml) was applied on top of a drop of the corresponding buffer deposited onto the substrate and allowed to adsorb for ∼10 min. These methods were described previously (1,12,13,36), and the efficiency of pulling in both substrates was comparable and the results obtained were identical.
To validate pFS-1, we only selected recordings that showed the spacer region of 50–80 nm (∼80 nm if the protein is being pulled from its termini), resulting from the stretching of the N2B fragment, followed by no more than six force peaks from the mechanical unfolding of the ubiquitin repeats (Fig. 2 d). For the carrier-guest strategy using hosted proteins, apart from the spacer region and several force peaks that originate from the unfolding of the ubiquitin repeats, the recordings should always show the force peak that originates from the unfolding of the carrier ubiquitin repeat (easily identified by its larger ΔLc value; Fig. 3 e) before the stretching of the guest protein (Fig. 4, a and d) (23,36,37).
All of the data reported here were analyzed with the use of Igor Pro 6.0 (Wavemetrics, Lake Oswego, OR) and are expressed as the mean value ± standard deviation.
Results and Discussion
pFS series of expression vectors
The pFS-1 is a cloning/expression vector that encodes for a fusion heteropolyprotein that facilitates the analysis of any protein of interest (but particularly for structured proteins) by AFM-SMFS. It is formed by several human ubiquitin repeats (UniProtKB/Swiss-Prot code P0CG47) and a fragment of the N2B protein from human cardiac titin (UniProtKB/Swiss-Prot code Q8WZ42). The former serves as a single-molecule marker, and the latter serves as a spacer that situates the force events of the polyprotein and those of the protein of interest at a distance from the proximal region of the force-extension recordings, which is usually contaminated (Figs. 1 and 2). This vector also contains a variety of restriction sites between the repeats that facilitate the cloning of any protein of interest into it (Fig. 2 a). Furthermore, it contains two terminal tags (one at each terminus) to optimize the purification of the full-length form of the recombinant protein. In a second version of the vector, pFS-2, one ubiquitin unit contains an MCS in a tolerant force-hidden loop into which proteins with low and/or complex/polymorphic mechanostability (e.g., some IDPs) can be cloned. This strategy protects the protein of interest behind the mechanical clamp of the ubiquitin carrier, facilitating the selection of unambiguous force recordings (Figs. 3 and 4).
Nanomechanics of pFS vectors
We performed AFM-SMFS on the purified pFS-1 and pFS-2 proteins to show their full potential (Figs. 2 d, 3 e, and 4). The N2B fragment was seen to behave as a random coil IDP, as does the full-length protein (18). It contains 204 amino acids, which upon stretching from its termini should display an extension of ∼81 nm (nominal extension, considering a gain in contour length of 0.4 nm per stretched amino acid) (4). Any force peak that appeared in this region was discarded from our analyses. Ubiquitin repeats showed an Fu of 189 ± 37 pN and a ΔLc of 23.4 ± 0.5 nm (n = 574; Fig. 4, b and c), which are, in principle, comparable to those previously reported (16). The slightly lower Fu values observed here may reflect the influence of the different linker sequences that flank ubiquitin in the pFS vectors (which are close to the mechanical clamp of the ubiquitin repeats and may therefore slightly affect their mechanical stability by steric/electrostatic effects) or possible unspecific interactions between modules in the polyprotein. These effects have been observed in other proteins (12,38–40). The ubiquitin repeat carrying the MCS in pFS-2 had an Fu of 185 ± 41 pN and a ΔLc of 28.3 ± 0.6 nm (n = 55), indicating that the mechanical stability of this module is not affected by insertion of the MCS. The gain in contour length of ∼5 nm is the result of the extra residues added by the MCS (14 amino acids × 0.4 nm/amino acid ≈ 5 nm (4)).
It should be noted that, in principle, the presence of a relatively long elastic linker in the fusion heteropolyprotein may alter the mechanostability of the protein under study (41). However, this effect is estimated to be a mere increase of 0.1–0.2 pN/nm (42). In our setup and experimental conditions, using I27 polyproteins (1), we estimated that this effect would result in an increase of 10 pN every 100 nm of extension (J. Oroz and M. Carrión-Vázquez, data not shown). Hence, the stretching of the N2B fragment present in the pFS vectors would result in an increase of 8–16 pN in the Fu of the protein under study, a force value that is roughly within the error range of the measurements. Thus, this effect can be considered negligible for most purposes.
Nanomechanics of a structured protein grafted in pFS-2: titin I27 module
To validate the carrier-guest strategy, we first used a protein module with a defined structure and well-known mechanical properties, the I27 module from human cardiac titin (UniProtKB/Swiss-Prot code Q8WZ42 (1)). As expected from our design, the carrier ubiquitin repeat of the resulting fusion protein (termed pFS-2+I27) unfolded before the force-protected I27 module did (Fig. 4 a) (23). This I27 module grafted within the ubiquitin carrier had an Fu of 203 ± 20 pN and a ΔLc of 27.2 ± 0.4 nm (n = 13; Fig. 4, b and c), in close agreement with previously reported values (1). Likewise, the carrier ubiquitin repeat had an Fu of 179 ± 46 pN and a ΔLc of 30.0 ± 0.9 nm (n = 13). This increase in ΔLc of ∼7 nm compared with a regular ubiquitin corresponds to the length of the remaining MCS (10 residues = 4 nm) (4) and the folded I27 included inside this carrier ubiquitin (the size of folded I27 is 3–4 nm (1)). Hence, the mechanical stability of both modules did not appear to be modified in the carrier-guest construct, demonstrating that the ubiquitin carrier can be used to graft folded proteins inside its fold with virtually no change in the mechanostabilities of both proteins. However, there appear to be limitations in the loading capabilities of the carrier that may compromise its mechanostability (J. Oroz and M. Carrión-Vázquez, unpublished results). In fact, as reported previously, the grafting of proteins with distant N-/C-termini may affect the fold of the carrier protein and lead to the mutual exclusiveness of the folding between the carrier and guest proteins (37,43,44). A possible solution for this negative effect would be to include long flexible linkers between both modules to reduce the steric effects that impede their folding (44). Indeed, our results showing the preserved fold for the carrier ubiquitin and the guest I27 (with distant N-/C-termini) in the pFS-2+I27 were somewhat surprising considering previous work (37). This unexpected success may be due to the inclusion of several glycine residues (44) in the MCS of the carrier ubiquitin. In the future, the extension of this glycine region may avoid possible problems with larger proteins.
Nanomechanics of a nonfibrillogenic IDP grafted in pFS-2: VAMP2
We next tested whether the carrier-guest strategy was suitable for carrying out an unambiguous nanomechanical analysis of IDPs. Accordingly, we cloned the cytoplasmic region (amino acids 1–94) of VAMP2 (a.k.a. synaptobrevin) from R. norvegicus (UniProtKB/Swiss-Prot code P63045), a component of the SNARE complex (45), within the carrier ubiquitin repeat. This nonfibrillogenic IDP was expected to behave as a random coil and thus shows no force peaks when stretched at the usual pulling velocity (18). As expected (23,37), the force-extension recordings had a force peak originated from the mechanical unfolding of the ubiquitin carrier, followed by the stretching of the guest VAMP2 with no mechanical resistance other than entropic elasticity. The ΔLc value obtained was 63 nm, which corresponds to the length of the unfolded carrier ubiquitin repeat plus that of synaptobrevin (∼26 nm from this carrier ubiquitin, as four residues from its MCS were removed for VAMP2 cloning, and ∼37 nm from the stretching of VAMP2; Fig. 4 d) (4). Moreover, the carrier ubiquitin repeat again displayed a mechanical behavior comparable to that of regular ubiquitin.
An alternative version of the pFS-2 in which titin I27 was used as a carrier module instead of ubiquitin, pFS-2/I27, was also used to host VAMP2 (cloned in an MCS formed by AgeI and SmaI restriction sites, located between residues A42 and A43 in the CD loop of I27), and gave comparable results (22). Although the FG loop is quite variable in length among titin Ig modules, and was successfully used to insert glycine residues (46), in practice the CD loop seemed to tolerate insertions better (H. Li and J. M. Fernández, Columbia University, personal communication, 2002).
Conclusion
The standard heteropolyprotein strategy that has been used to date for AFM-SMFS relies on the use of several repeats of a marker protein arranged in series, such that the unfolding of a protein with lower mechanostability than the marker occurs at the beginning of the force-extension recordings (Figs. 1–5). However, this strategy has severe drawbacks for analyzing low-mechanical stability proteins or IDPs by AFM-SMFS (Fig. 1) (11).
Figure 5.
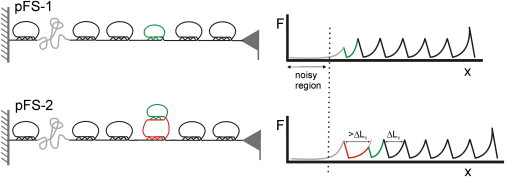
Advantages of the pFS family of vectors for AFM-SMFS: the spacer and the carrier guest strategies. When the protein of interest has a lower mechanical stability (small ring, top) than the single-molecule marker (larger rings, black), the use of the standard heteropolyprotein strategy, with the mechanical clamps of the protein marker arranged in series, often results in data contaminated with spurious force peaks that originate from nonspecific sources in the noisy region of the recordings (proximal to substrate). The use of the N2B region allows the data from the protein of interest to appear away from this region. The pFS-1 (left panel shows its mechanical representation) is sufficient for proteins with unique or simple patterns of force peaks. In the case of proteins with complex or polymorphic patterns of force peaks, the force peak of the “carrier” (larger ring) in the pFS-2 (cartoon shows its mechanical representation) serves to announce the latter appearance of the events of the protein of interest (combined with the N2B strategy allows the carrier to appear away from the proximal region). Thus, using the carrier-guest strategy, the mechanical determinants of the protein under study (small ring connected to the larger ring) are force-hidden behind the mechanical clamp of the carrier, so that the protein under study will always be stretched after (though not necessarily immediately after) the carrier protein, and thus will be at a distance from the noisy proximal region of the recording. The ubiquitin carrier protein can readily be identified in the force-extension recordings because it has a larger ΔLc value (>ΔLc in the figure) than that of a regular ubiquitin repeat (black). Note that by using a standard heteropolyprotein strategy, the features from the protein of interest can appear in the noisy region.
We developed a family of cloning/expression vectors (pFS) for general use in AFM-SMFS (Figs. 2, 3, and 5). The expression of any protein in this vector produces a fusion heteropolyprotein that can be unequivocally analyzed by AFM-SMFS. Furthermore, the expression and purification protocols are optimized such that very pure preparations of the full-length protein can be easily obtained. This vector contains several elements that make this robust and powerful approach the system of choice for typical AFM-SMFS applications. pFS-2 provides a new, to our knowledge, strategy (the carrier-guest approach) that is extremely useful for the unequivocal nanomechanical analysis of proteins with low mechanical stability, and particularly for proteins with complex/polymorphic mechanostability, such as fibrillogenic IDPs (Fig. 4) (22).
We also present a proof-of-concept to demonstrate the advantages of using these new vectors and the carrier-guest strategy to unambiguously analyze low-mechanical-stability proteins and IDPs (Figs. 4 and 5) (12,23). The importance of AFM-SMFS for studying IDPs is highlighted by the causal role played by amyloidogenic IDPs in several diseases (21). Here, we analyzed an IDP, the VAMP2 protein (45), that is not fibrillogenic or related to any amyloidogenic disease. This protein is assumed to be a random coil, and accordingly it offered no mechanical resistance in AFM-SMFS. Of more importance, the use of the pFS-2 vector also allows the unambiguous mechanical analysis of fibrillogenic IDPs, specifically those that are known to be causally related to neurodegenerative diseases (i.e., neurotoxic proteins) and to adopt different conformations (47). This analysis revealed that these proteins display a broad mechanical polymorphism, ranging from abundant low-mechanostability conformers to low-abundance conformers with very high mechanostability (22). Of interest, the unequivocal characterization of the conformational polymorphism and the discovery of hypermechanostable conformers in those proteins could only be achieved by means of the carrier-guest strategy described here. However, and particularly when analyzing the nanomechanics of proteins with a tendency to oligomerize (e.g., neurotoxic proteins), the strategy presented here only guarantees the stretching of single molecules. The analyzed molecule could still be involved in a series of interactions (with the AFM elements, substrate or cantilever tip, and other surrounding IDP molecules forming oligomers). To discard this possibility, one should employ additional controls, such as using inhibitors of the oligomerization process (22) or performing refolding experiments in buffer that is devoid of IDPs.
Acknowledgments
We thank R. Scheller for the pGEX-KGVAMP2 clone. We also thank the members of M.C.-V.'s laboratory for discussions and critical comments on the manuscript.
M.C.-V. designed the research, analyzed the results, and wrote the final draft of the paper. J.O. performed the experiments, analyzed the results, and wrote the first draft of the article. R.H. performed the experiments related to VAMP2.
The authors declare a competing financial interest: M.C.-V., J.O., and R.H. are the co-inventors on an international patent application (No. P 201031846, PCT/ES2011/070867) covering the results contained in this article. Any potential income generated by exploitation of the patent rights and received by their employer, the Consejo Superior de Investigaciones Científicas (CSIC), shall be shared with these authors according to Spanish law and the regulations of the CSIC.
This work was funded by grants from the Ministerio de Ciencia e Innovación (BIO2007-67116), the Consejería de Educación de la Comunidad de Madrid (S-0505/MAT/0283), and the CSIC (200620F00) to M.C.-V. J.O. and R.H. received fellowships from the Consejería de Educación de la Comunidad de Madrid and the Fundación Ferrer (Severo Ochoa fellowship), respectively.
References
- 1.Carrión-Vazquez M., Oberhauser A.F., Fernandez J.M. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog. Biophys. Mol. Biol. 2000;74:63–91. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 2.Yang G., Cecconi C., Bustamante C. Solid-state synthesis and mechanical unfolding of polymers of T4 lysozyme. Proc. Natl. Acad. Sci. USA. 2000;97:139–144. doi: 10.1073/pnas.97.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bustamante C., Marko J.F., Smith S. Entropic elasticity of λ-phage DNA. Science. 1994;265:1599–1600. doi: 10.1126/science.8079175. [DOI] [PubMed] [Google Scholar]
- 4.Ainavarapu S.R., Brujic J., Fernandez J.M. Contour length and refolding rate of a small protein controlled by engineered disulfide bonds. Biophys. J. 2007;92:225–233. doi: 10.1529/biophysj.106.091561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li H., Oberhauser A.F., Fernandez J.M. Multiple conformations of PEVK proteins detected by single-molecule techniques. Proc. Natl. Acad. Sci. USA. 2001;98:10682–10686. doi: 10.1073/pnas.191189098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Best R.B., Li B., Clarke J. Can non-mechanical proteins withstand force? Stretching barnase by atomic force microscopy and molecular dynamics simulation. Biophys. J. 2001;81:2344–2356. doi: 10.1016/S0006-3495(01)75881-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dietz H., Rief M. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc. Natl. Acad. Sci. USA. 2004;101:16192–16197. doi: 10.1073/pnas.0404549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steward A., Toca-Herrera J.L., Clarke J. Versatile cloning system for construction of multimeric proteins for use in atomic force microscopy. Protein Sci. 2002;11:2179–2183. doi: 10.1110/ps.0212702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.García-Manyes S., Brujić J., Fernández J.M. Force-clamp spectroscopy of single-protein monomers reveals the individual unfolding and folding pathways of I27 and ubiquitin. Biophys. J. 2007;93:2436–2446. doi: 10.1529/biophysj.107.104422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.García-Manyes S., Dougan L., Fernández J.M. Direct observation of an ensemble of stable collapsed states in the mechanical folding of ubiquitin. Proc. Natl. Acad. Sci. USA. 2009;106:10534–10539. doi: 10.1073/pnas.0901213106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandal M., Valle F., Samorì B. Conformational equilibria in monomeric α-synuclein at the single-molecule level. PLoS Biol. 2008;6:e6. doi: 10.1371/journal.pbio.0060006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oroz J., Valbuena A., Carrión-Vázquez M. Nanomechanics of the cadherin ectodomain: “canalization” by Ca2+ binding results in a new mechanical element. J. Biol. Chem. 2011;286:9405–9418. doi: 10.1074/jbc.M110.170399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valbuena A., Oroz J., Carrión-Vázquez M. On the remarkable mechanostability of scaffoldins and the mechanical clamp motif. Proc. Natl. Acad. Sci. USA. 2009;106:13791–13796. doi: 10.1073/pnas.0813093106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Best R.B., Brockwell D.J., Clarke J. Force mode atomic force microscopy as a tool for protein folding studies. Anal. Chim. Acta. 2003;479:87–105. [Google Scholar]
- 15.Rounsevell R., Forman J.R., Clarke J. Atomic force microscopy: mechanical unfolding of proteins. Methods. 2004;34:100–111. doi: 10.1016/j.ymeth.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 16.Carrión-Vazquez M., Li H., Fernandez J.M. The mechanical stability of ubiquitin is linkage dependent. Nat. Struct. Biol. 2003;10:738–743. doi: 10.1038/nsb965. [DOI] [PubMed] [Google Scholar]
- 17.Finley D., Bartel B., Varshavsky A. The tails of ubiquitin precursors are ribosomal proteins whose fusion to ubiquitin facilitates ribosome biogenesis. Nature. 1989;338:394–401. doi: 10.1038/338394a0. [DOI] [PubMed] [Google Scholar]
- 18.Li H., Linke W.A., Fernandez J.M. Reverse engineering of the giant muscle protein titin. Nature. 2002;418:998–1002. doi: 10.1038/nature00938. [DOI] [PubMed] [Google Scholar]
- 19.Hossain M.D., Furuike S., Kinosita K., Jr. The rotor tip inside a bearing of a thermophilic F1-ATPase is dispensable for torque generation. Biophys. J. 2006;90:4195–4203. doi: 10.1529/biophysj.105.079087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rief M., Gautel M., Gaub H.E. Reversible unfolding of individual titin immunoglobulin domains by AFM. Science. 1997;276:1109–1112. doi: 10.1126/science.276.5315.1109. [DOI] [PubMed] [Google Scholar]
- 21.Fink A.L. Natively unfolded proteins. Curr. Opin. Struct. Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Hervás R., Oroz J., Carrión-Vázquez M. Common features at the start of the neurodegeneration cascade. PLoS Biol. 2012 doi: 10.1371/journal.pbio.1001335. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng Q., Li H. Domain insertion effectively regulates the mechanical unfolding hierarchy of elastomeric proteins: toward engineering multifunctional elastomeric proteins. J. Am. Chem. Soc. 2009;131:14050–14056. doi: 10.1021/ja903589t. [DOI] [PubMed] [Google Scholar]
- 24.Leake M.C., Grützner A., Linke W.A. Mechanical properties of cardiac titin's N2B-region by single-molecule atomic force spectroscopy. J. Struct. Biol. 2006;155:263–272. doi: 10.1016/j.jsb.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 25.Ferraro D.M., Hope E.K., Robertson A.D. Site-specific reflex response of ubiquitin to loop insertions. J. Mol. Biol. 2005;352:575–584. doi: 10.1016/j.jmb.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 26.Ferraro D.M., Ferraro D.J., Robertson A.D. Structures of ubiquitin insertion mutants support site-specific reflex response to insertions hypothesis. J. Mol. Biol. 2006;359:390–402. doi: 10.1016/j.jmb.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 27.Ferraro D.M., Robertson A.D. Predicting the magnitude of the reflex response to insertions in ubiquitin. J. Mol. Biol. 2008;375:764–772. doi: 10.1016/j.jmb.2007.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Calakos N., Bennett M.K., Scheller R.H. Protein-protein interactions contributing to the specificity of intracellular vesicular trafficking. Science. 1994;263:1146–1149. doi: 10.1126/science.8108733. [DOI] [PubMed] [Google Scholar]
- 29.Miroux B., Walker J.E. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 30.Sambrook J., Fritsch E.F., Maniatis T. 2nd ed. Cold Spring Harbor; New York: 1989. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- 31.Went H.M., Benítez-Cardoza C.G., Jackson S.E. Is an intermediate state populated on the folding pathway of ubiquitin? FEBS Lett. 2004;567:333–338. doi: 10.1016/j.febslet.2004.04.089. [DOI] [PubMed] [Google Scholar]
- 32.Schlierf M., Li H., Fernández J.M. The unfolding kinetics of ubiquitin captured with single-molecule force-clamp techniques. Proc. Natl. Acad. Sci. USA. 2004;101:7299–7304. doi: 10.1073/pnas.0400033101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valbuena A., Oroz J., Carrión-Vázquez M. Quasi-simultaneous imaging/pulling analysis of single polyprotein molecules by atomic force microscopy. Rev. Sci. Instrum. 2007;78:113707. doi: 10.1063/1.2794732. [DOI] [PubMed] [Google Scholar]
- 34.Florin E.-L., Rief M., Gaub H.E. Sensing specific molecular interactions with the atomic force microscope. Biosens. Bioelectron. 1995;10:895–901. [Google Scholar]
- 35.Marko J.F., Siggia E.D. Stretching DNA. Macromolecules. 1995;28:8759–8770. [Google Scholar]
- 36.Oroz J., Hervás R., Carrión-Vázquez M. Unequivocal single-molecule force spectroscopy of intrinsically disordered proteins. In: Uversky V.N., Dunker A.K., editors. Intrinsically Disordered Proteins: Vol. I. Experimental Techniques. Springer-Verlag; Heidelberg, Germany: 2012. In press. [Google Scholar]
- 37.Peng Q., Li H. Direct observation of tug-of-war during the folding of a mutually exclusive protein. J. Am. Chem. Soc. 2009;131:13347–13354. doi: 10.1021/ja903480j. [DOI] [PubMed] [Google Scholar]
- 38.Politou A.S., Gautel M., Pastore A. Immunoglobulin-type domains of titin are stabilized by amino-terminal extension. FEBS Lett. 1994;352:27–31. doi: 10.1016/0014-5793(94)00911-2. [DOI] [PubMed] [Google Scholar]
- 39.Scott K.A., Steward A., Clarke J. Titin; a multidomain protein that behaves as the sum of its parts. J. Mol. Biol. 2002;315:819–829. doi: 10.1006/jmbi.2001.5260. [DOI] [PubMed] [Google Scholar]
- 40.Rounsevell R.W.S., Steward A., Clarke J. Biophysical investigations of engineered polyproteins: implications for force data. Biophys. J. 2005;88:2022–2029. doi: 10.1529/biophysj.104.053744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Evans E., Ritchie K. Strength of a weak bond connecting flexible polymer chains. Biophys. J. 1999;76:2439–2447. doi: 10.1016/S0006-3495(99)77399-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zinober R.C., Brockwell D.J., Smith D.A. Mechanically unfolding proteins: the effect of unfolding history and the supramolecular scaffold. Protein Sci. 2002;11:2759–2765. doi: 10.1110/ps.0224602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Radley T.L., Markowska A.I., Loh S.N. Allosteric switching by mutually exclusive folding of protein domains. J. Mol. Biol. 2003;332:529–536. doi: 10.1016/s0022-2836(03)00925-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cutler T.A., Mills B.M., Loh S.N. Effect of interdomain linker length on an antagonistic folding-unfolding equilibrium between two protein domains. J. Mol. Biol. 2009;386:854–868. doi: 10.1016/j.jmb.2008.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellena J.F., Liang B., Tamm L.K. Dynamic structure of lipid-bound synaptobrevin suggests a nucleation-propagation mechanism for trans-SNARE complex formation. Proc. Natl. Acad. Sci. USA. 2009;106:20306–20311. doi: 10.1073/pnas.0908317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carrión-Vazquez M., Marszalek P.E., Fernandez J.M. Atomic force microscopy captures length phenotypes in single proteins. Proc. Natl. Acad. Sci. USA. 1999;96:11288–11292. doi: 10.1073/pnas.96.20.11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uversky V.N. In: Protein Folding and Misfolding: Neurodegenerative Diseases. Ovádi J., Orosz F., editors. Springer-Verlag; Berlin-Heidelberg, Germany: 2009. pp. 21–75. [Google Scholar]
- 48.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]