

Abstract
Despite advances in medical and surgical therapy, cancer kills more than half a million people in the United States annually, and the majority of these patients succumb to metastatic disease. The traditional approach to treating systemic disease has been the use of cytotoxic chemotherapy. However, chemotherapy is rarely curative and toxicity is often dose limiting. In addition, the effects of chemotherapy are nonspecific, targeting both malignant and normal tissues. As a result, recent efforts increasingly have focused on developing agents that target specific molecules in tumor cells in order to both improve efficacy and limit toxicity. This review summarizes the history and current use of targeted molecular therapy for cancer, with a special emphasis on recently developed inhibitors of Focal Adhesion Kinase (FAK).
Keywords: Angiogenesis, carcinogenesis, focal adhesion kinase, targeted therapeutics, tyrosine kinase
INTRODUCTION
More than 550,000 people in the United States die from cancer annually, and the majority of these succumb to metastatic disease [1]. The traditional approach to treating systemic disease has been the use of cytotoxic chemotherapy. However, chemotherapy is rarely curative and toxicity is often dose limiting. Moreover, the effects of chemotherapy are nonspecific, targeting both malignant and normal tissues. As a result, recent efforts increasingly have focused on developing agents that target specific molecules in tumor cells in order to both improve efficacy and limit toxicity [2].
TYROSINE KINASE INHIBITORS
One of the first classes of molecules to be suggested as a potential target for anticancer therapy was the tyrosine kinases (TKs). As their name implies, these enzymes catalyze the phosphorylation of tyrosine kinase moieties on a variety of other signaling proteins [3]. TKs can be classified as either receptor or non-receptor kinases. Receptor tyrosine kinases (RTKs) are membrane spanning proteins that generally function in pairs. Activation induces dimerization, and dimerization then triggers a phosphorylation cascade involving the cytosolic non-receptor tyrosine kinases (Fig. 1) [3–6]. Non-receptor tyrosine kinases relay intracellular signals by additional protein phosphorylation [6]. Many processes involved in tumor progression and metastasis, including proliferation, angiogenesis, migration and cell survival, are influenced by activation of RTKs and the subsequent signal cascade [3, 5]. Moreover, mutations can alter the expression or activation of TKs and thereby lead to uncontrolled activation in cancer cells [7]. Of the fifty-eight known RTKs, approximately 30% have been found to be either mutated or unregulated in cancer [4, 7, 8]. In addition, several RTKs are known to play important roles in tumorigenesis, and have become effective therapeutic targets. For example, c-kit is unregulated in gastrointestinal stromal tumors (GISTs) and acute myelogenous leukemia (AML), HER2 is elevated in breast cancer, and RET is altered the MEN disorders [7]. These and other RTKs have long been known to play a role in cancer progression and, as such, inhibition of TKs has been at the forefront of targeted cancer therapy.
Fig. 1.

Activation of an RTK induces dimerization and downstream phosphorylation of non-receptor tyrosine kinases. Major signaling cascades include the RAS/RAF/MAPK pathway, MEK pathway, and the PI3kinase/AKT pathway.
(Adapted by permission from Macmillan Publishers Ltd: Nature Reviews Cancer, Zhu & Parada, Nature Reviews Cancer, 2002, 2: 616–626, copyright 2002.)
The recognition that TKs play a central role in many tumorigenic processes prompted the development of a wide variety of TK inhibitors (TKIs). These agents function by targeting the receptor or the ligand that promotes the downstream signal cascade [3]. TKIs can be classified based upon the targeted molecule or the targeted signal cascade. Increasingly, however, these agents are classified based upon molecular structure; this classification divides TKIs into the following groups: (1) monoclonal antibodies, (2) small molecule kinase inhibitors, and (3) multitargeted kinase inhibitors (Table 1).
Table 1.
Tyrosine Kinase Inhibitors [2]
| Monoclonal Antibodies | Imitinib |
|---|---|
| Trastuzumab | |
| Cetuximab | |
| Panitumumab | |
| Small Molecule Inhibitors | Gefitinib |
| Erlotinib | |
| Multitargeted Inhibitors | Lapatinib |
| Dasatinib | |
| Sunitinib | |
| Ax+itinib | |
| Vandetanib |
Monoclonal Antibodies
Monoclonal antibodies were at the forefront of TKI development. The first TKI developed for cancer therapy was imatinib mesylate, designed to treat chronic myelogenous leukemia (CML). The development of this drug was based upon the observation that most patients with CML possess the t(9;22) translocation mutation (Philadelphia chromosome) that fuses the Abl tyrosine kinase to the Bcr gene on chromosome 22 [3, 9]. This mutation then results in the production of two tyrosine kinases, p190 and p210, both of which are involved in hematopoetic stem cell proliferation and inhibition of apoptosis [3]. Imatinib, therefore, was designed to block the ATP binding site on these proteins, hindering activation and downstream signaling. This prototypical TKI revolutionized the treatment of CML and, as such, laid the ground work for the development of a wide array of similar TKIs [10].
In addition to blocking ATP binding in p190 and p210, imatinib was also found to inhibit the c-kit and PDGFR tyrosine kinases. After demonstrated success in treating CML, imatinib was then tested in patients with tumors possessing mutations in c-kit and PDGFR [11]. Gastrointestinal stromal tumors (GISTs), for example, are notoriously resistant to cytotoxic chemotherapy, but are well known to express c-kit. Imatinib therapy proved to be highly successful in these unusual tumors and has significantly altered the approach to treating patients with GIST [12]. Currently, there are several clinical trials designed to test the efficacy of imatinib in other solid tumors including colorectal cancer, ovarian cancer, prostate cancer and thyroid cancer [13]. Nevertheless, despite successes with this agent, prolonged use of imatinib has resulted in the development of resistant tumors. The most common mechanism of resistance appears to involve mutations in the kinase domain of bcr-abl that then prevent binding of the drug [14]. As a result, current research is focusing on overcoming this limitation.
The epidermal growth factor receptor family of TKs has also provided a number of targets for TK inhibition. Epidermal growth factor receptor 2 (EGFR2, ErbB2), also known as the human epidermal receptor 2 (HER2) for example, can bind a wide array of growth factors, thereby inducing dimerization and stimulating downstream activation of either the RAS/MAPK, PI3 kinase/Akt/mTOR, or other signaling pathways, that promote cell proliferation and migration and inhibit apoptosis [15, 16], [15] HER2 is over expressed in many malignancies, and between 20–30% of advanced breast cancers express this receptor [15, 17]. Trastuzumab is a humanized monoclonal antibody that inhibits activation of the HER2 receptor has been shown to affect breast cancer cells by inducing cell cycle arrest at G1 via up regulation and translocation of p27 from cytosol to nuclei [15, 18, 19], Trastuzumab has demonstrated efficacy against both early and late stage breast cancer, and is currently the standard of care for HER2 positive tumors [20, 21].
In addition, trials are underway pairing trastuzumab with more traditional chemotherapeutic agents in an attempt to both improve efficacy and to decrease toxicity [20]. The recent phase III ToGA trial has suggested that trastuzumab may be a reasonable adjunct to capecitabine and cisplatin or fluorouracil and cisplatin for patients with HER2-positive gastric or gastroesophageal junction cancer [22, 23]. Trials are also underway to test the efficacy of trastuzumab in combination with other targeted therapies. For example, trastuzumab-DM1 is a combination of the monoclonal antibody and the maytansine-derivative, DM1, a drug that interferes with microtubule formation. Pre-clinical studies demonstrate efficacy in lapatinib and trastuzumab-refractory breast cancer cells, and this combination is currently in phase II clinical trials for the treatment of breast cancer [24–27].
Another TKI that has proven useful is cetuximab, a monoclonal antibody that targets epidermal growth factor receptors [5, 28]. The history of cetuximab development is interesting. Initially, a murine monoclonal antibody (called 225) that could bind and inhibit EGFR was found to increase apoptosis and cell cycle arrest in G1 [29]. Experimental data were promising, however concern about the use of a mouse monoclonal antibody in the human population, and the potential for anti-mouse antibody response, prompted chimerization of the antibody with human IgG1 [30, 31]. The resulting chimeric antibody (C225, cetuximab) has subsequently proven to be both safe and efficacious in a number of settings [31, 32]. Cetuximab in conjunction with cytotoxic chemotherapy improves progression free survival and overall response rate in chemoresistant metastatic colorectal cancer [5, 33]. In 2004, cetuximab either in combination with irinotecan or as a single agent was approved by the FDA for the treatment of metastatic colorectal cancers that express EGFR and are refractory to irinotecan- or oxaliplatin-based therapies [5, 34, 35]. Another interesting finding with cetuximab has been the identification of KRAS mutation status as a predictor of response to therapy. As cetuximab increasingly was used to treat refractory metastatic colorectal cancer, it became clear that this agent is most effective in a subset of patients whose tumors possessed wild-type KRAS [33, 36–38]. In contrast, tumors possessing a KRAS mutation responded poorly to cetuximab [39–41]. As a result, current practice requires KRAS testing for patients under consideration for treatment with cetuximab [42, 43]. Finally, cetuximab in combination with chemotherapy has proven useful in several other malignancies, including head and neck squamous cell carcinoma, and is under investigation for use in other cancers. Additional EGFR inhibitors (panitumumab, pertuzumab) also are being studied and hold promise [43–48].
Small Molecule TKIs
The second class of TKIs includes several small molecules that inhibit activation of these enzymes. For example, Gefitinib is a small molecule inhibitor that targets the epidermal growth factor receptor 1 (EGFR1/HER1) by inhibiting autophosphorylation [27, 49, 50] Gefitinib was originally approved for the treatment of non-small cell lung cancer in 2003. However, a variety of later trials have shown mixed response [51–55]. Erolotinib, another small molecule inhibitor of EGFR autophosphorylation, has had somewhat greater success. In a large trial treating patients with advanced non-small cell lung cancer (stage IIIB or IV), erlotinib therapy prolonged progression free and overall survival, and is currently considered second line therapy for treating non-small cell lung cancer [56–59]. Erlotinib also has proven efficacy in proved in combination with gemcitabine for treating pancreatic cancer [59].
Multitargeted Inhibitors
The third class of TKIs, multitargeted inhibitors, includes a variety of agents with varied mechanisms of action. The uniting factor among these molecules is their ability to inhibit multiple TKs, and, with hope, to overcome the development of resistance. For example, several molecules are designed to inhibit not only EGFR/HER2 activation, for example, but also downstream cell signaling molecules such as the src family of kinases, and proteins such as c-kit, HER2 and PDGFR [60]. Lapatinib is an EGFR and HER2 inhibitor that possesses extended binding time to EGFR that prolongs its inhibitory effect. This agent has been studied alone and in combination for treating breast cancer. Although efficacy as a single agent was limited, in combination with cytotoxic chemotherapy, lapatinib does improve outcome. For example, in patients with metastatic breast cancer that over express HER2, treatment with lapatinib plus capecitabine markedly improved progression free survival and overall disease progression [27]. Similarly, lapatinib and letrozole have proven useful in postmenopausal women with advanced breast cancers that over express HER2 and are hormone receptor positive [61]. Another multi-targeted TKIs, dasatinib, has demonstrated increased inhibitory activity against bcr-abl compared to imatinib in vitro and is currently approved for the treatment of CML in patients who have developed resistance to imatinib [10, 60, 62, 63]. Finally, additional multi-targeted agents are under development. For example, neratinib inhibits both EGFR (ErbB1), HER2 (ErbB2), and HER4 (ErbB4) [64]; clinical trials are underway [13, 65].
Several of the multitargeted TKIs are more difficult to classify because they target a variety of kinases and downstream signaling cascades. For example, some of these molecules inhibit angiogenesis by blocking the vascular endothelial growth factor receptor (VEGFR), while also inhibiting tyrosine kinases such as c-kit and HER2. Sunitinib, for example, inhibits c-kit, PDGFR-α, CSF-1 receptor, and VEGFR-1, 2 and 3 [27]. The FDA approved of its use in advanced renal cell carcinoma after it demonstrated efficacy by improving progression free survival and overall survival compared to interferon-α [66, 67]. Sunitinib also has been used successfully to treat GIST tumors that have developed resistance to imatinib, and is FDA approved for use in that setting [66, 68, 69]. Sunitinib also has been studied for treating refractory thyroid cancer [70] and metastatic breast cancer [71]. However, not unexpectedly, one consequence of targeting so many different TKs has been increased toxicity. For example, in the study treating metastatic breast cancer, dose modification was necessary in 56% of patients due to adverse effects [71]. Similarly, in a study combining sunitinib with cyclophosphomide and methotrexate, of the 15 patients studied, three developed neutropenia and five developed mucositis [27]. Another phase I dose-escalation study combining sunitinib and capecitabine in patients with solid tumors, five grade 3 adverse effects emerged: abdominal pain, mucosal inflammation, fatigue, neutropenia and hand-foot syndrome [72]. Similar side effects have been reported in other clinical trials, and one trial was terminated early because of the high incidence of neutropenia, febrile neutropenia and fatigue [27]. Interestingly, in one study of efficacy against hepatocellular carcinoma, the neutropenia and skin toxicities seemed to correlate with response to therapy, overall survival and time to tumor progression, suggesting that those toxicity may be a marker for tumor response [73].
Several other multitargeted TKIs are currently under investigation. Axitinib inhibits all VEGF receptors (1, 2 and 3), PDGFR-β and c-kit [27]. Clinical trials continue to investigate the use axitinib for the treatment of a variety of solid tumors including, but not limited to, renal cell carcinoma, hepatocellular carcinoma, colorectal cancer, lung cancer, and melanoma [13]. Vandetanib inhibits EGFR, the kinase domain of VEGFR2, and RET autophosphorylation that is currently in the early stages of clinical investigation [27, 74–78]. At present, the indications for use of these agents, both alone or in combination with other drugs, is evolving and awaits the results of clinical trials.
TKIs represent some of the earliest efforts at targeted therapeutics and the development of monoclonal antibodies, small molecule inhibitors, and multitargeted TKIs has laid the ground work for the development of other biologic agents. In addition, these agents have taught us that in some cases biomarkers like KRAS mutation and EGFR overexpression can predict response. As such, TKIs were some of the first agents to be used in a truly individualized approach to cancer therapy.
ANGIOGENESIS INHIBITORS
Angiogenesis is one of the earliest events in tumor growth and is characterized by tumor microvascular formation [79]. Once a tumor grows beyond the limit of oxygen diffusion (approximately 2mm3), the hypoxic environment triggers the release of pro-angiogenic factors that in turn stimulate new blood vessel formation. The pro-angiogenic factors released include, vascular endothelial growth factors (VEGFA, B, C, D and E), basic fibroblast growth factor (bFGF), IL-8, placenta-like growth factors (PLGF-1 and 2), neurophilins (NRP1 and NRP2), transforming growth factors (TGF), fibroblast growth factor (FGF), and platelet derived growth factor (PDGF) [5, 79, 80]. Although all of these molecules represent potential anti-angiogenic targets, VEGF and its receptors, VEGFRs, have received the most attention. VEGFR stimulation activates intracellular PLC_ (phospholipase C-γ), PKC (protein kinase C) and the MAPK pathway, PI3 kinase, Akt/PKB (protein kinase B), NFκB, endothelial nitric oxide syntheses, and inhibits pro-apoptotic proteins, thereby inducing endothelial cell survival, proliferation and migration, and increasing vasodilation, vascular remodeling and vessel permeability [81]. In normal tissues, VEGFR2 appears to be the major inducer of angiogenesis, whereas the relatively weak signal cascade of VEGFR1 may actually decrease angiogenesis. VEGFR3 is involved in embryologic and post-natal angiogenesis and lymphangiogenesis. In cancer, VEGFR1 and 2 appear to be the receptors most involved in tumor angiogenesis [5]. Both VEGF and VEGFRs have emerged as logical targets for drugs seeking to inhibit angiogenesis [42, 79].
Bevacizumab is an anti-VEGFA antibody that has been widely studied and utilized. This agent reduces the amount of effective circulating VEGFA and thereby prevents VEGF receptor activation [42]. Clinical efficacy of this agent was first demonstrated in metastatic colorectal cancer. In this setting, bevacizumab in combination with irinotecan, 5FU and leukovorin (FOLFIRI) induced higher response rates and prolonged progression free and overall survival compared to traditional chemotherapy [82]. As a result, bevacizumab is currently utilized for both first and second line therapy in stage IV colorectal cancer [83, 84]. Nevertheless, as with most other biologic agents, subsequent experience with this drug has yielded mixed results. The BRiTE (Bevacizumab Regimens’ Investigation of Treatment Effects) study, an observational cohort study of 1,953 patients with metastatic colorectal cancer from 248 United State sites between 2004 and 2005, examined the efficacy of bevacizumab combined with chemotherapy as first line treatment for metastatic colorectal cancer. At 20 months, progression free survival was observed in 22% of patients who had received bevacizumab, while disease progression was seen in 79% of the patients. In addition 66% of patients died during the study period, and 12% either withdrew from the study or were lost to follow-up [85]. Similarly, in a phase III study by Stathopoulos in 2010, median overall survival did not differ between groups that received bevacizumab with chemotherapy (irinotecan, 5FU, leukovorin) and groups who received chemotherapy alone [86]. At present, bevacizumab is widely used for treating metastatic colorectal cancer, but its ultimate utility remains to be seen.
The efficacy of bevacizumab has subsequently been tested in other tumor types, including pancreatic and hepatocellular carcinoma, renal cell carcinoma, neuroendocrine tumors, non-small cell lung cancer, and breast, brain and ovarian cancers, and, as was the case with colorectal cancer, results have been promising but mixed. [13] In non-small cell lung cancer, effects on overall survival were inconsistent, but the drug appeared to slow the progression of the disease [87]. In vitro data suggest efficacy in combination with docetaxal for treatment of breast and prostate cancers [88]. In combination therapy with paclitaxel for metastatic breast cancer, bevacizumab improved response rates compared to paclitaxel alone [89]. Moreover, in a Phase II study of bevacizumab with erlotinib for treating recurrent malignant glioma, progression free survival was not significantly improved in patients who received bevacizumab [90]. To date, the FDA has approved bevacizumab in combination with carboplatin and paclitaxel for the treatment of non-small cell lung cancer, and in combination with interferon for the treatment of metastatic renal cell carcinoma [84, 91, 92]. It has also been approved for use as a second-line single agent for the treatment of glioblastoma [84]. In 2008, bevacizumab was approved as first-line treatment in combination with paclitaxel for metastatic HER2 negative breast cancer [84]. However, the results of more recent analyses of several clinical trials have failed to show any survival advantage in breast cancer patients who receive bevacizumab compared to chemotherapy alone. As a result, the Oncologic Drugs Advisory Committee of the U.S. FDA has made the controversial recommendation that approval be withdrawn for first-line treatment for metastatic breast cancer [93]. The FDA is currently reviewing the data on this drug, and the decision regarding possible revocation of approval are pending [94].
Toxicity has also been of concern with bevacizumab. While the most common toxicities are not severe (hepatotoxicity, proteinuria, diarrhea, myelosuppression, fatigue, infection, pain, nausea/vomiting, and hypokalemia), more rare but serious problems have been observed [87, 89, 90, 95]. For example, the BRiTE study identified new onset or worsening hypertension in 22% of patients and hypertension was implicated in eight serious adverse events. Additional less common but serious adverse events included postoperative wound healing complications (4%), grade 3–4 bleeding (2.2%), arterial thromboembolic events (2%), and gastrointestinal perforation (2%). Significant postoperative wound healing complications occurred following major surgery and in patients in whom surgery was undertaken within two weeks of the last bevacizumab dose [85]. These adverse events has led to an FDA drug warning and safety labeling update concerning these adverse effects [84].
Although bevacizumab has been most widely studied, other anti-angiogenic agents are under development. For example, Aflibercept (VEGF Trap) is a molecule in which the ligand binding domains of the VEGFR1 and VEGFR2 proteins are fused with human IgG1, forming a decoy receptor that binds all VEGFs and placental growth factor [27]. While this agent appears promising in the preclinical setting, clinical safety and efficacy have not yet been proven [27].
CELL CYCLE INHIBITORS
Cell cycle perturbations are also known to play a role in cancer progression, therefore the regulators of these processes also represent viable anti cancer targets. The cell cycle is regulated by a variety of cyclins and cyclin dependent kinases (CDKs; Fig. 2), and the CDKs, in turn, are regulated by several naturally occurring inhibitors (CDKIs) that prevent CDK binding to cyclins. Progression through the cell cycle is permitted to occur when the CDKIs do not prevent CDK/cyclin interactions. For example, cyclinE/CDK2 complex formation promotes cell entry into S phase, and CyclinB/CDK1 allows mitosis to occur [96, 97]. Overall, nine CDKs have been described and these molecules interact with twenty possible cyclins (A-T) [96]. In addition, there is evidence that CDK or cyclin production is disrupted in several malignancies, including melanoma, lung, breast and colorectal cancers [96, 98, 99].
Fig. 2.
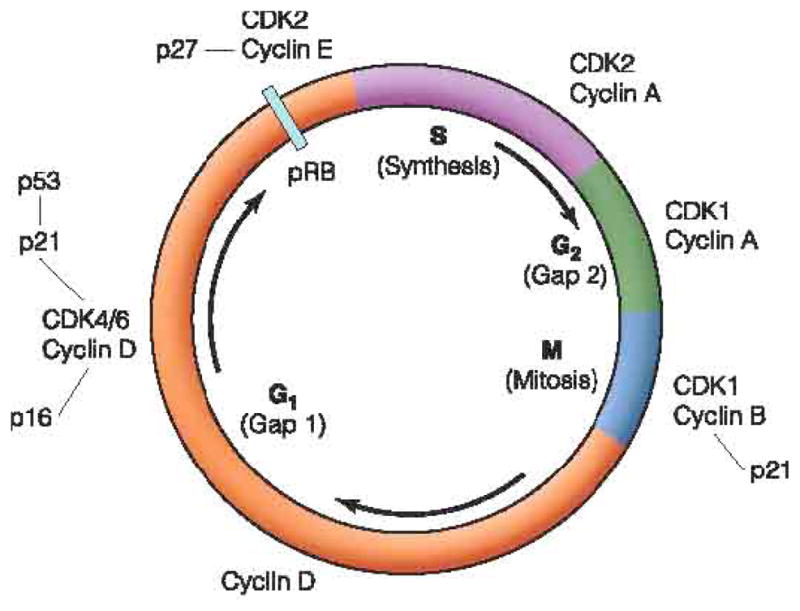
Progression through each phase of the cell cycle is regulated by cyclin/CDK interactions.
(Copyright of Renaic, with kind permission of Renaic Corporation and Sysmex Corporation.)
The existence of naturally occurring CDKIs provided a logical starting point for anti-cancer drug development. For example, flavopiridol is an analogue of a naturally occurring flavinoid CDKI [100]. Flavopiridol acts on the G1 and S phases of the cell cycle via inhibition of CDKs 2, 4, 6, and 9, and has demonstrated early success in vitro by inducing arrest of cellular growth and inducing apoptosis in chronic lymphocytic leukemia and colorectal cancer cells [101, 102]. Currently, flavopiridol is useful for treating recurrent CLL, where it has been shown to induce a clinical response in patients with high-risk genomic features [103]. In addition, when given with cytosine arabinoside and mitoxantrone, flavopiridol induced complete remission in 67% of patients with AML who had poor risk features (older age, unfavorable genetic mutations, or secondary AML) [104]. Phase I trials indicate drug tolerability and phase II trials continue to test this drug’s efficacy in a variety of cancers [105].
Roscovitine is a similar CDKI that inhibits cyclinE/CDK2, cyclinB/CDK1, cyclinH/CDK7, and cyclinT1/CDK9 and prevents progression through S, G2 and M phases of the cell cycle [96]. In vitro studies also indicate that this drug induces apoptosis by activating p53, suppressing NF-κB, and down-regulating Mcl-1 (an anti-apoptosis protein) in multiple myeloma cells [106–108]. It also inhibits mitosis by decreasing production of promoters of cell division, such as Aurora-A/B, CDC25C, polo-like kinase, and WEEL [109]. Preclinical studies have demonstrated the efficacy of this agent in a variety of cancer cell lines. A phase II clinical trial of roscovitine for the treating resistant non-small cell lung cancer has recently been completed, and phase I investigation of its clinical safety in treating any solid tumors resistant to standard treatment is underway [13].
In addition to flavopiridol and roscovitine, and increasing number of both naturally occurring and synthetic CDKIs are currently undergoing preclinical and clinical evaluation Table 2. E7070, AZD5438, SNS-032 (BMS-387032), bryostatin-1, PD 0332991, and SCH 727965 are CDKIs that are at various stages of development. Although many of these agents look promising, safety and efficacy are not yet well understood [96].
Table 2.
Cell Cycle Inhibitors [2]
| Cell Cycle Inhibitor | Mechanism of Action | Effects | Clinical Trials* |
|---|---|---|---|
| flavopiridol | Inhibits CDK 2, 4, 6, 9 | G1/S arrest | Phase I/II (hematologic and several solid tumors) |
| Roscovitine | Inhibits cyclinE/CDK2, cyclinB/CDK1, cyclinH/CDK7, cyclinT1/CDK9; p53 activation, NF-κB, decreases Mcl-1 | S/G2/M arrest | Phase I/II (solid tumors) |
| E7070 | Inhibits CDK2, cyclin E | G1/S progression | Phase I/II (several solid tumors, melanoma) |
| SNS-032 | Inhibits CDK 2, 7, 9, VEGF | Inhibits progression and transcription, angiogenesis | Phase I (advanced B-cell lymphomas, several solid tumors) |
| bryostatin-1 | Affects protein kinase C, CDK2; induces p21 | Cell cycle modulation | Phase I/II (combination therapy for hematologic and several solid tumors) |
| AZD5438 | Inhibits CDK1, 2, 9 | Affects all phases of cell cycle | Trials terminated dues to safety concerns |
INDUCERS OF APOPTOSIS
In addition to aberrant cell cycle regulation, cancer cells often display decreased rates of apoptosis compared to non-neoplastic cells. Two major pathways are involved in this process of programmed cell death: an extrinsic pathway, mediated by death receptors such as FAS, TNF, and TRAIL, and an intrinsic pathway that is mitochondrial and apoptosome-mediated. Cancer therapies that target the inappropriate loss of control over apoptosis have been developed to induce the intrinsic and extrinsic pathways either independently or together [2].
Apoptosis inducers are generally classified based on their mechanism of action. Bortezomib, a boronic acid dipeptide, was the first pro-apoptotic agent used in cancer treatment. Bortezomib promotes apoptosis by inhibiting the 26S proteosome, thereby inhibiting degradation of a number of cellular proteins that are involved in the cell cycle, transcription, and tumor suppression [110]. Early work suggested that 26S proteosome inhibition promoted apoptosis in multiple myeloma cells, [110] and the APEX trial suggested superior efficacy of bortezomib over dexamethasone for refractory multiple myeloma [111]. Additional trials have suggested that bortezomib may augment the activity of more traditional chemotherapeutic agents. In multiple myeloma, the addition of bortezomib to melphalan and prednisone slowed time to disease progression, increased the incidence of partial and complete response, prolonged the duration of response, and improved three year survival compared to melphalan and prednisone alone [112].
Suberoylanilide hydroxamic acid (SAHA) is another inducer of apoptosis that inhibits histone deacetylase [113]. SAHA and other histone deacetylase inhibitors down regulate TNF_ receptor-1 expression and activation of NF-κB, thereby inhibiting tumorigenesis [114]. SAHA has been shown to be both safe and efficacious for treating cutaneous T cell lymphoma, and this agent is currently approved for use in patients who have failed previous treatment (third line therapy) [115]. Additional clinical investigations are underway, including a phase III trial of SAHA use against advanced mesothelioma [116].
The TRAIL1 and TRAIL 2 pathways also have been suggested to be promising pro-apoptotic targets. TRAILR1 and TRAILR2 are believed to be involved in apoptosis via FADD and caspase-8 and/or -10 recruitment, and/or by activation of other signaling pathways involving NF-κB, MAPK, PI3 kinase and Akt. Mapatumumab (HGS-ETR1) and lexatumumab (HGS-ETR2) are 2 monoclonal antibodies that agonistically bind the TRAILR1 and TRAILR2 receptors, respectively [117]. The role of mapatumumab and lexatumumab in combination with other agents has been supported by the observation of a synergistic effect with either antibody and cisplatin or bortezomib in non-small cell lung cancer cells and malignant mesothelioma cells, or with radiotherapy in colorectal cancer cells [118–120]. Clinical trials are underway for both mapatumumab and lexatumumab [13].
The promising results with bortezomib, SAHA, and TRAIL inhibitors has prompted the development of a variety of other pro- apoptotic agents that are currently under investigation (Table 3). TLK286 is a modified glutathione analogue that is metabolized by glutathione S transferase (GST) in the tumor cell cytosol, yielding a molecule that is toxic to the cell. Because GST is over expressed in many resistant tumor cells, TLK286 may be an ideal therapeutic option in these refractory tumors [121, 122]. Preliminary data suggest that TLK286 has little efficacy as a single agent, but combination with other agents may be useful [123–128]. FTI R115777 is a methyl quinolone farnesyl transferase inhibitor (FTI) that induces apoptosis by yet another mechanism. FTI R115777 is thought to induce cell death by disrupting the mitochondrial membrane and inducing caspase-9 activation [129]. 7AAG is a inhibitor of heat shock protein HSP90, a molecule that is frequently over expressed in tumor cells [130]. This agent has been thoroughly studied in vitro and in vivo, and affects a variety of cancer cell types by inducing apoptosis, decreasing growth, potentiating the effects of known chemotherapeutic drugs, and sensitizing tumors to radiotherapy [131]. Clinical safety and efficacy await the completion of clinical trials.
Table 3.
Inducers of Apoptosis [2]
| Inducers of Apoptosis | Mechanism of Action | Effects | Clinical Trials* |
|---|---|---|---|
| bortezomib | Inhibits 26S proteosome | Decreased degradation of cell cycle and transcription proteins | FDA approved as first-line treatment of multiple myeloma, and as second line treatment of mantle cell lymphoma. |
| suberoylanilide hydroxamic acid (SAHA) | Inhibits histone deacetylase | Suppress tumorigenic gene expression | FDA approved as third-line treatment of cutaneous T cell lymphoma; Phase III (advanced mesothelioma) |
| TLK286 | Formation of a toxic metabolite | Induces apoptosis | Phase III (refractory gynecologic malignancies), combination therapy |
| ONYX015 | Adenovirus; invades and replicates in p53 mutated cells, inactivates p53 gene | Lyses p53 mutated tumor cells | Phase I/II (combination therapy for solid tumors). |
| FTI-R115777 | Blocks farnesylation of Ras, disrupts mitochondrial membrane, induces caspase-9 activation | Inhibit cell growth and induce apoptosis | Phase III (colorectal cancer and AML; no significant improvement in progression frees survival or overall survival, respectively). Phase I/II (pediatric leukemias, glioma) |
| 17AAG | Inhibits HSP90 | Inhibit cell motility and invasion | Phase I/II (multiple myeloma) |
| mapatumumab | TRAILR1 agonist | Inhibit TRAIL ligand binding and apoptotic signal cascade | Phase I/II (several solid tumors) |
| lexatumumab | TRAILR2 agonist |
FAK INHIBITORS
Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase that increasingly has been implicated in cancer progression, and as such, appears to be a valuable target for anti-cancer agents. FAK is a 125kDa protein that was identified at adhesion sites between cells and the extracellular environment. It is encoded by a gene located on chromosome 8 and houses three domains, the amino-N-terminal domain, the central catalytic domain and the carboxy-C-terminal domain. FAK activation relies upon autophosphorylation of the unique Y-397 site that is found in the N-terminal domain. Y-397 has a number of interesting properties. This region binds a number of signaling proteins such as src, PI-3 kinase, and Grb-7. It also binds EGFR, VEGFR, and p53 and other molecules that are critical for carcinogenesis [132]. The interaction between Y-397 and p53 is particularly intriguing. It is well known that p53 is mutated in many cancers; interestingly, p53 can inhibit FAK expression, and FAK can inhibit p53 expression. In breast cancer cells, p53 mutation is highly correlated with FAK over expression [133]. FAK also activates proteins that promote cell motility and migration, invasion, survival, angiogenesis, lymphangiogenesis, and proliferation, such as paxillin and talin [132]. (Fig. 3). From a clinical standpoint, FAK is upregulated in several types of cancer including ovarian, thyroid, head and neck, lung, kidney, brain, hepatocellular, pancreatic, colorectal, breast [134, 135], cervical [135], and prostate [136] cancers, as well as melanoma, neuroblastoma, osteosarcoma, and sarcoma [137, 138]. The potential role of FAK activation cancer growth and progression has led to the development of several therapeutic agents targeting this molecule [132] (Table 4).
Fig. 3.
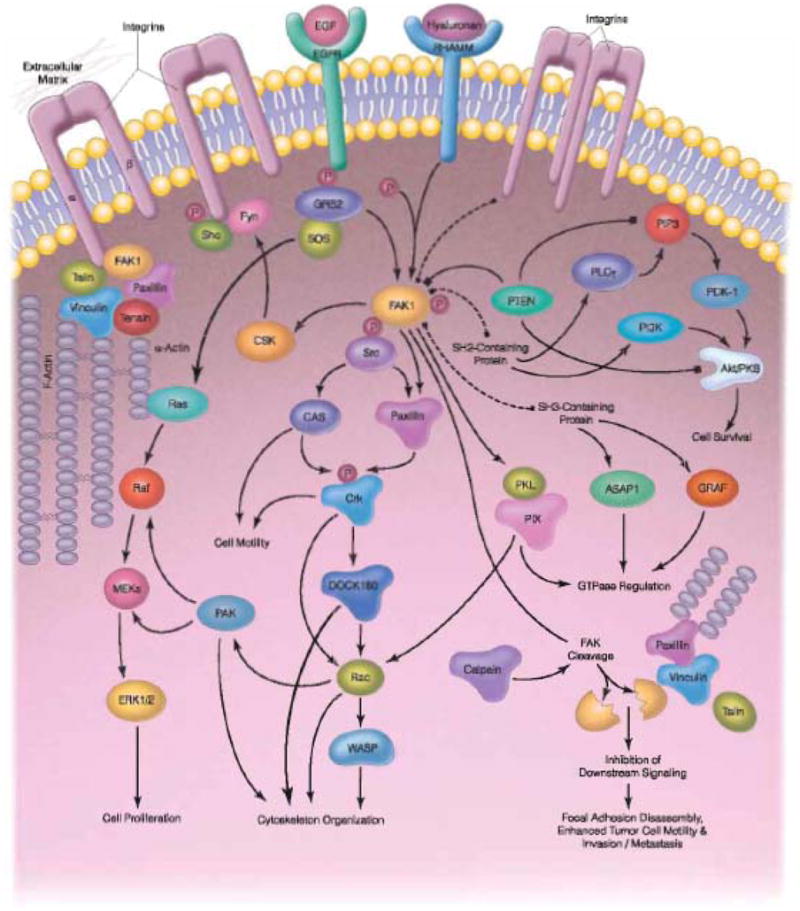
FAK plays a key role in a signaling cascade that can lead to some of the tumorigenic properties of cancer cells. FAK activation induces cell proliferation, motility, survival, invasion and metastasis.
SABiosciences. www.sabiosciences.com
Table 4.
FAK Inhibitors
| Inhibitor Name | Chemical Name | Structure | |
|---|---|---|---|
| PF 573,228 (PF-228) | 6-(4-(3-(methylsulfonyl) benzylamino)-5- (trifluoromethyl)pyrimidin- 2-ylamino)-3, 4-dihydro quinolin-2(1H)-one |
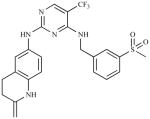
|
Pfizer |
| PF562, 271-26 (PF-271) | N-Methyl-N-(3- {[2-(2-oxo-2,3-dihydro-1H-indol- 5-ylamino)-5-trifluoromethyl- pyrimidin-4-ylamino]-methyl}- pyridin-2-yl)-methanesulfonamide |
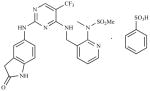
|
Pfizer |
| NVP-226 | (2-[5-Chloro-2-[2-methoxy-4- (4-morpholinyl)phenylamino] pyrimidin-4-ylamino]-N- methylbenzamide |
|
Novartis |
| Y15* | 1,2,4,5-Benzenetetraamine tetrahydrochloride |
 · 4HH2CI |
|
| PND-1186** | 2,4-diamino-pyridine-based; X=bond or (C1-C3)alkyl comprising 0–1 heteroatom selected from the group consisting of N, O, S(O) and S(O)2, the (C1-C3)alkyl is substituted with 0–1 hydroxy, halo, (C1-C3)alkoxy, (C1-C3)alkylamino or (C1-C3)2dialkylamino groups. R1 and R2 are 5–12 membered Mono-, bi- or polycyclic, aromatic or partially aromatic rings. R3 is a trifluoromethyl, halo, nitro or cyano; salt, tautomer, solvate, hydrate, or aprodrug. |
|
Reported by Golubovskaya et al., J. Med. Chem, V 51, p7405–7416, 2008, ref. in (15).
Reported by Tanjoni et al., Cancer Biol. Ther., 2010, V 15, 2010, ref in (17).
With FAK as a potential target for anti-cancer therapy, the initial attempts at inhibition focused upon down-regulation of FAK expression. Initial studies demonstrated that silencing FAK by transfection with dominant negative C-terminal FAK-CD decreased adhesion, colonization, and in vivo tumor growth by breast cancer cells [139]. Similarly, transfection with FAK-silenced RNA (FAK-siRNA) increased rounding, decreased growth and decreased in vivo tumorigenesis [139]. An attempt to increase the anti-tumorigenic affect of FAK inhibition next focused on the combined inhibition of FAK and its downstream effector src. Inhibition of src with PP2 in combination with adenoviral FAK-CD resulted in increased apoptosis compared to either molecule alone [140]. These initial experiments proved that inhibition of FAK expression could decrease cellular functions associated with cancer growth and progression, providing scientific rationale for further development of FAK inhibitors.
Based upon the observed interaction between FAK and p53, another approach involved activation of p53 (therefore decreasing FAK) or elimination of p53-deficient cells. This indirect approach involved the introduction of retroviruses that express p53 into rapidly dividing p53-deficient cells. For example, AD5CMV-p53 is an adenoviral vector encoding p53 that was shown to replenish p53 in p53 deficient tumor cells. This approach inhibited lung cancer cell growth both in vitro and in vivo [141]. Clinical phase I and II trials have subsequently demonstrated that this agent is both safe and effective against esophageal squamous cell carcinoma [142, 143]. Nevertheless, concern about the safety of a viral vector and about the logistics of production and storage persist [144].
Concerns about the reliability of gene transfection and the safety of a viral vector have lead to a third approach to FAK inhibition. A variety of small molecule inhibitors have been developed with the goal of specifically blocking FAK kinase activity. The earliest small molecule inhibitor of FAK, TAE226, blocks the ATP binding site and inhibits FAK phosphorylation at both Y397 and Y861, a site that is involved with VEGF signaling. This molecule showed promise in preclinical in vitro and in vivo trials, and most recently, was found to inhibit angiogenesis in models of human colon cancer [145]. The experimental success of TAE then led to the development of additional small molecule inhibitors. Like TAE226, PF-562,271 blocks the ATP binding site of FAK and of Pyk2 (protein-rich tyrosine kinase 2), another nonreceptor tyrosine kinase known to bind and activate src [146, 147]. PF-562,271 inhibited in vivo breast cancer cell growth and metastasis in a murine model, and decreased the in vivo growth of prostate cancer cells and in vitro growth of lung cancer cells [146, 148–150]. It also has exhibited synergy with sunitinib in the inhibition of hepatocellular tumor growth in vivo [151]. Phase I clinical trials employing PF-562,271 for treating prostate, pancreatic, and head and neck cancers, have recently been completed. Another inhibitor, PF-573,228, works similarly and has been found to inhibit breast cancer cell migration [148]. Interestingly, this agent in combination with tamoxifen decreased proliferation in ER positive breast cancer cells [152]. Other FAK inhibitors include PF-04554878, which is currently in Phase I clinical trials for the treatment of advanced non-hematologic malignancies, and GSK2256098 which has completed Phase I trials [13].
Additional small molecule inhibitors of FAK have taken advantage of other mechanisms of blocking FAK activation. PND-1186, for example, is thought to inhibit ATP activity by substituting a pyridine ring for the pyramidine ring found in other ATP-binding FAK inhibitors [153, 154]. Early studies show that this molecule increases breast cancer cell apoptosis, inhibits in vivo breast cancer cell growth and metastasis, and inhibits ascites and peritoneal seeding in models of ovarian cancer [153, 155].
Although the early FAK inhibitors have shown significant promise in anti-tumorigenic activity, the nonspecific nature of the ATP binding in both FAK and in other tyrosine kinases in particular has raised concerns about potential toxicity. A solution, therefore, would be to target a site that is highly specific to FAK. The Y397 site appears to offer just such an opportunity. Recently, several small molecule inhibitors that directly target the Y397 autophosphorylation site have been developed [156]. One such molecule, 1,2,4,5-benzenetetraamine tetrahydrochloride (Y15), binds specifically to the Y397 site, does not affect ATP binding or Pyk-2, and effectively inhibits FAK activation. In vitro, Y15 increased detachment and inhibited adhesion of breast cancer cells and inhibited tumorigenesis in vivo [156]. Additional studies have demonstrated the efficacy of Y15 in inhibiting pancreatic and neuroblastoma tumor growth in vivo [157–159]. In addition, when combined with gemcitabine, these two agents demonstrated synergy in inhibiting pancreatic cancer cell growth [159].
CONCLUSION
Despite decades of experience with cytotoxic chemotherapy, no agent or combination of agents has yet to cure cancer. As the biologic underpinnings of carcinogenesis are better understood, targeting molecular therapeutics to those molecules and pathways that promote cancer growth has become an increasingly attractive adjunct. Beginning with TKIs, an increasing number of biologic agents have shown promise both in the laboratory and in clinical trials. Angiogenesis inhibitors have been widely studied and are included in multidrug regimens for treating a number of malignancies. Cell cycle inhibitors and inducers of apoptosis increasingly are showing promise. Finally, inhibition of FAK, especially by small molecule inhibitors such as Y15, may prove to be both efficacious and, with hope, nontoxic. Going forward, both cancer research and cancer therapy will no doubt rely heavily on these exciting molecular targets and their inhibitors.
Acknowledgments
We would like to thank Ms. Amanda J. Ellis for assistance with preparation of the manuscript. Dr. Vita Golubovskaya is supported by the Susan G. Komen for the Cure grant.
ABBREVIATIONS
- CDK
Cyclin dependent kinase
- EGFR
Epidermal growth factor receptor
- FAK
Focal adhesion kinase
- RTK
Receptor tyrosine kinase
- TKI
TK inhibitor
- TK
Tyrosine kinase
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular endothelial growth factor receptor
Footnotes
CONFLICT OF INTEREST
Dr. Vita Golubovskaya discloses potential conflict of interest with CureFAKtor Pharmaceuticals.
References
- 1.American Cancer Society. Cancer Facts & Figures 2010. American Cancer Society; Atlanta: 2010. [Google Scholar]
- 2.Heffler M, Golubovskaya V, Bullard Dunn KM. Evolving molecular therapies and their applications to surgical oncology. ACS Surg Principles Pract. 2010 (in press) [Google Scholar]
- 3.Arora A, Scholar EM. Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther. 2005;315(3):971–9. doi: 10.1124/jpet.105.084145. [DOI] [PubMed] [Google Scholar]
- 4.Robinson DR, Yi-Mi Q, Su-Fang L. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 5.Arnold D, Seufferlein T. Targeted treatments in colorectal cancer: state of the art and future perspectives. Gut. 2010;59:838–858. doi: 10.1136/gut.2009.196006. [DOI] [PubMed] [Google Scholar]
- 6.Taniguchi T. Cytokine signaling through nonreceptor tyrosine kinases. Science. 1995;268(5208):251–255. doi: 10.1126/science.7716517. [DOI] [PubMed] [Google Scholar]
- 7.Amit I, Wides R, Yarden Y. Evolvable signaling networks of receptor tyrosine kinases: relevance of robustness to malignancy and to cancer therapy. Mol Syst Biol. 2007;3:151. doi: 10.1038/msb4100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Bacco F, Fassetta M, Rasola A. Receptor tyrosine kinases as targets for cancer therapy. Cancer Ther. 2004;2:317–328. [Google Scholar]
- 9.Popenoe DW, Schaefer-Rego K, Mears JG, Bank A, Leibowitz D. Frequent and extensive deletion during the 9,22 translocation in CML. Blood. 1986;68(5):1123–1128. [PubMed] [Google Scholar]
- 10.Agrawal M, Garg RJ, Cortes J, Quintas-Cardama A. Tyrosine kinase inhibitors: the first decade. Curr Hematol Malig Rep. 2010;5(2):70–80. doi: 10.1007/s11899-010-0045-y. [DOI] [PubMed] [Google Scholar]
- 11.Sleijfer S, Seynaeve C, Wiemer E, Verweij J. Practical aspects of managing gastrointestinal stromal tumors. Clin Colorectal Cancer. 2006;6(Suppl 1):S18–23. doi: 10.3816/ccc.2006.s.003. [DOI] [PubMed] [Google Scholar]
- 12.Cassier PA, Blay JY. Imatinib mesylate for the treatment of gastrointestinal stromal tumor. Expert Rev Anticancer Ther. 2008;10(5):623–34. doi: 10.1586/era.10.33. [DOI] [PubMed] [Google Scholar]
- 13.Clinical Trialsgov. www.clinicaltrials.gov.
- 14.Lee TS, Ma W, Zhang X, Giles F, Cortes J, Kantarjian H, Albitar M. BCR-ABL alternative splicing as a common mechanism for imatinib resistance: evidence from molecular dynamics simulations. Mol Cancer Ther. 2008;7(12):3834–3841. doi: 10.1158/1535-7163.MCT-08-0482. [DOI] [PubMed] [Google Scholar]
- 15.Hudis CA. Trastuzumab- mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 16.Yarden Y, Sliwkowski MX. Untangling the ErbB signaling network. Nat Rev. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 17.Krop IE, Muralidhar B, Jones SF, Holden SN, Yu W, Girish S, Tibbitts J, Yi JH, Sliwkowski MX, Jacobson F, Lutzker SG, Burris HA. Phase I study of trastuzumab-DM1, and HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clinic Oncol. 2010;28(16):2698–2704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 18.Yakes FM, Chinraranalab W, Ritter CA, King W, Seelig S, Arteaga CL. Herceptin-induced inhibition of phosphatidylinositol-3 kinase and Akt is required for antibody-mediated effects of p27, cyclin D1, and antitumor action. Cancer Res. 2002;62(14):4132–4141. [PubMed] [Google Scholar]
- 19.Le XF, Pruefer F, Bast RC., Jr HER2-targeting antibodies modulate the cyclin-dependent kinase inhibitor p27Kip1 via multiple signaling pathways. Cell Cycle. 2005;4(1):87–95. doi: 10.4161/cc.4.1.1360. [DOI] [PubMed] [Google Scholar]
- 20.Chang HR. Trastuzumab-based neoadjuvant therapy in patients with HER2-positive breast cancer. Cancer. 2010;116(12):2856–2867. doi: 10.1002/cncr.25120. [DOI] [PubMed] [Google Scholar]
- 21.Christofanilli M. Novel targeted therapies in inflammatory breast cancer. Cancer. 2010;116(Suppl 11):2837–2839. doi: 10.1002/cncr.25172. [DOI] [PubMed] [Google Scholar]
- 22.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, Aprile G, Kulikov E, Hill J, Lehle M, Ruschoff J, Kang YK. Trastuzumab in combination with chemotherapy versus chemotherapy alone for the treatment of HER2-positive advanced gastric or gastro-esophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376(9742):687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 23.Cancer Research Highlights. Breast cancer drug helps patients with gastric cancer. NCI Cancer Bull. 2009;6(11) [Google Scholar]
- 24.Juntilla TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-1090-x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 25.Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, Blättler WA, Lambert JM, Chari RV, Lutz RJ, Wong WL, Jacobson FS, Koeppen H, Schwall RH, Kenkare-Mitra SR, Spencer SD, Sliwkowski MX. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 26.Vogel CL, Burris HA, Limentani S, Borson R, O’Shaughnessy J, Vukelja S, Agresta S, Klenke B, Birkner M, Rugo HS. A phase II study of trastuzumab-DM1 (T0DM1), a HER2 antibody-drug conjugate (ADC), in patients with HER2+ metastatic breast cancer (MBC): final results. J Clinic Oncology, 2009 ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2009;27(15S):1017. [Google Scholar]
- 27.Alvarez RH, Valero V, Hortobagyi GN. Emerging targeted therapies for breast cancer. J Clin Oncol. 2010;28(20):3366–3379. doi: 10.1200/JCO.2009.25.4011. [DOI] [PubMed] [Google Scholar]
- 28.Jonker D, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebbutt NC, van Hazel G, Wierzbicki R, Langer C, Moore MJ. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357(20):2040–2048. doi: 10.1056/NEJMoa071834. [DOI] [PubMed] [Google Scholar]
- 29.Normanno N, Bianco C, De Luca A, Maiello MR, Salomon DS. Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr Relat Cancer. 2003;10(1):1–21. doi: 10.1677/erc.0.0100001. [DOI] [PubMed] [Google Scholar]
- 30.Shin DM, Donato NJ, Perez-Soler R, Shin HJ, Wu JY, Zhang P, Lawhorn K, Khuri FR, Glisson BS, Myers J, Clayman G, Pfister D, Falcey J, Waksal H, Mendelsohn J, Hong WK. Epidermal growth factor receptor-targeted therapy with C225 and cisplatin in patients with head and neck cancer. Clin Cancer Res. 2001;7(5):1204–13. [PubMed] [Google Scholar]
- 31.Goldstein NI, Prewett M, Zuklys K, Rockwell P, Mendelsohn J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin Cancer Res. 1995;1:1311–1318. [PubMed] [Google Scholar]
- 32.Merlano M, Russi E, Benasso M, Corvo R, Colantonio I, Vigna-Taglianti R, Bacigalupo A, Numico G, Crosetto N, Gasco M, Lo Nigro C, Vitiello R, Violante S, Garrone O. Cisplatin-based chemoradiation plus cetuximab in locally advanced head and neck cancer: a phase II clinical study. Ann Oncol. 2010 doi: 10.1093/annonc/mdq412. [Epbu ahead of print] [DOI] [PubMed] [Google Scholar]
- 33.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 34.FDA approves Erbitux for colorectal cancer. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2004/ucm108244.htm.
- 35.Cetuximab. http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm129241.htm.
- 36.Van Cutsem E, Nowacki M, Lang I, Cascinu S, Shchepotin I, Maurel J, Rougier P, Cunningham D, Nippgen J, Kohne C. Randomized phase II study of irinotecan and 5-FU/FA with or without cetuximab in the first-line treatment of patients with metastatic colorectal cancer (mCRC): the CRYSTAL trial. Abstract. J Clinic Oncol; ASCO Annual Meeting Proceedings (Post-Meeting Edition); 2007; 2007. [Google Scholar]
- 37.Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, Zampino M, Donea S, Ludwig H, Zubel A, Koralewski P. Cetuximab plus 5-FU/FA/oxaliplatin (FOLFOX-4) versus FOLFOX-4 in the first-line treatment of metastatic colorectal cancer (mCRC): OPUS, a randomized phase II study. Abstract. J Clinic Oncol; ASCO Annual Meeting Proceedings (Post-Meeting Edition); 2007; 2007. [Google Scholar]
- 38.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that express the epidermal growth factor receptor. J Clin Oncol. 2004;22(7):1201–1208. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 39.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, Burris HA, III, Poplin EA, Hidalgo M, Baselga J, Clark EA, Mauro DJ. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25(22):2330–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 40.Tejpar S, Peeters M, Humblet Y, Vermorken JB, De Hertogh G, De Roock W, Nippgen J, von Heydebreck A, Stroh C, Van Cutsem E. Relationship of efficacy with KRAS status (wild type versus mutant) in patients with irinotecan-refractory metastatic colorectal cancer (mCRC) treated with irinotecan (q2w) and escalating doses of cetuximab (q1w): the EVEREST experience (preliminary data) J Clinic Oncol, 2008 ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2008;26(15S):4001. [Google Scholar]
- 41.Bokemeyer C, Bondarenko I, Hartmann JT, De Braud FG, Volovat C, Nippgen J, Stroh C, Celik I, Koralewski P. KRAS status and efficacy of first-line treatment of patients with metastatic colorectal cancer (mCRC) with FOLFOX with or without cetuximab: The OPUS experience. J Clinic Oncol, 2008 ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2008;26(15S):4000. [Google Scholar]
- 42.Winder T, Lenz H-J. Molecular predictive and prognostic markers in colon cancer. Cancer Treat Rev. 2010 doi: 10.1016/j.ctrv.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 43.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;27(12):2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 44.Lynch DH, Yang XD. Therapeutic potential of ABX-EGF: a fully human anti-epidermal growth factor receptor monoclonal antibody for cancer treatment. Semin Oncol. 2002;29(1 Suppl 4):47–50. doi: 10.1053/sonc.2002.31522. [DOI] [PubMed] [Google Scholar]
- 45.DiNicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 46.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Taberno J, Macarulla T, Di Fiore F, Gangloff AO, Ciardello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations of the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11(8):753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 47.Nahta R, Hung M-C, Esteva FJ. The HER-2-targeting antibodies trastuzumab and pertuzumab synergistically inhibit the survival of breast cancer cells. Cancer Res. 2004;64:2343–2346. doi: 10.1158/0008-5472.can-03-3856. [DOI] [PubMed] [Google Scholar]
- 48.Agus DB, Gordon MS, Taylor C, Natale RB, Karlan B, Mendelson DS, Press MF, Allison DE, Sliwkowski MX, Lieberman G, Kelsey SM, Fyfe G. Phase I clinical study of per-tuzumab, a novel HER dimerization inhibitor, in patients with advanced cancer. J Clin Oncol. 2005;23(11):2534–2543. doi: 10.1200/JCO.2005.03.184. [DOI] [PubMed] [Google Scholar]
- 49.Normanno N, Tejpar S, Morgillo F, De Luca A, Van Cutsem E. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat Rev Clin Oncol. 2009;6:519–527. doi: 10.1038/nrclinonc.2009.111. [DOI] [PubMed] [Google Scholar]
- 50.Burton A. What went wrong with Iressa? Lancet Oncol. 2002;3(12):708. doi: 10.1016/s1470-2045(02)00938-5. [DOI] [PubMed] [Google Scholar]
- 51.Information for healthcare professionals: gefitinib (marketed Iressa) FDA Alert. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/DrugSafetyInformationforHeathcareProfessionals/ucm085197.htm.
- 52.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 53.Ibrahim EM. Frontline gefitinib in advanced non-small cell lung cancer: Meta-analysis of published randomized trials. Ann Thoracic Med. 2010;5(3):153–160. doi: 10.4103/1817-1737.65047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Price KA, Azzoli CG, Krug LM, Pietanza MC, Rizvi NA, Pao W, Kris MG, Riely GJ, Heelan RT, Arcila ME, Miller VA. Phase II trial of gefitinib and everolimus in advanced non-small cell lung cancer. J Thoracic Oncol. 2010;5(10):1623–1629. doi: 10.1097/JTO.0b013e3181ec1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kitazaki T, Oka M, Nakamura Y, Tsurutani J, Doi S, Yasunaga M, Takemura M, Yabuuchi H, Soda H, Kohno S. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer. 2005;49(3):337–343. doi: 10.1016/j.lungcan.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 56.Shepherd FA, Pereira JR, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, Campos D, Maoleekoonpiroj S, Smylie M, Martins R, van Kooten M, Dediu M, Findlay B, Tu D, Johnston D, Bezjak A, Clark G, Santabarbara P, Seymour L. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 57.Cohen MH, Johnson JR, Chen YF, Sridhara R, Pazdur R. FDA drug approval summary: erlotinib (Tarceva) tablets. Oncologist. 2005;12(7):461–466. doi: 10.1634/theoncologist.10-7-461. [DOI] [PubMed] [Google Scholar]
- 58.Erlotinib. http://www.fda.gov/AboutFDA/CentersOffices/CDER/ucm209058.htm.
- 59.FDA approval for erlotinib hydrochloride. http://www.cancer.gov/cancertopics/druginfo/fda-erlotinib-hydrochloride.
- 60.O’Hare T, Walters DK, Stoffregen EP, Sherbenou DW, Heinrich MC, Deininger MW, Druker BJ. Combined Abl inhibitor therapy for minimizing drug resistance in chronic myeloid leukemia: Src/Abl inhibitors are compatible with imatinib. Clin Cancer Res. 2005;11(19 pt 1):6987–6993. doi: 10.1158/1078-0432.CCR-05-0622. [DOI] [PubMed] [Google Scholar]
- 61.FDA approval for lapatinib ditosylate.
- 62.Baccarini M, Kantarjian HM, Apperley JF. Efficacy of dasatinib (Sprycel) in patients with chronic phase chronic myelogenous leukemia resistance to or intolerant of imatinib: updated results of the CA180013 START-C Phase II study (abstract No. 164) Blood. 2006;108(11 pt 1):53. [Google Scholar]
- 63.Cortes J, Kim DW, Martinelli G, Rutchie E, Roy L, Coutre S, Corm S, Hamerschlak N, Tang JL, Hochhaus A, Khoury HJ, Brummendorf TH, Michallet M, Rege-Cambrin G, Gambacorti-Passerini C, Radich JP, Ernst T, Zhu C, Van Tornout JM, Talpaz M. Efficacy and safety of dasatinib in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blast phase. Leukemia. 2008;22(12):2176–1283. doi: 10.1038/leu.2008.221. [DOI] [PubMed] [Google Scholar]
- 64.Wong KK, Fracasso PM, Bukowski RM, Lynch TJ, Munster PN, Shapiro GI, Janne PA, Eder JP, Maughton MJ, Ellis MJ, Jones SF, Mekhail T, Zacharchuk C, Vermette J, Abbas R, Quinn S, Powell C, Burris HA. A phase I study with neratinib (HKI-272): an irreversible pan ErbB receptor tyrosine kinase inhibitor, in patients with solid tumors. Clin Cancer Res. 2009;15(7):2552–2558. doi: 10.1158/1078-0432.CCR-08-1978. [DOI] [PubMed] [Google Scholar]
- 65.Burstein HJ, Sun Y, Dirix LY, Paridaens R, Tan AR, Awada A, Ranade A, Jiao S, Schwartz G, Abbas R, Powell C, Turnbull K, Vermette J, Zacharchuk C, Badwe R. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J Clin Oncol. 2010;28(8):1301–1307. doi: 10.1200/JCO.2009.25.8707. [DOI] [PubMed] [Google Scholar]
- 66.FDA approval for sunitinib malate. http://www.cancer.gov/cancertopics/druginfo/fda-sunitinib-malate.
- 67.Oudard S, Beuselinck B, Decoene J, Albers P. Sunitinib for the treatment of metastatic renal cell carcinoma. Cancer Treat Rev. 2010 doi: 10.1016/j.ctrv.2010.08.005. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 68.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Hein-rich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failed imatinib; a randomised controlled trial. Lancet. 2006;368:1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 69.Casali PG, Garrett CR, Blackstein ME, Shah M, Verweij J, McArthur G, Judson I, Li J, Baum CM, Demetri GD. Updated results from a phase III trial of sunitinib in GIST patients (pts) for whom imatinib (IM) therapy has failed due to resistance or intolerance. J Clin Oncol, 2006 ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2006;24(18S):9513. [Google Scholar]
- 70.Carr LL, Mankoff D, Goulart BH, Eaton KD, Capell PT, Kell EM, Bauman J, Martins RG. Phase II study of sunitinib in FDG-PET positive, differentiated thyroid cancer and metastatic medullary carcinoma of thyroid with functional imaging correlation. 2010 doi: 10.1158/1078-0432.CCR-10-0994. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, Lehman M, Adams BJ, Bello CL, De-Primo SE, Baum CM, Miller KD. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with and anthracy-cline and a taxane. J Clin Oncol. 2008;26(11):1810–1816. doi: 10.1200/JCO.2007.14.5375. [DOI] [PubMed] [Google Scholar]
- 72.Sweeney CJ, Chiorean EG, Verschraegen CF, Lee FC, Jones S, Royce M, Tye L, Liau KF, Bello A, Chao R, Burris HA. A phase I study of sunitinib plus capecitabine in patients with advanced solid tumors. J Clin Oncol. 2010 doi: 10.1200/JCO.2009.26.9696. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhu AX, Duda DG, Ancukiewicz M, di Tomaso E, Clark JW, Miksad R, Fuchs CS, Ryan DP, Jain RK. Exploratory analysis of early toxicity of sunitinib in advanced hepatocellular carcinoma patients: kinetics and potential biomarker value. Clin Cancer Res. 2010 doi: 10.1158/1078-0432.CCR-10-0515. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kovacs MJ, Reece DE, Marcellus D, Meyer RM, Mathews S, Dong RP, Eisenhauer E. A phase II study of ZD6474 (Zactima), a selective inhibitor of VEGFR and EGFR tyrosine kinase in patients with relapsed multiple myeloma--NCIC CTG IND.145. Invest New Drugs. 2006;24(6):529–535. doi: 10.1007/s10637-006-9022-7. [DOI] [PubMed] [Google Scholar]
- 75.Arnold AM, Seynour L, Smylie M, Ding K, Ung Y, Findlay B, Lee CW, Djurfeldt M, Whitehead M, Ellis P, Goss G, Chan A, Meharchand J, Alam Y, Gregg R, Butts C, Langmuir P, Shepherd F. Phase II study of vandetanib or placebo in small-cell lung cancer patients after complete or partial response to induction chemotherapy with or without radiation therapy: National Cancer Institute of Canada Clinical Trials Group Study BR.20. J Clin Oncol. 2007;25(27):4278–4284. doi: 10.1200/JCO.2007.12.3083. [DOI] [PubMed] [Google Scholar]
- 76.Heymach JV, Johnson BE, Prager D, Csada E, Roubec J, Pesek M, Spasova I, Belani CP, Bodrogi I, Gadgeel S, Kennedy SJ, Hou J, Herbst RS. Randomised, placebo-controlled phase II study of vandetanib plus docetaxel in previously treated non small-cell lung cancer. J Clin Oncol. 2007;25(27):4270–4277. doi: 10.1200/JCO.2006.10.5122. [DOI] [PubMed] [Google Scholar]
- 77.Horti J, Widmark A, Stenzi A, Federico MH, Abratt RP, Sanders N, Pover GM, Bodrogi I. A randomized, double-blind, placebo-controlled phase II study of vandetanib plus docetaxel/prednisolone in patients with hormone-refractory prostate cancer. Cancer Biother Radiopharm. 2009;24(2):175–180. doi: 10.1089/cbr.2008.0588. [DOI] [PubMed] [Google Scholar]
- 78.Natale RB, Bodkin D, Govindan R, Sleckman BG, Rizvi NA, Capo A, Germonpre P, Eberhardt WE, Stockman PK, Kennedy SJ, Ranson M. Vandetanib versus gefitinib in patients with advanced non-small-cell lung cancer: results from a two-part, double-blind, randomised phase II study. J Clin Oncol. 2009;27(15):2523–2529. doi: 10.1200/JCO.2008.18.6015. [DOI] [PubMed] [Google Scholar]
- 79.Kesisis G, Broxterman H, Giaccone G. Angiogenesis inhibitors. Drug selectivity and target specificity. Curr Pharmacol Des. 2007;13(27):2795–2809. doi: 10.2174/138161207781757033. [DOI] [PubMed] [Google Scholar]
- 80.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliff P, Moons L, Jain RK, Collen D, Keshert E. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394(6692):485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 81.Gotnik K, Verheul HMW. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13:1–14. doi: 10.1007/s10456-009-9160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevaciaumab plus irinotecan, fluorouracil, and leukovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 83.Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus FOLFOX4 as a second-line treatment of colorectal cancer. Oncologist. 2007;12(3):356–361. doi: 10.1634/theoncologist.12-3-356. [DOI] [PubMed] [Google Scholar]
- 84.FDA approval for bevacizumab. http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab.
- 85.Kozloff M, Yood MU, Berlin J, Flynn PJ, Kabbinavar FF, Purdie DM, Ashby MA, Dong W, Sugrue MM, Grothey A. Clinical outcomes associated with bevacizumab-containing treatment of metastatic colorectal cancer: the BRiTE observational cohort study. Oncologist. 2009;14:862–870. doi: 10.1634/theoncologist.2009-0071. [DOI] [PubMed] [Google Scholar]
- 86.Stathopoulos GP, Batziou C, Trafalis D, Kountantos J, Batzios S, Stathopoulos J, Legakis J, Armakolas A. Treatment of colorectal cancer with and without bevacizumab: a phase III study. Oncology. 2010;78(5–6):376–381. doi: 10.1159/000320520. [DOI] [PubMed] [Google Scholar]
- 87.Dingemans AM, de Langen AJ, van den Boogaart V, Marcus JT, Backes WH, Scholtens HT, van Tinteren H, Hoekstra OS, Pruim J, Brans B, Thunnissen FB, Smit EF, Groen HJ. First-line erlotinib and bevacizumab in patient with locally advanced and/or metastatic non-small cell lung cancer: a phase II study including molecular imaging. Ann Oncol. 2010 doi: 10.1093/annonc/mdq391. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 88.Ortholan C, Durivault J, Hannoun-Levi JM, Guyot M, Bourcier C, Ambrosetti D, Safe S, Pages G. Bevacizumab/docetaxal association is more efficient that docetaxal alone in reducing breast and prostate cancer cell growth: a new paradigm for understanding the therapeutic effect of combined treatment. Eur J Cancer. 2010 doi: 10.1016/j.ejca.2010.07.021. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 89.Kountourakis P, Doufexis D, Maliou S, Karagiannis A, Kardara E, Margari C, Sykoutri D, Tzovaras A, Ardvanis A. Bevacizumab combined with two-weekly paclitaxel as first-line therapy for metastatic breast cancer. Anticancer Res. 2010;30(7):3969–3971. [PubMed] [Google Scholar]
- 90.Sathornsumetee S, Desjardins A, Vrendenburgh JJ, McLendon RE, Marcello J, Herndon JE, Mathe A, Hamilton M, Rich JN, Norfleet JA, Gururangan S, Friedman HS, Reardon DA. Phase II trial of bevacizumab and erlotinib in patients with recurrent malignant glioma. Neuro-oncology. 2010 doi: 10.1093/neuonc/noq099. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cohen MH, Gootenberg J, Keegan P, Pazdur R. FDA drug approval summary: bevacizimab (Avastin) plus carboplatin and paclitaxel as first-line treatment of advanced/metastatic recurrent nonsquamous non-small cell lung cancer. Oncologist. 2007;12(6):713–718. doi: 10.1634/theoncologist.12-6-713. [DOI] [PubMed] [Google Scholar]
- 92.Summers J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma. Oncologist. 2010;15(1):104–111. doi: 10.1634/theoncologist.2009-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chustecka Z. Will the FDA revoke bevacizumab’s approval for breast cancer? http://www.medscape.com/viewarticle/725509.
- 94.Chustecka Z. FDA delays decision on breast cancer indication for bevacizumab. http://www.medscape.com/viewarticle/728972.
- 95.Hurvitz SA, Allen HJ, Moroose RL, Chan D, Hagenstad C, Applebaum SH, Patel G, Hu EH, Ryba N, Lin LS, Wang H, Glaspy J, Slamon DJ, Kabbinavar F. A phase II trial of decetaxel with bevacizumab as first-line therapy for HER2-negative metastatic breast cancer (TORI B01) Clin Breast Cancer. 2010;10(4):307–312. doi: 10.3816/CBC.2010.n.040. [DOI] [PubMed] [Google Scholar]
- 96.Dickson MA, Schwartz GK. Development of cell-cycle inhibitors for cancer therapy. Curr Oncol. 2009;16(2):36–43. doi: 10.3747/co.v16i2.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cheng T. Cell cycle inhibitors in normal and tumor stem cells. Oncogene. 2004;23:7256–7266. doi: 10.1038/sj.onc.1207945. [DOI] [PubMed] [Google Scholar]
- 98.Sutherland RL, Musgrove EA. Cyclin E and prognosis in patients with breast cancer. N Engl J Med. 2002;347(20):1546–1547. doi: 10.1056/NEJMNEJMp020124. [DOI] [PubMed] [Google Scholar]
- 99.Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove EA, Sutherland RL. Expression and amplification of cyclin genes in human breast cancer. Oncogene. 1993;8(8):2127–2133. [PubMed] [Google Scholar]
- 100.Lechpammer M, Xu X, Ellis FH, Bhattacharaya N, Shapir GI, Loda M. Flavopiridol reduces malignant transformation of the esophageal mucosa in p27 knockout mice. Oncogene. 2005;224:1683–1688. doi: 10.1038/sj.onc.1208375. [DOI] [PubMed] [Google Scholar]
- 101.Semenov IAC, Roginskaya V, Chauhan D, Corey SJ. Growth inhibition and apoptosis of myeloma cells by the CDK inhibitor flavopiridol. Leukemia Res. 2002;26(3):271–280. doi: 10.1016/s0145-2126(01)00103-5. [DOI] [PubMed] [Google Scholar]
- 102.Ambrosinin G, Seelman SL, Qin LX, Schwartz GK. The cyclin-dependent kinase inhibitor flavopiridol potentiates the effects of topoisomerase I poisons by suppressing Rad51 expression in a p53-dependent manner. Cancer Res. 2008;68(7):2312–2320. doi: 10.1158/0008-5472.CAN-07-2395. [DOI] [PubMed] [Google Scholar]
- 103.Lin TS, Ruppert AS, Johnson AJ, Fischer B, Heerema NA, Andritsos LA, Blum KA, Flynn JM, Jones JA, Hu W, Moran ME, Mitchell SM, Smith LL, Wagner AJ, Raymond CA, Schaaf LJ, Phelps MA, Villalona-Calero MA, Grever MR, Byrd JC. Phase II study of flavopiridol in relapsed chronic lymphocytic leukemia demonstrating high response rates in genetically high-risk disease. J Clin Oncol. 2009;27(35):6012–8. doi: 10.1200/JCO.2009.22.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Karp JE, Blackford A, Smith BD, Alino K, Seung AH, Bolanos-Meade J, Greer JM, Carraway HE, Gore SD, Jones RJ, Levis MJ, McDevitt MA, Doyle LA, Wright JJ. Clinical activity of sequential flavopiridol, cytosine arabinoside and mitoxantrone for adults with newly diagnosed, poor-risk acute myelogenous leukemia. Leukemia Res. 2010;34(7):877–882. doi: 10.1016/j.leukres.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rathkopf D, Dickson MA, Feldman DR, Carvajal RD, Shah MA, Wu N, Lefkowitz R, Gonen M, Cane LM, Dials HJ, Winkelmann JL, Bosl GJ, Schwartz GK. Phase I study of flavopiridol with oxaliplatin and fluorouracil/leucovorin in advanced solid tumors. Clin Cancer Res. 2009;15(23):7405–11. doi: 10.1158/1078-0432.CCR-09-1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dey A, Wong ET, Cheok CF, Tergaonkar V, Lane DP. R-Roscovitine simultaneously targets both the p53 and NF-kappaB pathways and causes potentiation of apoptosis: implications in cancer therapy. Cell Death Differ. 2008;15(2):263–273. doi: 10.1038/sj.cdd.4402257. [DOI] [PubMed] [Google Scholar]
- 107.Raje N, Kumar S, Hideshima T, Roccaro A, Ishitsuka K, Yasui H, Shiraishi N, Chauhan D, Munshi NC, Green SR, Anderson KC. Seliciclib (CYC202 of R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood. 2005;106(3):1042–1047. doi: 10.1182/blood-2005-01-0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.MacCallum DE, Melville J, Frame S, Watt K, Anderson S, Gianella-Borradori A, Lane DP, Green SR. Seliclib (CYC202, R-roscovitine) induces cell death in multiple myeloma cell by inhibition of RNA polymerase II-dependent transcription and down-regulation of Mcl-1. Cancer Res. 2005;65(12):5399–5407. doi: 10.1158/0008-5472.CAN-05-0233. [DOI] [PubMed] [Google Scholar]
- 109.Whittaker SR, Te Poele RH, Chan F, Linardopoulos S, Walton MI, Garrette MD, Workman P. The cyclin-dependent kinase inhibitor seliciclib (R-roscovitine; CYC202) decreases the expression of mitotis control genes and prevents entry into mitosis. Cell Cycle. 2007;6(24):3114–3131. doi: 10.4161/cc.6.24.5142. [DOI] [PubMed] [Google Scholar]
- 110.Richardson PG, Hideshima T, Anderson KC. Bortezomib (PS-341): a novel, first-in-class proteosome inhibitor for the treatment of multiple myeloma and other cancers. Cancer Control. 2003;10(5):361–369. doi: 10.1177/107327480301000502. [DOI] [PubMed] [Google Scholar]
- 111.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Bladé J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352(24):2487–2496. doi: 10.1056/NEJMoa043445. [DOI] [PubMed] [Google Scholar]
- 112.San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shilberg O, Kropff M, Spicka I, Petrucci MT, Palumbo A, Samilova OS, Dmoszynska A, Abdulkadyrov KM, Schots R, Jiang B, Mateos MV, Anderson KC, Esseltine DL, Liu K, Cakana A, van de Velde H, Richardson PG. Bortezomib plus melphalan and prednisone for the initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 113.Richon VM, Zhou X, Rifkind RA, Marks PA. Histone deacetylase inhibitors: development of suberoylanilide hydroxamic acid (SAHA) for the treatment of cancers. Blood Cells, Mol Dis. 2001;27(1):260–264. doi: 10.1006/bcmd.2000.0376. [DOI] [PubMed] [Google Scholar]
- 114.Imre G, Gekeler V, Leja A, Beckers T, Boehm M. Histone deacetylase inhibitors suppress the inducibility of nuclear factor-KB by tumor necrosis factor-a receptor-1 down-regulation. Cancer Res. 2006;66:5409–5418. doi: 10.1158/0008-5472.CAN-05-4225. [DOI] [PubMed] [Google Scholar]
- 115.Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007;12(10):1247–1252. doi: 10.1634/theoncologist.12-10-1247. [DOI] [PubMed] [Google Scholar]
- 116.Paik PK, Krug LM. Histone deacetylase inhibitors in malignant pleural mesothelioma: preclinical rationale and clinical trials. J Thorac Oncol. 2010;5(2):275–279. doi: 10.1097/JTO.0b013e3181c5e366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Johnstone RW, Frew AJ, Smyth MJ. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat Rev, Cancer. 2008;8(10):782–798. doi: 10.1038/nrc2465. [DOI] [PubMed] [Google Scholar]
- 118.Luster TA, Carrell JA, McCormick K, Sun D, Humphreys R. Mapatumumab and lexatumumab induce apoptosis in TRAIL-R1 and TRAIL-R2 antibody-resistant NSCLC cell lines when treated in combination with bortezomib. Mol Cancer Ther. 2009;8(2):292–302. doi: 10.1158/1535-7163.MCT-08-0918. [DOI] [PubMed] [Google Scholar]
- 119.Belyanskaya LL, Marti TM, Hopkins-Donaldson S, Kurtz S, Felley-Bosco E, Stahel RA. Human agonistic TRAIL receptor antibodies Mapatumumab and Lexatumumab induce apoptosis in malignant mesothelioma and act synergistically with cisplatin. Mol Cancer. 2007;6:66. doi: 10.1186/1476-4598-6-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Marini P, Denzinger S, Schiller D, Kauder S, Welz S, Humphreys R, Daniel PT, Jendrossek V, Budach W, Belka C. Combined treatment of colorectal tumours with agonistic TRAIL receptor antibodies HGS-ETR1 and HGS-ETR2 and radiotherapy: enhanced effects in vitro and dose-dependent growth delay in vivo. Oncogene. 2006;25(37):5145–5154. doi: 10.1038/sj.onc.1209516. [DOI] [PubMed] [Google Scholar]
- 121.Tew K. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994;54:4313–4320. [PubMed] [Google Scholar]
- 122.Lewis A, Hayes JD, Wolf R. Glutathione and glutathione-dependent enzymes in ovarian adenocarcinoma cell lines derived from a patient before and after the onset of drug resistance: intrinsic differences and cell cycle effects. Carcinogenesis. 1988;9(7):1283–1287. doi: 10.1093/carcin/9.7.1283. [DOI] [PubMed] [Google Scholar]
- 123.Rosen LS, Laxa B, Boulos L, Wiggins L, Keck JG, Jameson AJ, Parra R, Patel K, Brown GL. Phase I study of TLK286 (Telcyta) administered weekly in advanced malignancies. Clin Cancer Res. 2004;10(11):3689–3698. doi: 10.1158/1078-0432.CCR-03-0687. [DOI] [PubMed] [Google Scholar]
- 124.Rosen LS, Brown J, Laxa B, Boulos L, Reiswig L, Henner WD, Lum RT, Schow SR, Maack CA, Kreck JG, Mascavage JC, Dombrowski JA, Gomez RF, Brown GL. Phase I study of TLK286 (glutathione S-transferase P1–1 activated glutathione analogue) in advanced refractory solid malignancies. Clin Cancer Res. 2003;9(5):1628–1638. [PubMed] [Google Scholar]
- 125.Kavanagh JJ, Gershenson DM, Choi H, Lewis L, Patel K, Brown GL, Garcia A, Spriggs DR. Multi-institutional phase 2 study of TLK286 (TELCYTA, a glutathione S-transferase P1–1 activated glutathione analog prodrug) in patients with platimun and paclitaxel refractory or resistant ovarian cancer. Inter J Gynecologic Cancer. 2005;15(4):593–600. doi: 10.1111/j.1525-1438.2005.00114.x. [DOI] [PubMed] [Google Scholar]
- 126.Kavanagh JJ, Levenbeck CF, Ramirez PT, Wolf JL, Moore CL, Jones MR, Meng L, Brown GL, Bast RC., Jr Phase 2 study of canfosfamide in combination with pegylated liposomal doxorubicin in platinum and paclitaxel resistant epithelial ovarian cancer. J Hematol Oncol. 2010;11(3):9. doi: 10.1186/1756-8722-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vergote I, Finkler N, del Campo J, Lohr A, Hunter J, Matei D, Kavanagh J, Vermorken JB, Meng L, Jones M, Brown G, Kave S. Phase 3 randomised study of canfosfamide (telcyta, TLK286) versus pegylated liposomal doxorubicin or topotecan as third-line therapy in patients with platinum-refractory or -resistant ovarian cancer. Eur J Cancer. 2009;45(13):2324–2332. doi: 10.1016/j.ejca.2009.05.016. [DOI] [PubMed] [Google Scholar]
- 128.Phase II randomized study of TLK286 versus doxorubicin HCl liposome or topotecan as thurd-line therapy in patients with platinnum-refractory or -resistant metastatic ovarian epithelial, fallopian tube, or primary peritoneal cancer. http://www.cancer.gov/search/ViewClinicalTrials.aspx?cdrid=353149&version=HealthProfessional&protocolsearchid=8293121.
- 129.Beaupre DM, Cepero E, Obeng EA, Boise LH, Lichtenheld MG. R115777 induces Ras-independent apoptosis of myeloma cells via multiple intrinsic pathways. Mol Cancer Ther. 2004;3:179–186. [PubMed] [Google Scholar]
- 130.Ferrarini M, Heltai S, Zocchi MR, Rugarli C. Unusual expression and localization of heat-shock proteins in human tumor cells. Abstract. Int J Cancer. 1992;51(4):613–619. doi: 10.1002/ijc.2910510418. [DOI] [PubMed] [Google Scholar]
- 131.Usmani SZ, Bona R, Li Z. 17 AAG for HSP90 inhibition in cancer--from bench to bedside. Curr Mol Med. 2009;9(5):654–664. doi: 10.2174/156652409788488757. [DOI] [PubMed] [Google Scholar]
- 132.Golubovskaya VM, Cance WG. Focal adhesion kinase and p53 signaling in cancer cells. Int Rev Cytol. 2007;263:103–153. doi: 10.1016/S0074-7696(07)63003-4. [DOI] [PubMed] [Google Scholar]
- 133.Golubovskaya VM, Conway-Dorsey K, Edmiston SN, Tse CK, Lark AA, Livasy CA, Moore D, Millikan RC, Cance WG. FAK overexpression and p53 mutations are highly correlated in human breast cancer. Int J Cancer. 2009;125(7):1735–1738. doi: 10.1002/ijc.24486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, Simkins S, Xu L. Immunohistochemical analysis of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000;6(6):2417–2423. [PubMed] [Google Scholar]
- 135.Oktay MH, Oktay K, Hamele-Bena D, Buyuk A, Koss LG. Focal adhesion kinase as a marker of malignant phenotype in breast and cervical carcinomas. Hum Pathol. 2003;34(3):240–245. doi: 10.1053/hupa.2003.40. [DOI] [PubMed] [Google Scholar]
- 136.Tremblay L, Hauck W, Aprikian AG, Begin LR, Chapdelaine S. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int J Cancer. 1996;68(2):164–171. doi: 10.1002/(sici)1097-0215(19961009)68:2<169::aid-ijc4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 137.Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of focal adhesion kinase gene and invasive cancer. Lancet. 1993;342(8878):1024–1025. doi: 10.1016/0140-6736(93)92881-s. [DOI] [PubMed] [Google Scholar]
- 138.Weiner TM, Liu ET, Craven RJ, Cance WG. Expression of growth factor receptors, the focal adhesion kinase, and other tyrosine kinases in human soft tissue tumors. Ann Surg Oncol. 1994;1(1):18–27. doi: 10.1007/BF02303537. [DOI] [PubMed] [Google Scholar]
- 139.Golubovskaya VM, Zheng M, Zhang L, Cance WG. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer. 2009;12(9):280. doi: 10.1186/1471-2407-9-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Golubovskaya VM, Gross S, Kaur AS, Wilson RI, Xu LH, Yang XH, Cance WG. Simultaneous inhibition of focal adhesion kinase and SRC enhances detachment and apoptosis in colon cancer cell lines. Mol Cancer Res. 2003;1(10):755–764. [PubMed] [Google Scholar]
- 141.Sakai R, Kagawa S, Yamasaki Y, Kojima T, Uno F, Hashimoto Y, Watanabe Y, Urata Y, Tanaka N, Fujiwara T. Pre-clinical evaluation of differentially targeting dual virotherapy for human solid cancer. Mol Cancer Ther. 2010;9(6):1884–1893. doi: 10.1158/1535-7163.MCT-10-0205. [DOI] [PubMed] [Google Scholar]
- 142.Wolf JK, Bodurka DC, Gano JB, Deavers M, Ramondetta L, Ramirez PT, Levenback C, Gershenson DM. A phase I study of Adp52 (INGN 201; ADVEXIN) for patients with platinum- and paclitaxel-resistant epithelial ovarian cancer. Gynecol Oncol. 2004;94(2):442–448. doi: 10.1016/j.ygyno.2004.05.041. [DOI] [PubMed] [Google Scholar]
- 143.Shimada H, Matsubara H, Shiratori T, Shimizu T, Miyazaki S, Okazumi S, Nabeta Y, Shuto K, Hayashi H, Tanizawa T, Nakatani Y, Nakasa H, Kitada M, Ochiai T. Phase I/II adenoviral p53 gene therapy for chemoradiation resistant advanced esophageal squamous cell carcinoma. Cancer Sci. 2006;97(6):554–561. doi: 10.1111/j.1349-7006.2006.00206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yoo GH, Moon J, Leblanc M, Lonardo F, Urba S, Kim H, Hanna E, Tsue T, Valentino J, Ensley J, Wolf G. A phase 2 trial of surgery with perioperative INGN 201 (Ad5CMV-p53) gene therapy followed by chemoradiotherapy for advanced, resectable squamous cell carcinoma of the oral cavity, oropharynx, hypopharynx, and larynx: report of the Southwest Oncology Group. Arch Otolaryngol Head Neck Surg. 2009;135(9):869–74. doi: 10.1001/archoto.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Schultze A, Decker S, Otten J, Horst AK, Vohwinkel G, Schuch G, Bokemeyer C, Loges S, Fiedler W. TAE226-mediated inhibition of focal adhesion kinase interferes with tumor angiogenesis and vasculogenesis. Investig New Drugs. 2009 doi: 10.1007/s10637-009-9326-5. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 146.Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, Coleman K, Yao L, Martinez-Alsina L, Lorenzen M, Berliner M, Lussion M, Patel N, Schmitt E, LaGreca S, Jani J, Wessel M, Marr E, Griffor M, Vajdos F. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68(6):1935–1944. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 147.Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383(6600):547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 148.Wendt MK, Schiemann WP. Therapeutic targeting of the focal adhesion complex prevents oncogenic TGF-beta signaling and metastasis. Breast Cancer Res. 2009;11(5):R68. doi: 10.1186/bcr2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bagi CM, Roberts GW, Andresen CJ. Dual focal adhesion kinase/Pyk2 inhibitor has positive effects on bone tumors: implications for bone metastases. Cancer. 2008;112(10):2113–2321. doi: 10.1002/cncr.23429. [DOI] [PubMed] [Google Scholar]
- 150.Sun H, Pisle S, Gardner ER, Figg WD. Bioluminescent imaging study: FAK inhibitor, PF-562,271, preclinical study in PC3M-luc-C6 local implant and metastasis xenograft models. Cancer Biol Ther. 2010;10(1):38–43. doi: 10.4161/cbt.10.1.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bagi CM, Christensen J, Cohem DP, Walter GR, Wilkie D, Swanson T, Tuthill T, Andresen CJ. Sunitinib and PF-562,271 (FAK/Pyk2 inhibitor) effectively block growth and recovery of human hepatocellular carcinoma in a rat xenograft model. Cancer Biol Ther. 2009;8(9):856–865. doi: 10.4161/cbt.8.9.8246. [DOI] [PubMed] [Google Scholar]
- 152.Hiscox S, Barnfather P, Hayes E, Bramble P, Christensen J, Nicholson RI, Barrett-Lee P. Inhibition of focal adhesion kinase suppresses the adverse phenotype of endocrine-resistant breast cancer cells and improves endocrine response in endocrine-sensitive cells. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-0857-4. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 153.Tanjoni I, Walsh C, Uryu S, Tomar A, Nam JO, Mielgo A, Lim ST, Liang C, Koenig M, Sun C, Patel N, Kwok C, McMahon G, Stupack DG, Schlaepher DD. PND- 1186 PAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010;9(10):764–777. doi: 10.4161/cbt.9.10.11434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schaller MD, Frisch SM. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010;9(10):791–793. doi: 10.4161/cbt.11729. [DOI] [PubMed] [Google Scholar]
- 155.Walsh C, Tanjoni I, Uryu S, Tomar A, Nam JO, Luo H, Phillips A, Patel N, Kwok C, McMahon G, Stupack DG, Schlaepfer DD. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther. 2010;9(10) doi: 10.4161/cbt.9.10.11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, Cance WG. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the Y397 site of focal adhesion kinase decreases tumor growth. J Med Chem. 2008;51:7405–7416. doi: 10.1021/jm800483v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zheng D, Golubovskaya V, Kurenova E, Wood C, Massol NA, Ostrov D. A novel strategy to inhibit FAK and IGF-1R decreases growth of pancreatic cancer xenografts. Mol Carcinogenesis. 2010;49(2):200–209. doi: 10.1002/mc.20590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Beierle EA, Ma X, Stewart J, Nyberg C, Trujillo A, Cance WG, Golubovskaya VM. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle. 2010;9(5):1005–1015. doi: 10.4161/cc.9.5.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Masasol NA, Magis A, Ostrov D, Cance WG, Golubovskaya VM. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle. 2009;8(15):2435–2443. doi: 10.4161/cc.8.15.9145. [DOI] [PMC free article] [PubMed] [Google Scholar]