

Abstract
Inflammation induces the NF-κB dependent protein A20 in human renal proximal tubular epithelial cells (RPTEC), which secondarily contains inflammation by shutting down NF-κB activation. We surmised that inducing A20 without engaging the pro-inflammatory arm of NF-κB could improve outcomes in kidney disease. We showed that hepatocyte growth factor (HGF) increases A20 mRNA and protein levels in RPTEC without causing inflammation. Upregulation of A20 by HGF was NF-κB/RelA dependent as it was abolished by overexpressing IκBα or silencing p65/RelA. Unlike TNFα, HGF caused minimal IκBα and p65/RelA phosphorylation, with moderate IκBα degradation. Upstream, HGF led to robust and sustained AKT activation, which was required for p65 phosphorylation and A20 upregulation. While HGF treatment of RPTEC significantly increased A20 mRNA, it failed to induce NF-κB dependent, pro-inflammatory MCP-1, VCAM-1, and ICAM-1 mRNA. This indicates that HGF preferentially upregulates protective (A20) over pro-inflammatory NF-κB dependent genes. Upregulation of A20 supported the anti-inflammatory effects of HGF in RPTEC. HGF pretreatment significantly attenuated TNFα-mediated increase of ICAM-1, a finding partially reversed by silencing A20. In conclusion, this is the first demonstration that HGF activates an AKT-p65/RelA pathway to preferentially induce A20 but not inflammatory molecules. This could be highly desirable in acute and chronic renal injury where A20-based anti-inflammatory therapies are beneficial.
Keywords: Inflammation, Cell signaling, Nuclear Factor kappa B, Hepatocyte Growth Factor, Acute Kidney Injury
Introduction
Renal inflammation is an invariable finding in acute and chronic kidney injury (Eddy, 2005; Liu, 2002; Remuzzi and Bertani, 1998). It is a critical initiating and aggravating factor in kidney damage (Akcay et al., 2009; Remuzzi et al., 1997). Specifically, RPTEC are central players in tubulointerstitial inflammation, being both producers of inflammatory cytokines and chemokines and early casualties of inflammation and apoptosis (Remuzzi et al., 1997; van Kooten et al., 1999). Clinical studies suggest that inflammation is a major aggravating factor in decline of renal function in patients with chronic kidney disease, or acute renal failure due to sepsis, acute graft rejection, or other causes (Akcay et al., 2009; Robertson and Kirby, 2003; Segerer et al., 2000). Accordingly, control of the inflammatory response has been beneficial in attenuating kidney injury in several animal models of chronic and acute renal failure (Tamada et al., 2003; Tashiro et al., 2003; Vielhauer et al., 2004). Clinically, more needs to be done to define optimal and safe anti-inflammatory therapies to prevent or treat acute kidney injury and forestall progression of chronic kidney disease.
Our laboratory has long focused on defining the functions and molecular targets of A20/TNFAIP3, a potent inhibitor of NF-κB activation and inflammation in many cells, including human RPTEC (Arvelo et al., 2002; Cooper et al., 1996; Grey et al., 1999; Kunter et al., 2005; Patel et al., 2006). A20 was first identified as a TNFα-inducible gene in endothelial cells (Opipari et al., 1990). Although originally characterized as an inhibitor of TNFα-induced apoptosis (Daniel et al., 2004; Opipari et al., 1992), its anti-apoptotic effect does not apply to RPTEC (Kunter et al., 2005). A20 is now recognized primarily as a central regulator of inflammation due to its ubiquitous inhibitory effect on NF-κB activation in response to a broad spectrum of pro-inflammatory mediators (Beyaert et al., 2000; Cooper et al., 1996; Longo et al., 2003). In previously published work, we demonstrated that overexpression of A20 in RPTEC strongly inhibits TNFα mediated up-regulation of the pro-inflammatory adhesion molecule ICAM-1 and the chemokine MCP-1, through inhibition of NF-κB activation (Kunter et al., 2005). A20 knockout mice die prematurely due to cachexia and massive multi-organ inflammation (Lee et al., 2000), demonstrating its fundamental role in the hierarchy of inflammatory responses. As a NFκB-dependent gene, A20 is part of a negative regulatory loop limiting NF-κB activation and inflammation (Bach et al., 1997; Krikos et al., 1992). Our laboratory is searching for means of inducing A20 to control inflammation without initiating an inflammatory response.
HGF is a multifunctional, pleoitropic protein with mitogenic, motogenic, and morphogenic effects in a wide variety of cells (Matsumoto and Nakamura, 1996). It is an important regulator of kidney function and a potent renoprotective agent (Matsumoto and Nakamura, 2001), preserving normal structure and function and accelerating recovery in experimental models of acute renal failure and ischemic injury (Kawaida et al., 1994; Miller et al., 1994). HGF also ameliorates chronic renal injury in a variety of models, including remnant kidney (Gong et al., 2004), unilateral ureteral obstruction (Mizuno et al., 2001) and diabetic nephropathy (Mizuno and Nakamura, 2004). These benefits are ascribed to its anti-apoptotic, anti-fibrotic (Liu, 2002), and anti-inflammatory effects in myriad pathologies including inflammatory bowel disease (Ohda et al., 2005), airway inflammation (Ito et al., 2005) and acute and chronic kidney disease (Gong et al., 2006a; Gong et al., 2006b; Kawaida et al., 1994). Some of the advantageous effects of HGF have been linked to its ability to induce (Muller et al., 2002) or inhibit NF-κB activation both in vitro (Gong et al., 2008; Min et al., 2005) and in vivo, including in models of inflammatory kidney diseases (Giannopoulou et al., 2008; Gong et al., 2006a; Gong et al., 2006b).
In this work, we questioned whether HGF treatment of RPTEC affects expression of the anti-inflammatory protein A20, and if so, how this relates to the anti-inflammatory effects of HGF and its ability to activate or inhibit NF-κB in these cells.
Material and Methods
Reagents
We purchased human recombinant tumor necrosis factor-alpha (TNFα) and hepatocyte growth factor (HGF) from R&D Systems (Minneapolis, MN, USA), and 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride (LY294002), gelatin and fetal bovine serum (FBS) from Sigma-Aldrich Co (St. Louis, MO, USA).
Cell culture
Primary human RPTEC were purchased from Lonza (Allendale, NJ, USA) and cultured in suggested medium: REBM/REGM bullet kit (Biowhittaker Inc, Walkersville, MD, USA) or RECGM2 (Promocell, Heidelberg, Germany), as described (Kunter et al., 2005). Confluent cells between passages 6–9, derived from 3 different single donor preparations were used in experiments.
Western blot analysis
RPTEC were incubated with HGF (50 ng/mL) or TNFα (100 U/mL) for varying time periods. In some experiments cells were pre-incubated with the PI3K/AKT inhibitor LY294002 for 30 min. Total protein cell lysates were recovered, and analyzed (Laemmli, 1970) by Western blot (WB), as described (Kunter et al., 2005) using: rabbit anti-IκB-α, mouse anti-βactin and rabbit anti-p65 antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), rabbit anti-phospho-p65 (Ser536; P-p65), rabbit anti-phospho-IκB-α (Ser32; P-IκBα), rabbit anti-phospho RelB (Ser552; P-RelB), and rabbit anti-phospho Glycogen Synthase 3 beta (Ser9; P-GSK3β) (Cell Signaling Technology, Danvers, MA, USA), chicken anti-TNFAIP3 (A20), and rabbit anti-A1 (Abcam Inc., Cambridge, MA, USA), and mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Calbiochem, San Diego, CA, USA). Membranes were incubated with appropriate secondary antibodies (Thermo Scientific, Rockford, IL, USA). Immunoblots were scanned and the band intensity was quantified by densitometry using NIH ImageJ 1.41 software.
Quantitative real time polymerase chain reaction (qPCR)
Messenger RNA was isolated from RPTEC using RNeasy mini kits (Qiagen, Valencia, CA, USA) and cDNA was synthesized using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). Quantitative PCR was performed to measure human p65, MCP-1, ICAM-1, VCAM-1, A1 and A20 mRNA levels (primer sequences available upon request) using iTaq Fast SYBR Green Supermix with ROX (Bio-Rad), and ABI 7500 Fast Real-time PCR system (Applied Biosystems, Inc., Foster City, CA, USA). Expression of target genes was normalized to that of the housekeeping gene or βactin.
Adenoviral-mediated gene transfer
Recombinant adenovirus (rAd.) vector expressing the porcine IκBα gene (ECI-6) (rAd.IκBα) was generated by C.J. Wrighton (Wrighton et al., 1996). Control rAd. β-galactosidase (rAd.βgal) was a kind gift from Dr. R. Gerald (University of Texas Southwestern Medical Center). rAd. were generated and tittered, as described (Ferran et al., 1998). RPTEC were transduced at a multiplicity of infection (MOI) of 50 plaque-forming units per cell (pfu/cell), as previously described (Kunter et al., 2005). Transgene expression (IκBα and βgal) was analyzed by Western blot and X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactoside) staining (Sanes et al., 1986).
SiRNA transfection
Predesigned p65/RelA siRNA, TNFAIP3 siRNA and AllStars negative control siRNA, as well as Hiperfect transfection reagent, were purchased from Qiagen. Transfections were carried out according to the manufacturer’s fast-forward transfection protocol. Experiments were performed 24–48 h following transfection. Knockdown of target genes was confirmed by qPCR and Wersten Blot.
Statistical analysis
Results are presented as means ± standard error of mean (SEM). Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by post-hoc Tukey multiple comparison test when F was significant, using Prism 5 for Mac software (GraphPad, Inc., La Jolla, CA, USA). Concentration-dependent effects were tested by Pearson correlation and linear regression analysis. Differences between groups were rated significant at a probability error (P) < 0.05.
RESULTS
HGF upregulates A20 mRNA and protein expression in RPTEC
To determine whether HGF regulates A20 expression in RPTEC, we treated human RPTEC cultures with HGF at concentrations ranging from 10 to 50 ng/mL for 2 to 24 h, and determined A20 protein levels by Western blot analysis. Our results indicated that HGF significantly upregulated A20 protein expression in a time and concentration dependent manner (Pearson (r)=0.9703, r2= 0.9415, p<0.05), with the maximum (up to 3-fold) observed 6 h after the addition of 50 ng/mL of HGF (Fig. 1A&B). Correspondingly, A20 mRNA levels peaked at 1 h and declined by 3 h, following HGF treatment as determined by quantitative real-time PCR (qPCR) (Fig. 1C). HGF-mediated upregulation of A20 mRNA was temporally comparable to that of TNFα (100U/mL, i.e. 5ng/mL), a bona-fide transcriptional activator of A20 (Kunter et al., 2005). Indeed, A20 mRNA levels also peaked at 1 h and declined by 3 h following addition of TNFα, (Fig. 1C). At these concentrations, A20 protein and mRNA levels were significantly higher following TNFα as compared to HGF (Fig. 1C&D). This reflects the use of TNFα concentration ranging 100-fold its effective dose (ED50). This TNFα dose is required to produce the pro-inflammatory effects of this cytokine in vitro (Gong et al., 2008; Lee et al., 2006). In fact, at comparable HGF (Rubin et al., 1991) and TNFα ED50 (Matthews et al., 1987) (2×ED50) TNFα (2 U/mL) failed to upregulate A20 or for the matter pro-inflammatory genes, as depicted in Figure 5, whereas HGF (50–60 ng/mL) led to maximal upregulation of A20 mRNA (Supplementary Fig. 1). Accordingly, all further experiments were performed using 50 ng/mL of HGF and 100 U/mL of TNFα.
Figure 1. HGF upregulates A20 mRNA and protein levels in RPTEC.

Western blots of RPTEC treated with A. HGF (50 ng/ml) for 2 to 24 h (time course); or B. HGF (10 to 50 ng/ml) for 6 h (dose curve). Cell lysates were run on PAGE, immunoblotted with anti-A20 antibody and re-probed with anti-GAPDH or anti-βactin antibody to correct for loading and allow semi-quantitative evaluation of the data by densitometry. Results of corrected densitometry, presented as percentage of control non-stimulated (NS) cells are expressed as mean± SEM of 4–5 independent experiments. C. Relative A20 mRNA levels measured by qPCR in human RPTEC cultures stimulated for 1 and 3 h with HGF (50 ng/ml) or TNFα (100 U/ml). Histograms represent the statistical analyzes of relative mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 4 independent experiments. D. Corresponding Western blot of RPTEC treated with HGF (50 ng/ml) or TNFα (100 U/ml) for 6 h; Results of corrected densitometry, presented as percentage of control non-stimulated (NS) cells are expressed as mean ± SEM of 4 independent experiments. *p<0.05, **p<0.01 and ***p< 0.001.
Figure 5. HGF does not upregulate pro-inflammatory cell surface adhesion molecules ICAM-1 and VCAM-1 and chemokine MCP-1 in RPTEC.

qPCR analysis was performed on total mRNA isolated from RPTEC cultures treated with HGF (25–200 ng/mL) or TNFα (5–200 U/mL) for 3h. Results demonstrate a significant concentration-dependent upregulation of MCP-1, VCAM-1 and ICAM-1 following treatment with TNF, while no significant upregulation of these genes was noted following treatment of RPTEC with HGF. Graph shows the statistical analyses of relative MCP-1, VCAM-1 and ICAM-1 mRNA levels after normalization by the housekeeping gene βactin. Results are expressed as mean ± SEM of 3–6 independent experiments. *p<0.05, **p< 0.01 and ***p< 0.001 when comparing with non-stimulated RPTEC; and ++p<0.01, +++p<0.001 when comparing RPTEC stimulated with HGF vs. TNFα.
HGF-induced upregulation of A20 is NF-κB-dependent
NF-κB activation is usually required to promote A20 transcription in response to pro-inflammatory stimuli (Bach et al., 1997; Krikos et al., 1992; Laherty et al., 1992; Laherty et al., 1993). However, activation of NF-κB by HGF remains controversial (Giannopoulou et al., 2008). Accordingly, we gauged the impact of inhibiting NF-κB by either overexpressing IκBα using recombinant adenovirus (rAd.) mediated gene transfer, or silencing the p65/RelA subunit of NF-κB, on HGF-mediated upregulation of A20. Our results showed that overexpression of IκBα significantly inhibited HGF-induced A20 protein and mRNA levels, as compared to non-transduced (NT) and rAd.βgal transduced RPTEC (Fig. 2A&B). A similar inhibition was also achieved in these conditions for TNFα-induced upregulation of A20. Moreover, knockdown of p65 using a specific siRNA, which decreased p65 mRNA levels by almost 80% and its protein levels by >90% (Supplementary Fig. 2), significantly decreased HGF-induced upregulation of A20 mRNA, while AllStars negative control siRNA did not (Fig. 2C). Altogether, these results indicate that HGF-induced upregulation of A20 in RPTEC requires NF-κB activation involving the p65/RelA subunit.
Figure 2. NF-κB is required for HGF-induced upregulation of A20 in RPTEC.

Overexpression of IκBα by means of rAd. mediated gene transfer (rAd.IκBα) significantly inhibits HGF (50 ng/ml), and TNFα (100 U/ml)-induced upregulation of A. A20 protein in human RPTEC, as shown by Western blot analysis 6 h following treatment. Cell lysates were immunoblotted with antibodies to A20, IκBα (to confirm transgene expression) and βactin (as loading control). Results shown are representative of 3 independent experiments.; and B. A20 mRNA levels, as evaluated by qPCR 1 h following treatment. Graphs represent the statistical analyzes of relative A20 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 4 independent experiments. A20 mRNA levels of non-stimulated (NS) RPTEC served as basal values. In both A and B, control cells were either non-transduced (NT) or transduced with the control adenovirus, rAd.βgal. *p<0.05 **p<0.01, and ***p< 0.001. C. p65/RelA silencing in RPTEC cultures significantly inhibits HGF (50 ng/ml) and TNFα (100 U/ml)-induced upregulation of A20 mRNA as shown by qPCR, 1 h following treatment. Graphs represent the statistical analyses of relative A20 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 3–5 independent experiments. Non-transfected (NT) or AllStars negative siRNA (Neg siRNA) served as controls. **p<0.01 and ***p<0.001.
HGF engages NF-κB signaling in a unique quantitative and qualitative manner
NF-κB activation by its primary activators, TNFα, IL-1 and LPS, occurs either through the canonical or alternative pathways. The former depends on activation of IκB kinase (IKK) subunits IKKβ and IKKα, IκBα phosphorylation, ubiquitination and degradation, p65/RelA phosphorylation, and translocation of the classical p50/p65 NF-κB heterodimer to the nucleus (Hacker and Karin, 2006). The latter requires specific activation of IKKα leading to p100 processing, followed by nuclear translocation of the p52:RelB NF-κB heterodimer (Hacker and Karin, 2006). Having already demonstrated the role of p65/RelA in inducing A20 transcription, we investigated whether HGF activates this subunit in RPTEC. Accordingly, we stimulated RPTEC with HGF (50 ng/mL) or TNFα (100 U/mL) as a positive control, and evaluated IκBα phosphorylation and degradation, and p65 phosphorylation by Western blot. Given our focus on early responses to HGF treatment, we did not evaluate its impact on phosphorylation and degradation of IκBβ. Indeed, IκBβ is classically responsible for delayed and persistent activation of NF-κB, in a stimulus and cell-type dependent manner that excludes TNFα; i.e TNFα does not usually cause degradation of IκBβ (Thompson et al., 1995). HGF stimulation resulted in a moderate, and transient phosphorylation of IκBα and p65, peaking within 5 min of HGF addition. This led to incomplete IκBα protein degradation within 15 min of adding HGF (Fig. 3). TNFα treatment, in contrast, led to significantly greater phosphorylation of IκBα and p65, followed by a complete degradation of IκBα within 15 min of adding TNFα (Fig. 3). Although the time course of phosphorylation/degradation of p65 and IκBα were fairly similar in HGF and TNFα-treated cells, these results highlight a significant quantitative difference between these two stimuli in engaging NF-κB signals. We also evaluated phosphorylation of RelB, a central player in the non-canonical pathway. Our results showed a robust phosphorylation of RelB at Ser-552 within 15 min of adding HGF, while we noted very mild phosphorylation of RelB following TNFα, at least within the studied time frame (Supplementary Fig. 3A). These latter results demonstrate a significant qualitative difference between TNFα and HGF in engaging NF-κB activation in RPTEC. However, in contrast to RelA, silencing of RelB that led to 80% knockdown of its RNA levels and >95% knockdown of its protein levels (Supplementary Fig. 2) did not impact HGF-induced upregulation of A20, indicating that it was not likely required for this process (Supplementary Fig. 3B).
Figure 3. HGF and TNFα differently activate NF-κB in RPTEC.
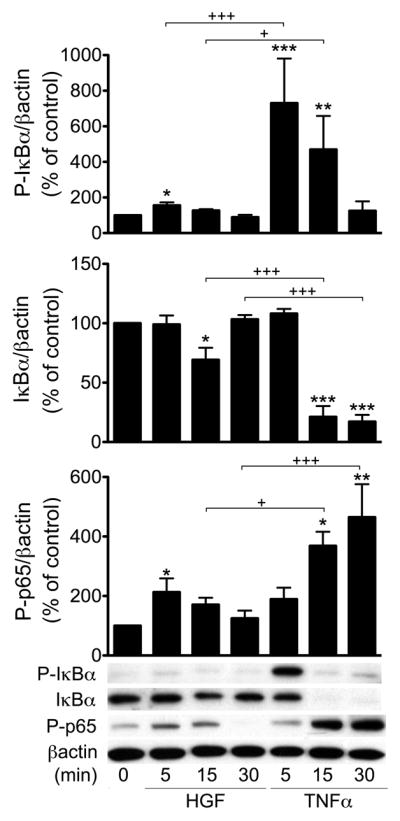
Cell lysates from RPTEC treated with HGF (50 ng/ml) or TNF (100 U/ml) for 5–30 min were immunoblotted with antibodies to phospho-IκBα (Ser32; P-IκBα), IκBα, phospho-65 (Ser536; P-p65). Results demonstrated significantly less IκBα phosphorylation and degradation, and less p65 phosphorylation in HGF-treated versus TNFα-treated RPTEC. Immunoblotting with the housekeeping protein βactin was used to correct for loading and allow semi-quantitative evaluation of the data by densitometry. Results of corrected densitometry, presented as percentage of control non-stimulated (NS) cells are expressed as mean ± SEM of 3–5 independent experiments. *p<0.05, **p<0.01 and ***p< 0.001 when comparing with non-stimulated RPTEC, and +p< 0.05, +++p<0.001 when comparing HGF vs. TNFα.
Activation of PI3K/AKT is required for the upregulation of A20 by HGF
To further analyze the molecular mechanisms upstream of IκBα and p65 phosphorylation that support HGF-induced upregulation of A20, we investigated which signaling pathway triggered by HGF is responsible for mediating NF-κB activation and increased A20 expression. The serine/threonine protein kinase, AKT, has been identified both as an activation target of the HGF/cMet signaling pathway in different cell types including RPTEC (Gong et al., 2005; Liu, 1999; Nakagami et al., 2001), and also as one of the possible upstream kinases in the NF-κB pathway (Ozes et al., 1999; Romashkova and Makarov, 1999). To investigate the role of AKT in HGF-induced activation of NF-κB and induction of A20, we treated RPTEC with 50ng/mL of HGF for 5 min to 2 h, and evaluated AKT phosphorylation at Ser-473 by Western blot. Our data indicate that HGF-induced phosphorylation of AKT was fast and sustained (Fig. 4A). Phosphorylation of GSK3β at Ser9, downstream of AKT activation, paralleled AKT phosphorylation in both its fastness and robustness in response to HGF (Fig. 4B). This result is in keeping with Gong et al’s report implicating AKT-dependent phosphorylation/inactivation of GSK3β in mediating the anti-inflammatory effects of HGF (Gong et al., 2008). In contrast, treatment of RPTEC with TNFα failed to phosphorylate AKT at Ser473, and GSK3β at Ser9, further highlighting the engagement of differential signaling pathways by HGF and TNFα Fig. 4B. Pretreatment of RPTEC with the PI3K inhibitor LY-294002 abolished phosphorylation of p65, the indicator of NF-κB activation (Fig. 4C), and significantly decreased A20 protein levels, downstream of p65 (Fig. 4D). These results indicate that AKT may directly activate IKKα and IKKβ to phosphorylate IκBα, and we confirmed by Western blot that HGF treatment of RPTEC phosphorylates IKKα/β, in an AKT-dependent manner, since this phosphorylation was blocked by pre-incubation with LY-294002 (Supplementary Fig. 4). Given low IκBα and high RelB phosphorylation in response to HGF, we surmise that the phospho IKKα/β band detected by Western blot is likely to be mainly composed of IKKα, the upstream kinase of RelB and less IKKβ, the upstream kinase of IκBα (Hacker and Karin, 2006). HGF-induced phosphorylation of RelB, was also AKT-dependent, as it was significantly blunted by LY-294002 (Supplementary Fig. 3C). Together, these results indicate that HGF-induced A20 upregulation in RPTEC occurs via an AKT/NF-κB-dependent pathway.
Figure 4. HGF-induced upregulation of A20 in RPTEC is AKT-dependent.

A. Cell lysates from RPTEC treated with HGF (50 ng/ml) for 5–120 min were immunoblotted with antibodies to phospho-AKT or total-AKT. Results showed fast, robust, and sustained phosphorylation of AKT following treatment with HGF. Results shown are representative of 3 independent experiments. B. HGF-induced phosphorylation of GSK3β, marking its inactivation by AKT, was significantly upregulated 10 and 30 min following treatment with HGF but not TNFα, as shown by Western blot analysis using a phospho-GSK3β antibody (P-GSK3β). C. HGF-induced phosphorylation of p65 was significantly reduced when RPTEC were pre-incubated with the 10 μM of the PI3K inhibitor, LY294002, for 30 min prior to adgding 50 ng/ml HGF for 15 min, as shown by Western blot analysis using a phospho-p65 specific antibody (P-p65). D. Similarly, HGF-induced upregulation of A20 was significantly inhibited when RPTEC were preincubated with 10 μM of LY294002 for 30 min prior to adding 50 ng/ml HGF for 6 h, as shown by Western blot analysis using an anti-A20 antibody. Cells lysates were also immunoblotted with anti P-AKT and AKT antibodies to control for LY294002 activity. In B, C, and D GAPDH or βactin immunoblots were used to correct for loading and allow semi-quantitative evaluation of the data by densitometry. Results of corrected densitometry, presented as percentage of control non-stimulated (NS) cells, are expressed as mean ± SEM of 4 (B&C), and 6–8 (D) independent experiments *p<0.05, ** p< 0.01. *** p<0.001.
HGF-induced activation of NF-κB in RPTEC does not cause pro-inflammatory responses
In most cell types, activation of NF-κB in response to inflammation concomitantly triggers pro-inflammatory (Lawrence, 2009; Pasparakis, 2009) and anti-inflammatory responses, including A20, which secondarily suppresses NF-κB activation (Bach et al., 1997; Lawrence and Fong, 2010). Accordingly, an inflammatory burst normally precedes accumulation of functional levels of A20. We questioned whether HGF-mediated activation of NF-κB would be accompanied by such a burst. Therefore, we investigated the effects of HGF on expression of the NF-κB dependent chemokine MCP-1, and adhesion molecules, VCAM-1 and ICAM-1 in RPTEC (Collins et al., 1995; Kunter et al., 2005). Cells were treated for 3h with varying concentrations of HGF (25–200 ng/mL) or TNFα (5–200 U/mL), and MCP-1, VCAM-1 and ICAM-1 mRNA levels were measured. HGF treatment did not significantly affect basal expression of these pro-inflammatory molecules, even at the highest HGF concentration (Fig. 5). In contrast, and in agreement with previously published data, TNFα increased MCP-1, VCAM-1 and ICAM-1 mRNA levels in a concentration-dependent manner (MCP-1, Pearson r=0.8761, r2=0.2351, p< 0.05; VCAM-1 Pearson r= 0.9516, r2= 0.61 P<0.0001; ICAM-1 Pearson r=0.9679, r2=0.644, p< 0.001) (Fig. 5). These results suggest that HGF may activate NF-κB in a manner that favors the transcription of anti-inflammatory and protective NF-κB dependent genes such as A20, but not that of the pro-inflammatory, NF-κB dependent genes MCP-1, VCAM-1 and ICAM-1. Our finding that HGF upregulates mRNA and protein levels of the NF-κB dependent anti-apoptotic Bcl family member, A1/Bfl1, in RPTEC supports this hypothesis (Kunter et al., 2005) (Supplementary Fig. 5).
HGF inhibits TNF-induced pro-inflammatory response in RPTEC, in part via an A20-dependent mechanism
As stated, HGF’s remarkable anti-inflammatory effect in RPTEC occurs partly through blocking NF-κB activation (Gong, 2008). To determine whether HGF affects TNFα-induced inflammatory responses in RPTEC, we examined the effects of pre-treatment with HGF on TNFα-induced ICAM-1 upregulation. Our results indicate that 4 h pre-treatment of RPTEC with HGF significantly inhibited TNFα-induced upregulation of ICAM-1 mRNA (Fig. 6). In contrast, simultaneous treatment of RPTEC with HGF and TNFα failed to reduce ICAM-1 upregulation by TNFα (data not shown), suggesting that HGF-dependent inhibition of TNFα-induced inflammatory response may require de novo protein synthesis. Having demonstrated that maximal HGF-induced upregulation of A20 occurs at a time similar to the HGF pre-treatment time required to achieve inhibition of TNFα-induced upregulation of ICAM-1, we questioned whether HGF-induced A20 contributed to the anti-inflammatory effect of this growth factor in RPTEC.
Figure 6. HGF-induced upregulation of A20 contributes to the anti-inflammatory effect of this growth factor in RPTEC.

RPTEC were transfected with A20 silencing RNA (A20 siRNA) or AllStars negative control siRNA (Neg siRNA). Twenty-four h later, cells were pre-treated with HGF (50 ng/mL) for 4 h, then stimulated with TNFα (100 U/mL) for 3h. RNA was extracted and ICAM-1 mRNA levels were determined by qPCR. Our results indicate that pre-treatment of RPTEC with HGF significantly attenuates TNFα-induced upregulation of ICAM-1 in non-transfected (NT) and AllStars negative control siRNA transfected RPTEC. A20 silencing significantly decreases the ability of HGF to inhibit TNFα-induced upregulation of ICAM-1. Moreover, A20 silencing significantly increases basal, as well as HGF, and TNFα-induced upregulation of ICAM-1 mRNA. Graphs shown represent the statistical analyses of relative ICAM-1 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 4–6 independent experiments. *p<0.05 when comparing RPTEC stimulated with TNFα vs. cells pre-incubated with HGF prior to TNFα. +p<0.05, ++<0.01, +++p<0.001 when comparing with corresponding values in NT RPTEC, and δ p<0.05 when comparing with corresponding values in AllStars negative control siRNA transfected RPTEC.
To test this hypothesis, we silenced A20 expression with a specific siRNA that knocked down HGF-induced A20 mRNA upregulation by >90% and TNFα-induced upregulation by almost 50% (Supplementary Fig. 6), and evaluated the impact of HGF upon TNFα-induced upregulation of ICAM-1 mRNA in both conditions. Our results indicated that A20 silencing significantly reduces HGF’s inhibitory effect upon TNFα-mediated upregulation of ICAM-1 mRNA (Fig. 6). This result demonstrates that A20 actively mediates the anti-inflammatory effects of HGF in RPTEC. Remarkably, A20 silencing significantly upregulated ICAM-1 mRNA levels in basal condition, and following HGF and TNFα treatment of RPTEC (Fig. 6), which highlights A20’s ability to maintain basal anti-inflammatory status, prevent an inflammatory response following HGF treatment, and control TNFα pro-inflammatory responses. In support of this latter, we demonstrate that A20 silencing in RPTEC significantly increases ICAM-1 and MCP1 basal but also TNFα-induced mRNA levels (Supplementary Fig. 7).
Discussion
Inflammation is a major pathological finding in a variety of chronic kidney diseases and acute renal failure (Segerer et al., 2000). In the latter, it manifests primarily in ischemia/reperfusion injury, often following renal transplantation (Gueler et al., 2004). Rapid activation of NF-κB follows ischemic events in the kidney, resulting in increased production of pro-inflammatory cytokines and chemokines, and culminating in vascular compromise and kidney damage (Cao et al., 2004; Linas et al., 1988). It is well appreciated that RPTEC are the most susceptible to this injury, which results in tubular necrosis. In fact, RPTEC contribute to their demise by acquiring a pro-inflammatory phenotype, promoting the local inflammatory milieu (Bonventre and Zuk, 2004). Therefore, controlling inflammation, particularly in RPTEC, is an attractive therapeutic strategy to reverse acute kidney injury, halt chronic disease, and protect renal allografts.
Our laboratory has explored the anti-inflammatory effect of A20 in different cell types. We have shown that this protein exerts potent NF-κB inhibitory effects in RPTEC, shutting down the upregulation of pro-inflammatory molecules such as MCP-1, and ICAM-1 (Kunter et al., 2005). We surmised that A20-based therapies could reduce inflammation, ultimately preventing renal dysfunction. As evidence, overexpression of A20 in rat kidneys using rAd-mediated gene transfer protects from acute tubular necrosis following renal ischemia (Lutz et al., 2008). However, translation of A20 gene-therapies to the clinic has proven difficult given the toxicity of these viral vectors (2002). Our laboratory, therefore, has been exploring means to induce A20 in RPTEC without prompting a concurrent inflammatory response (Bach et al., 1997).
Our results presented herein demonstrate that this goal could be achieved by HGF. Treatment of RPTEC with HGF promoted A20 transcription and protein expression in a time and concentration-dependent manner. HGF-induced A20 transcription in human primary RPTEC, echoes similar results obtained in the mammary MCF7 tumor cell line (Leroy et al., 2006). Interestingly, HGF also induced A20 transcription in primary human coronary artery endothelial cell (EC) but not smooth muscle cells despite similar expression of the HGF receptor, c-Met, in these cells (Nakamura et al., 1995) (Supplementary Fig. 8). This indicates a cell-type specific effect of HGF that could be exploited for achieving differential therapeutic effects. Remarkably, our data also demonstrates that HGF upregulates A20 in RPTEC without upregulating the pro-inflammatory molecules MCP-1, VCAM-1 and ICAM-1. This held true even at concentrations 4-fold higher than those upregulating A20 expression. In complete contrast, pro-inflammatory mediators like TNFα upregulated MCP-1, VCAM-1 and ICAM-1 concurrently with A20.
As stated earlier, A20 is a NF-κB dependent gene activated through the canonical NF-κB pathway in response to pro-inflammatory cytokines. We questioned whether NF-κB activation was also required for HGF-mediated activation of A20. Our results showed unequivocally that it was NF-κB dependent, since over-expression of the NF-κB repressor IκBα and silencing of p65 inhibited HGF-dependent A20 upregulation. However, HGF-triggered signaling differed qualitatively and quantitatively from that of TNFα. HGF treatment of RPTEC induced very mild IκBα and p65 phosphorylation paralleling slower and significantly lower IκBα degradation, compared to TNFα. These results are in agreement with previous results obtained in the MLP29 liver cell line (Muller et al., 2002), and with earlier studies implicating HGF/cMet signaling in activating the canonical NF-κB pathway (Fan et al., 2005). In contrast, HGF promoted quicker and stronger phosphorylation of RelB Ser-552 than TNFα. HGF-mediated phosphorylation of RelB was likely not implicated in A20 upregulation, since RelB silencing did not affect it. This result demonstrates qualitative differences between HGF and TNFα engagement of non-canonical NF-κB signaling molecules. Phosphorylation of Ser-552 of RelB is a potential tag for its degradation (Marienfeld et al., 2001), which could protect from kidney injury since RelB silencing in the kidney was shown to prevent renal ischemia/reperfusion injury in mice (Feng et al., 2009). However, we did not observe any loss of RelB following its phosphorylation by HGF in RPTEC, at least within the time limit of our experiments (data not shown).
Having established that HGF-induced upregulation of A20 relied on the activation of the canonical NF-κB pathway, we searched for the pertinent upstream kinase (Dejardin, 2006; Scheidereit, 2006). AKT is a recognized activator of both canonical and non-canonical NF-κB pathways (Gustin et al., 2006; Ozes et al., 1999; Sizemore et al., 1999). It is also an activation target of HGF/cMet in different cell types (Khwaja et al., 1998; Kroening et al., 2010; Muller et al., 2002; Suzuki et al., 2000). We identified AKT as key to NF-κB activation and subsequent A20 upregulation in HGF-treated RPTEC. Indeed, inhibition of PI3K/AKT by LY294002 blocked phosphorylation of ReIB and abolished HGF-mediated IKKα/β and p65-Ser536 phosphorylation, thereby inhibiting A20 upregulation. These novel data in primary human RPTEC parallel AKT’s role in protecting breast cancer, glioma and canine kidney cell lines from apoptosis (Fan et al., 2005). However, they contradict results showing that ERK1/2 and p38 MAPK, but not AKT, are required for HGF-induced activation of NF-κB in the MLP29 liver cell line (Muller et al., 2002), which again emphasizes cell type specificity. Interestingly, HGF treatment of RPTEC led to stronger and more prolonged AKT phosphorylation when TNFα almost did not impact AKT phosphorylation levels in RPTEC, further underlining the differences between HGF and TNFα signaling in these cells (Fig. 4B).
Furthermore, pre-incubation of RPTEC with HGF for 4 h significantly prevented TNFα-induced upregulation of ICAM-1 expression, while simultaneous exposure to both HGF and TNFα failed to do so, likely indicating the need for de novo synthesis of an anti-inflammatory protein by HGF. These findings agree with previous studies showing that HGF decreases TNFα-mediated upregulation of the NFκB-dependent pro-inflammatory chemokines, RANTES and MCP-1 and adhesion molecule E-Selectin in the human RPTEC cell line, HKC8, and in human umbilical vein EC (Gong et al., 2006b; Gong et al., 2008). However, in contrast with our data, this inhibitory effect of HGF could be achieved with short (30 min) or no pre-incubation period, suggesting an alternative mechanism. In fact, our results indicate that in primary human RPTEC, the anti-inflammatory effect of HGF depends significantly on its ability to upregulate A20, given 4 h pre-treatment, i.e. to allow for de novo synthesis of the A20 protein. Whereas, Gong et al. showed that the anti-inflammatory effect of HGF in HKC8 cells correlated with an AKT-dependent inhibition of GSK3β activity, which inhibits p65/RelA phosphorylation at the GSK3β target residue Ser-468, an event required for the activation of NF-κB dependent pro-inflammatory MCP-1 and RANTES, but not protective IκBα and Bcl-2 (Gong et al., 2006b; Gong et al., 2008). Since this effect is post-translational, it understandably did not require prolonged pre-incubation with HGF. In accordance with these data, we confirm that HGF inactivates GSK3β as a result of its phosphorylation by AKT, which could certainly contribute to the anti-inflammatory effect of HGF, i.e prevent HGF-induced upregulation of MCP-1, VCAM-1, and ICAM-1. We are also exploring whether GSK3β inactivation is implicated in HGF-induced A20 upregulation in RPTEC.
Furthermore, A20 silencing significantly increased peak and basal expression levels of ICAM-1, used a surrogate for inflammation, despite pretreatment of RPTEC with HGF prior to TNFα, further confirming the importance of A20 in maintaining a non-inflammatory homeostatic state in RPTEC. Altogether, these data stress the diversity of seemingly independent HGF targets, underscoring its profound anti-inflammatory effects.
HGF treatment of RPTEC also led to upregulation of A1, an NF-κB dependent anti-apoptotic Bcl family member, that, as we have shown earlier, exerts potent anti-apoptotic effects in RPTEC (Kunter et al., 2005). This result highlights HGF’s ability to activate NF-κB in a way that promotes transcriptional upregulation of anti-inflammatory and anti-apoptotic but not pro-inflammatory genes, creating an enduring protection against a later inflammatory insult. These results support data from Bendinelli et al. showing that 24 h treatment with HGF decreases NF-κB transactivation in chondrocytes by increasing IκBα expression (Bendinelli et al., 2010). However, these authors did not show whether or not this was related to an initial activation of NF-κB, as we have for A20.
In summary, this study unravels and characterizes a novel, HGF-induced pathway that upregulates the potent anti-inflammatory protein A20 in human RPTEC without concurrent up-regulation of pro-inflammatory molecules. Furthermore, it shows that this upregulation of A20 contributes to HGF’s anti-inflammatory capacity. While it requires NF-κB activation, this process differs qualitatively, quantitatively and kinetically from that initiated by TNFα. Work is underway to further delineate these differences, which could help us identify novel therapeutic targets. Importantly, these data reconcile conflicting literature demonstrating both activation and inhibition of NF-κB by HGF/cMet signaling. We clearly show the bimodal ability of HGF in human primary RPTEC, to activate the protective arm of NF-κB and enhance the anti-inflammatory defenses of the cell, preventing full-blown responses to later inflammatory insults.
Clinically, these data unveil the possibility of increasing renal levels of A20 without causing inflammation allowing for the prevention of acute and chronic kidney diseases. It would also be ideal for kidney preconditioning to avoid ischemic and inflammatory injury in the peri-transplant period, thereby reducing primary non-function and incidence of acute rejection. HGF-based therapies are already available, having been implemented in experimental kidney disease and renal transplant models (Gong et al., 2004; Mizuno et al., 2001; Mizuno and Nakamura, 2004; Yamada, 2005; Zhang et al., 2008); and in early clinical trials treating kidney disease and ischemic ulcers of vascular diseases (Shigematsu et al., 2010). The fact that HGF also induces A20 in EC is likely to further improve renal protection, especially in the context of ischemic injury. However, we must acknowledge the potential carcinogenic effects of HGF (Comoglio et al., 2008) and its cell-type specificity. With these caveats, we aim to better delineate the molecular targets of HGF and isolate differences in affecting NF-κB activation pathways, to ultimately identify more selective and safer therapies. We envision preferential upregulation of the anti-inflammatory protein A20 as highly significant in this search.
Supplementary Material
qPCR analysis of A20 mRNA was performed on total mRNA isolated from RPTEC cultures treated with HGF 60 and 300ng/mL, or TNFα 2 and 10 U/mL, which corresponds to 2 and 10×ED50 concentrations of these molecules, respectively. Results demonstrate that HGF, but not TNFα, significantly increases A20 mRNA at the 2×ED50 concentration. HGF-induced upregulation of A20 mRNA reaches a plateau above this concentration, while TNFα demonstrate a dose dependent response, as measured up to 100 U/mL. Graphs shown represent the statistical analyses of relative A20 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 5 independent experiments. *p<0.05, **p< 0.01 and ***p< 0.001.
{kind=link}
RPTEC were transfected with p65 siRNA, RelB siRNA or AllStars negative control siRNA (Neg siRNA) for 48 h. Total cellular mRNA and protein were recovered and p65 and RelB mRNA and protein were measured by qPCR (expressed as percentage of non-transfected (NT) control cells after normalization by βactin) and Western blot analysis, respectively. We noted 75% to 80% reduction of mRNA levels and 90–95% reduction of protein levels (as depicted on representative Western blots) of both targeted genes. qPCR results are expressed as mean ± SEM of 4–5 independent experiments. ***p<0.01.
{kind=link}
A. Representative Western blot analysis using a phospho-RelB (P-RelB) specific antibody demonstrates significantly greater phosphorylation of RelB in RPTEC cultures 15 min following HGF (50 ng/mL), as compared to TNFα (100 U/mL) treatment. βactin immunoblotting was used to correct for loading and allow semi-quantitative evaluation of the data by densitometry. Results of corrected densitometry, presented as percentage of control non-stimulated (NS) cells are expressed as mean ± SEM of 3 independent experiments. *p<0.05 compared to NS cells B. RPTEC were transfected with RelB silencing RNA (RelB siRNA) 48 h, then stimulated with HGF (50 ng/mL) or TNFα (100 U/mL) for 1h, and A20 mRNA was evaluated by qPCR. Results indicate that RelB knockdown does not affect HGF-induced upregulation of A20 mRNA. Non-transfected (NT) and AllStars negative control siRNA (Neg siRNA) transfected cells were used as controls. Graphs shown represent the statistical analyses of relative A20 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 3–5 independent experiments. ***p<0.001 compared to NS cells within each siRNA treatment group. Pre-incubation with 10 μM of the PI3K inhibitor LY294002 for 30 min, prior to treatment with 50 ng/mL of HGF for 15 min (C) or 100U/mL of TNFα for 30 min (D) significantly decreased phosphorylation of RelB, as shown by Western blot analysis using a phospho-RelB antibody. Immunoblotting with βactin antibody was used to correct for loading and allow semi-quantitative evaluation of the data by densitometry. Results of corrected densitometry, presented as percentage of NS cells are depicted in the graph, and expressed as mean ± SEM of 3–4 independent experiments. *p<0.05; **p<0.01; ***p<0.001
{kind=link}
Representative Western blot of RPTEC treated with HGF (50 ng/mL) show IKKα/β phosphorylation 5 min following HGF treatment. Phosphorylation of IKKα/β was abrogated when RPTEC were pre-incubated with LY294002. Immunoblotting with βactin antibody served as loading control. Results shown are representative of 3 independent experiments.
{kind=link}
A. HGF (50 ng/mL) or TNFα (100 U/mL) treatment for 1 h significantly upregulate A1 mRNA levels in RPTEC, within of treatment, as measured by qPCR. Graphs shown represent the statistical analyses of relative A1 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 3 independent experiments. *p<0.05; ***p<0.001 compared to non-stimulated (NS) cells. B. This translates into a substantial increase in A1 protein levels 6h after treatment with either HGF or TNFα, as demonstrated by Western blot analysis using an anti-human A1 antibody. Immunoblotting with a GAPDH antibody served as loading control. C. HGF-induced A1 protein upregulation shows a time dependent curve that peaks 6 h, and is still sustained 24 h following addition of 50ng/mL of HGF, as shown by Western blot analysis. Immunoblotting with anti-GAPDH antibody served as loading control. Results are representative of 3 independent experiments
{kind=link}
RPTEC were either non-tranfected (NT) or transfected with A20 silencing RNA (A20 siRNA) or AllStars negative control siRNA (Neg siRNA) for 24 h. Cells were then pre-treated with HGF (50 ng/mL) for 4h followed by TNFα (100 U/mL) for 3h, and total RNA was extracted. A20 mRNA levels were measured by qPCR, normalized by βactin. Results show a total (100% inhibition) of HGF-induced and a significant 50% reduction of TNFα-induced A20 mRNA levels in A20 siRNA transfected RPTEC. Results are expressed as mean ± SEM of 4–6 independent experiments. *p<0.05, **p<0.01 when comparing RPTEC with corresponding values in NT, and δp<0.05 when comparing with corresponding values in AllStars siRNA transfected RPTEC.
{kind=link}
RPTEC were transfected with A20 silencing RNA (A20 siRNA) or AllStars negative control siRNA (Neg siRNA). Twenty-four h later, cells were stimulated with TNFα (100 U/mL) for 3h. RNA was extracted and MCP-1 and ICAM-1 mRNA levels were determined by qPCR. Our results indicate that A20 silencing significantly increases basal, and TNFα-induced upregulation of MCP-1 and ICAM-1 mRNA. Graphs shown represent the statistical analyses of relative MCP-1 and ICAM-1 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 5–7 independent experiments. *p<0.05, **p< 0.01 and ***p< 0.001.
{kind=link}
HCAEC and HCASMC were purchased from Lonza, cultured according to the manufacturer’s recommendations, and used between passages 4–8. Western blots of A. HCAEC treated with HGF (25 ng/mL) for 4 and 6 h or B. HCASMC treated with HGF (50 ng/mL) for 3 and 6 h. Cell lysates were run on PAGE, immunoblotted with anti-human A20 antibody and re-probed with anti-GAPDH or anti-βactin antibody to confirm equal loading. Results demonstrate significant upregulation of A20 in EC but not SMC. Data shown are representative of 3 independent experiments. C. Relative A20 mRNA levels measured by qPCR in HCASMC cultures stimulated for 1 h with HGF (25 to 200 ng/mL) or TNFα (100 U/mL), used as control, showed that lack of A20 upregulation in these cells in response to HGF was likely transcriptional. Indeed, we did not detect any increase in A20 mRNA levels after HGF. Histograms represent the statistical analyzes of relative mRNA levels after normalization with the house keeping gene βactin. Results are expressed as mean ± SEM of 2 independent experiments.
{kind=link}
Acknowledgments
Funding: This research was supported by NIH grants RO1 HL080130, RO1 DK063275 and Juvenile Diabetes Research Foundation grant 1-2007-567 to CF. EM is the recipient of a fellowship award from the National Council for Scientific and Technological Development (CNPq), Brasil. SMD, JJS and CP are the recipients of T32 NRSA grant HL00734.
Literature Cited
- 1.Assessment of adenoviral vector safety and toxicity: report of the National Institutes of Health Recombinant DNA Advisory Committee. Hum Gene Ther. 13(1):3–13. doi: 10.1089/10430340152712629. [DOI] [PubMed] [Google Scholar]
- 2.Akcay A, Nguyen Q, Edelstein CL. Mediators of inflammation in acute kidney injury. Mediators Inflamm. 2009;2009:137072. doi: 10.1155/2009/137072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arvelo MB, Cooper JT, Longo C, Daniel S, Grey ST, Mahiou J, Czismadia E, Abu-Jawdeh G, Ferran C. A20 protects mice from galactosmine/lipopolysaccharide acute toxic lethal hepatitis. Hepatology. 2002;35:535–543. doi: 10.1053/jhep.2002.31309. [DOI] [PubMed] [Google Scholar]
- 4.Bach FH, Hancock WW, Ferran C. Protective genes expressed in endothelial cells: a regulatory response to injury. Immunol Today. 1997;18:483–486. doi: 10.1016/s0167-5699(97)01129-8. [DOI] [PubMed] [Google Scholar]
- 5.Bendinelli P, Matteucci E, Dogliotti G, Corsi MM, Banfi G, Maroni P, Desiderio MA. Molecular basis of anti-inflammatory action of platelet-rich plasma on human chondrocytes: mechanisms of NF-kappaB inhibition via HGF. J Cell Physiol. 2010;225(3):757–766. doi: 10.1002/jcp.22274. [DOI] [PubMed] [Google Scholar]
- 6.Beyaert R, Heyninck K, van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-κB-dependent gene expression and apoptosis. Biochem Pharmac. 2000;60:1143–1151. doi: 10.1016/s0006-2952(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 7.Bonventre JV, Zuk A. Ischemic acute renal failure: An inflammatory disease? Kidney Int. 2004;66(2):480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 8.Cao CC, Ding XQ, Ou ZL, Liu CF, Li P, Wang L, Zhu CF. In vivo transfection of NF-kappaB decoy oligodeoxynucleotides attenuate renal ischemia/reperfusion injury in rats. Kidney Int. 2004;65(3):834–845. doi: 10.1111/j.1523-1755.2004.00463.x. [DOI] [PubMed] [Google Scholar]
- 9.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules - NF-κB and cytokine-inducible enhancers. FASEB Journal. 1995;9(10):899–909. [PubMed] [Google Scholar]
- 10.Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008;7(6):504–516. doi: 10.1038/nrd2530. [DOI] [PubMed] [Google Scholar]
- 11.Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-κB-dependent mechanism. J Biol Chem. 1996;271:18068–18073. doi: 10.1074/jbc.271.30.18068. [DOI] [PubMed] [Google Scholar]
- 12.Daniel S, Arvelo MB, Patel VI, Longo CR, Shrikhande G, Shukri T, Mahiou J, Sun DW, Mottley C, Grey ST, Ferran C. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood. 2004;104(8):2376–2384. doi: 10.1182/blood-2003-02-0635. [DOI] [PubMed] [Google Scholar]
- 13.Dejardin E. The alternative NF-kappaB pathway from biochemistry to biology: pitfalls and promises for future drug development. Biochem Pharmacol. 2006;72(9):1161–1179. doi: 10.1016/j.bcp.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Eddy AA. Progression in chronic kidney disease. Adv Chronic Kidney Dis. 2005;12(4):353–365. doi: 10.1053/j.ackd.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 15.Fan S, Gao M, Meng Q, Laterra JJ, Symons MH, Coniglio S, Pestell RG, Goldberg ID, Rosen EM. Role of NF-kappaB signaling in hepatocyte growth factor/scatter factor-mediated cell protection. Oncogene. 2005;24(10):1749–1766. doi: 10.1038/sj.onc.1208327. [DOI] [PubMed] [Google Scholar]
- 16.Feng B, Chen G, Zheng X, Sun H, Zhang X, Zhang ZX, Xiang Y, Ichim TE, Garcia B, Luke P, Jevnikar AM, Min WP. Small interfering RNA targeting RelB protects against renal ischemia-reperfusion injury. Transplantation. 2009;87(9):1283–1289. doi: 10.1097/TP.0b013e3181a1905e. [DOI] [PubMed] [Google Scholar]
- 17.Ferran C, Stroka DM, Badrichani AZ, Cooper JT, Wrighton CJ, Soares M, Grey ST, Bach FH. A20 inhibits NF-kappaB activation in endothelial cells without sensitizing to tumor necrosis factor-mediated apoptosis. Blood. 1998;91(7):2249–2258. [PubMed] [Google Scholar]
- 18.Giannopoulou M, Dai C, Tan X, Wen X, Michalopoulos GK, Liu Y. Hepatocyte growth factor exerts its anti-inflammatory action by disrupting nuclear factor-kappaB signaling. Am J Pathol. 2008;173(1):30–41. doi: 10.2353/ajpath.2008.070583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong R. Multi-target anti-inflammatory action of hepatocyte growth factor. Curr Opin Investig Drugs. 2008;9(11):1163–1170. [PubMed] [Google Scholar]
- 20.Gong R, Rifai A, Dworkin LD. Activation of PI3K-Akt-GSK3beta pathway mediates hepatocyte growth factor inhibition of RANTES expression in renal tubular epithelial cells. Biochem Biophys Res Commun. 2005;330(1):27–33. doi: 10.1016/j.bbrc.2005.02.122. [DOI] [PubMed] [Google Scholar]
- 21.Gong R, Rifai A, Dworkin LD. Anti-inflammatory effect of hepatocyte growth factor in chronic kidney disease: targeting the inflamed vascular endothelium. J Am Soc Nephrol. 2006a;17(9):2464–2473. doi: 10.1681/ASN.2006020185. [DOI] [PubMed] [Google Scholar]
- 22.Gong R, Rifai A, Dworkin LD. Hepatocyte growth factor suppresses acute renal inflammation by inhibition of endothelial E-selectin. Kidney Int. 2006b;69(7):1166–1174. doi: 10.1038/sj.ki.5000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong R, Rifai A, Ge Y, Chen S, Dworkin LD. Hepatocyte growth factor suppresses proinflammatory NFkappaB activation through GSK3beta inactivation in renal tubular epithelial cells. J Biol Chem. 2008;283(12):7401–7410. doi: 10.1074/jbc.M710396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong R, Rifai A, Tolbert EM, Biswas P, Centracchio JN, Dworkin LD. Hepatocyte growth factor ameliorates renal interstitial inflammation in rat remnant kidney by modulating tubular expression of macrophage chemoattractant protein-1 and RANTES. J Am Soc Nephrol. 2004;15(11):2868–2881. doi: 10.1097/01.ASN.0000141962.44300.3A. [DOI] [PubMed] [Google Scholar]
- 25.Grey ST, Arvelo MB, Hasenkamp W, Bach FH, Ferran C. A20 inhibits cytokine-induced apoptosis and nuclear factor κB-dependent gene activation in islets. J Exp Med. 1999;190:1135–1145. doi: 10.1084/jem.190.8.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gueler F, Gwinner W, Schwarz A, Haller H. Long-term effects of acute ischemia and reperfusion injury. Kidney Int. 2004;66(2):523–527. doi: 10.1111/j.1523-1755.2004.761_11.x. [DOI] [PubMed] [Google Scholar]
- 27.Gustin JA, Korgaonkar CK, Pincheira R, Li Q, Donner DB. Akt regulates basal and induced processing of NF-kappaB2 (p100) to p52. J Biol Chem. 2006;281(24):16473–16481. doi: 10.1074/jbc.M507373200. [DOI] [PubMed] [Google Scholar]
- 28.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006(357):re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 29.Ito W, Kanehiro A, Matsumoto K, Hirano A, Ono K, Maruyama H, Kataoka M, Nakamura T, Gelfand EW, Tanimoto M. Hepatocyte growth factor attenuates airway hyperresponsiveness, inflammation, and remodeling. Am J Respir Cell Mol Biol. 2005;32(4):268–280. doi: 10.1165/rcmb.2004-0058OC. [DOI] [PubMed] [Google Scholar]
- 30.Kawaida K, Matsumoto K, Shimazu H, Nakamura T. Hepatocyte growth factor prevents acute renal failure and accelerates renal regeneration in mice. Proc Natl Acad Sci U S A. 1994;91(10):4357–4361. doi: 10.1073/pnas.91.10.4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khwaja A, Lehmann K, Marte BM, Downward J. Phosphoinositide 3-kinase induces scattering and tubulogenesis in epithelial cells through a novel pathway. J Biol Chem. 1998;273(30):18793–18801. doi: 10.1074/jbc.273.30.18793. [DOI] [PubMed] [Google Scholar]
- 32.Krikos A, Laherty CD, Dixit VM. Transcriptional activation of the TNFα-inducible zinc finger protein, A20, is mediated by κB elements. Journal of Biological Chemistry. 1992;267(25):17971–17976. [PubMed] [Google Scholar]
- 33.Kroening S, Neubauer E, Wullich B, Aten J, Goppelt-Struebe M. Characterization of connective tissue growth factor expression in primary cultures of human tubular epithelial cells: modulation by hypoxia. Am J Physiol Renal Physiol. 2010;298(3):F796–806. doi: 10.1152/ajprenal.00528.2009. [DOI] [PubMed] [Google Scholar]
- 34.Kunter U, Daniel S, Arvelo MB, Choi J, Shukri T, Patel VI, Longo CR, Scali ST, Shrikhande G, Rocha E, Czismadia E, Mottley C, Grey ST, Floege J, Ferran C. Combined expression of A1 and A20 achieves optimal protection of renal proximal tubular epithelial cells. Kidney Int. 2005;68(4):1520–1532. doi: 10.1111/j.1523-1755.2005.00564.x. [DOI] [PubMed] [Google Scholar]
- 35.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 36.Laherty CD, Hu HM, Opipari AW, Wang F, Dixit VM. The Epstein-Barr virus LMP1 gene product induces A20 zinc finger protein expression by activating NF-κB. J BiolChem. 1992;267:24157–24160. [PubMed] [Google Scholar]
- 37.Laherty CD, Perkins ND, Dixit VM. Human T cell leukemia virus type I Tax and phorbol 12-myristate 13-acetate induce expression of the A20 zinc finger protein by distinct mechanisms involving nuclear factor κB. J Biol Chem. 1993;268:5032–5039. [PubMed] [Google Scholar]
- 38.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lawrence T, Fong C. The resolution of inflammation: anti-inflammatory roles for NF-kappaB. Int J Biochem Cell Biol. 2010;42(4):519–523. doi: 10.1016/j.biocel.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 40.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289(5488):2350–2354. doi: 10.1126/science.289.5488.2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee HT, Kim M, Jan M, Emala CW. Anti-inflammatory and antinecrotic effects of the volatile anesthetic sevoflurane in kidney proximal tubule cells. Am J Physiol Renal Physiol. 2006;291(1):F67–78. doi: 10.1152/ajprenal.00412.2005. [DOI] [PubMed] [Google Scholar]
- 42.Leroy C, Deheuninck J, Reveneau S, Foveau B, Ji Z, Villenet C, Quief S, Tulasne D, Kerckaert JP, Fafeur V. HGF/SF regulates expression of apoptotic genes in MCF-10A human mammary epithelial cells. Ann N Y Acad Sci. 2006;1090:188–202. doi: 10.1196/annals.1378.021. [DOI] [PubMed] [Google Scholar]
- 43.Linas SL, Shanley PF, Whittenburg D, Berger E, Repine JE. Neutrophils accentuate ischemia-reperfusion injury in isolated perfused rat kidneys. Am J Physiol. 1988;255(4 Pt 2):F728–735. doi: 10.1152/ajprenal.1988.255.4.F728. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y. Hepatocyte growth factor promotes renal epithelial cell survival by dual mechanisms. Am J Physiol. 1999;277(4 Pt 2):F624–633. doi: 10.1152/ajprenal.1999.277.4.F624. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y. Hepatocyte growth factor and the kidney. Curr Opin Nephrol Hypertens. 2002;11(1):23–30. doi: 10.1097/00041552-200201000-00004. [DOI] [PubMed] [Google Scholar]
- 46.Longo CR, Arvelo MB, Patel VI, Daniel S, Mahiou J, Grey ST, Ferran C. A20 protects from CD40-CD40 ligand-mediated endothelial cell activation and apoptosis. Circulation. 2003;108(9):1113–1118. doi: 10.1161/01.CIR.0000083718.76889.D0. [DOI] [PubMed] [Google Scholar]
- 47.Lutz J, Luong le A, Strobl M, Deng M, Huang H, Anton M, Zakkar M, Enesa K, Chaudhury H, Haskard DO, Baumann M, Boyle J, Harten S, Maxwell PH, Pusey C, Heemann U, Evans PC. The A20 gene protects kidneys from ischaemia/reperfusion injury by suppressing pro-inflammatory activation. J Mol Med. 2008;86(12):1329–1339. doi: 10.1007/s00109-008-0405-4. [DOI] [PubMed] [Google Scholar]
- 48.Marienfeld R, Berberich-Siebelt F, Berberich I, Denk A, Serfling E, Neumann M. Signal-specific and phosphorylation-dependent RelB degradation: a potential mechanism of NF-kappaB control. Oncogene. 2001;20(56):8142–8147. doi: 10.1038/sj.onc.1204884. [DOI] [PubMed] [Google Scholar]
- 49.Matsumoto K, Nakamura T. Emerging multipotent aspects of hepatocyte growth factor. J Biochem. 1996;119(4):591–600. doi: 10.1093/oxfordjournals.jbchem.a021283. [DOI] [PubMed] [Google Scholar]
- 50.Matsumoto K, Nakamura T. Hepatocyte growth factor: renotropic role and potential therapeutics for renal diseases. Kidney Int. 2001;59(6):2023–2038. doi: 10.1046/j.1523-1755.2001.00717.x. [DOI] [PubMed] [Google Scholar]
- 51.Matthews N, Neale ML, Jackson SK, Stark JM. Tumour cell killing by tumour necrosis factor: inhibition by anaerobic conditions, free-radical scavengers and inhibitors of arachidonate metabolism. Immunology. 1987;62(1):153–155. [PMC free article] [PubMed] [Google Scholar]
- 52.Miller SB, Martin DR, Kissane J, Hammerman MR. Hepatocyte growth factor accelerates recovery from acute ischemic renal injury in rats. Am J Physiol. 1994;266(1 Pt 2):F129–134. doi: 10.1152/ajprenal.1994.266.1.F129. [DOI] [PubMed] [Google Scholar]
- 53.Min JK, Lee YM, Kim JH, Kim YM, Kim SW, Lee SY, Gho YS, Oh GT, Kwon YG. Hepatocyte growth factor suppresses vascular endothelial growth factor-induced expression of endothelial ICAM-1 and VCAM-1 by inhibiting the nuclear factor-kappaB pathway. Circ Res. 2005;96(3):300–307. doi: 10.1161/01.RES.0000155330.07887.EE. [DOI] [PubMed] [Google Scholar]
- 54.Mizuno S, Matsumoto K, Nakamura T. Hepatocyte growth factor suppresses interstitial fibrosis in a mouse model of obstructive nephropathy. Kidney Int. 2001;59(4):1304–1314. doi: 10.1046/j.1523-1755.2001.0590041304.x. [DOI] [PubMed] [Google Scholar]
- 55.Mizuno S, Nakamura T. Suppressions of chronic glomerular injuries and TGF-beta 1 production by HGF in attenuation of murine diabetic nephropathy. Am J Physiol Renal Physiol. 2004;286(1):F134–143. doi: 10.1152/ajprenal.00199.2003. [DOI] [PubMed] [Google Scholar]
- 56.Muller M, Morotti A, Ponzetto C. Activation of NF-kappaB is essential for hepatocyte growth factor-mediated proliferation and tubulogenesis. Mol Cell Biol. 2002;22(4):1060–1072. doi: 10.1128/MCB.22.4.1060-1072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakagami H, Morishita R, Yamamoto K, Taniyama Y, Aoki M, Matsumoto K, Nakamura T, Kaneda Y, Horiuchi M, Ogihara T. Mitogenic and antiapoptotic actions of hepatocyte growth factor through ERK, STAT3, and AKT in endothelial cells. Hypertension. 2001;37(2 Part 2):581–586. doi: 10.1161/01.hyp.37.2.581. [DOI] [PubMed] [Google Scholar]
- 58.Nakamura Y, Morishita R, Higaki J, Kida I, Aoki M, Moriguchi A, Yamada K, Hayashi S, Yo Y, Matsumoto K, et al. Expression of local hepatocyte growth factor system in vascular tissues. Biochem Biophys Res Commun. 1995;215(2):483–488. doi: 10.1006/bbrc.1995.2490. [DOI] [PubMed] [Google Scholar]
- 59.Ohda Y, Hori K, Tomita T, Hida N, Kosaka T, Fukuda Y, Miwa H, Matsumoto T. Effects of hepatocyte growth factor on rat inflammatory bowel disease models. Dig Dis Sci. 2005;50(5):914–921. doi: 10.1007/s10620-005-2664-z. [DOI] [PubMed] [Google Scholar]
- 60.Opipari AJ, Boguski MS, Dixit VM. The A20 cDNA induced by tumor necrosis factor alpha encodes a novel type of zinc finger protein. J Biol Chem. 1990;265:14705–14708. [PubMed] [Google Scholar]
- 61.Opipari AJ, Hu HM, Yabkowitz R, Dixit VM. The A20 zinc finger protein protects cells from TNF cytotoxicity. J Biol Chem. 1992;267:12424–12427. [PubMed] [Google Scholar]
- 62.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401(6748):82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 63.Pasparakis M. Regulation of tissue homeostasis by NF-kappaB signalling: implications for inflammatory diseases. Nat Rev Immunol. 2009;9(11):778–788. doi: 10.1038/nri2655. [DOI] [PubMed] [Google Scholar]
- 64.Patel VI, Daniel S, Longo CR, Shrikhande GV, Scali ST, Czismadia E, Groft CM, Shukri T, Motley-Dore C, Ramsey HE, Fisher MD, Grey ST, Arvelo MB, Ferran C. A20, a modulator of smooth muscle cell proliferation and apoptosis, prevents and induces regression of neointimal hyperplasia. Faseb J. 2006;20(9):1418–1430. doi: 10.1096/fj.05-4981com. [DOI] [PubMed] [Google Scholar]
- 65.Remuzzi G, Bertani T. Pathophysiology of progressive nephropathies. N Engl J Med. 1998;339(20):1448–1456. doi: 10.1056/NEJM199811123392007. [DOI] [PubMed] [Google Scholar]
- 66.Remuzzi G, Ruggenenti P, Benigni A. Understanding the nature of renal disease progression. Kidney Int. 1997;51(1):2–15. doi: 10.1038/ki.1997.2. [DOI] [PubMed] [Google Scholar]
- 67.Robertson H, Kirby JA. Post-transplant renal tubulitis: the recruitment, differentiation and persistence of intra-epithelial T cells. Am J Transplant. 2003;3(1):3–10. doi: 10.1034/j.1600-6143.2003.30102.x. [DOI] [PubMed] [Google Scholar]
- 68.Romashkova JA, Makarov S. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–89. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 69.Rubin JS, Chan AM, Bottaro DP, Burgess WH, Taylor WG, Cech AC, Hirschfield DW, Wong J, Miki T, Finch PW, et al. A broad-spectrum human lung fibroblast-derived mitogen is a variant of hepatocyte growth factor. Proc Natl Acad Sci U S A. 1991;88(2):415–419. doi: 10.1073/pnas.88.2.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanes JR, Rubenstein JL, Nicolas JF. Use of a recombinant retrovirus to study post-implantation cell lineage in mouse embryos. EMBO J. 1986;5(12):3133–3142. doi: 10.1002/j.1460-2075.1986.tb04620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scheidereit C. IkappaB kinase complexes: gateways to NF-kappaB activation and transcription. Oncogene. 2006;25(51):6685–6705. doi: 10.1038/sj.onc.1209934. [DOI] [PubMed] [Google Scholar]
- 72.Segerer S, Nelson PJ, Schlondorff D. Chemokines, chemokine receptors, and renal disease: from basic science to pathophysiologic and therapeutic studies. J Am Soc Nephrol. 2000;11(1):152–176. doi: 10.1681/ASN.V111152. [DOI] [PubMed] [Google Scholar]
- 73.Shigematsu H, Yasuda K, Iwai T, Sasajima T, Ishimaru S, Ohashi Y, Yamaguchi T, Ogihara T, Morishita R. Randomized, double-blind, placebo-controlled clinical trial of hepatocyte growth factor plasmid for critical limb ischemia. Gene Ther. 2010;17(9):1152–1161. doi: 10.1038/gt.2010.51. [DOI] [PubMed] [Google Scholar]
- 74.Sizemore N, Leung S, Stark GR. Activation of phosphatidylinositol 3-kinase in response to interleukin-1 leads to phosphorylation and activation of the NF-kappaB p65/RelA subunit. Mol Cell Biol. 1999;19(7):4798–4805. doi: 10.1128/mcb.19.7.4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Suzuki A, Hayashida M, Kawano H, Sugimoto K, Nakano T, Shiraki K. Hepatocyte growth factor promotes cell survival from fas-mediated cell death in hepatocellular carcinoma cells via Akt activation and Fas-death-inducing signaling complex suppression. Hepatology. 2000;32(4 Pt 1):796–802. doi: 10.1053/jhep.2000.17738. [DOI] [PubMed] [Google Scholar]
- 76.Tamada S, Nakatani T, Asai T, Tashiro K, Komiya T, Sumi T, Okamura M, Kim S, Iwao H, Kishimoto T, Yamanaka S, Miura K. Inhibition of nuclear factor-kappaB activation by pyrrolidine dithiocarbamate prevents chronic FK506 nephropathy. Kidney Int. 2003;63(1):306–314. doi: 10.1046/j.1523-1755.2003.00714.x. [DOI] [PubMed] [Google Scholar]
- 77.Tashiro K, Tamada S, Kuwabara N, Komiya T, Takekida K, Asai T, Iwao H, Sugimura K, Matsumura Y, Takaoka M, Nakatani T, Miura K. Attenuation of renal fibrosis by proteasome inhibition in rat obstructive nephropathy: possible role of nuclear factor kappaB. Int J Mol Med. 2003;12(4):587–592. [PubMed] [Google Scholar]
- 78.Thompson JE, Phillips RJ, Erdjument-Bromage H, Tempst P, Ghosh S. I kappa B-beta regulates the persistent response in a biphasic activation of NF-kappa B. Cell. 1995;80(4):573–582. doi: 10.1016/0092-8674(95)90511-1. [DOI] [PubMed] [Google Scholar]
- 79.van Kooten C, Daha MR, van Es LA. Tubular epithelial cells: A critical cell type in the regulation of renal inflammatory processes. Exp Nephrol. 1999;7(5–6):429–437. doi: 10.1159/000020622. [DOI] [PubMed] [Google Scholar]
- 80.Vielhauer V, Berning E, Eis V, Kretzler M, Segerer S, Strutz F, Horuk R, Grone HJ, Schlondorff D, Anders HJ. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int. 2004;66(6):2264–2278. doi: 10.1111/j.1523-1755.2004.66038.x. [DOI] [PubMed] [Google Scholar]
- 81.Wrighton CJ, Hofer-Warbinek R, Moll T, Eytner R, Bach FH, de Martin R. Inhibition of endothelial cell activation by adenovirus-mediated expression of I kappa B alpha, an inhibitor of the transcription factor NF-kappa B. J Exp Med. 1996;183(3):1013–1022. doi: 10.1084/jem.183.3.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamada YIaK. HGF gene transfer increases kidney graft survival. Kidney International. 2005;68:1971–1972. [Google Scholar]
- 83.Zhang J, Yang J, Liu Y. Role of Bcl-xL induction in HGF-mediated renal epithelial cell survival after oxidant stress. Int J Clin Exp Pathol. 2008;1(3):242–253. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
qPCR analysis of A20 mRNA was performed on total mRNA isolated from RPTEC cultures treated with HGF 60 and 300ng/mL, or TNFα 2 and 10 U/mL, which corresponds to 2 and 10×ED50 concentrations of these molecules, respectively. Results demonstrate that HGF, but not TNFα, significantly increases A20 mRNA at the 2×ED50 concentration. HGF-induced upregulation of A20 mRNA reaches a plateau above this concentration, while TNFα demonstrate a dose dependent response, as measured up to 100 U/mL. Graphs shown represent the statistical analyses of relative A20 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 5 independent experiments. *p<0.05, **p< 0.01 and ***p< 0.001.
RPTEC were transfected with p65 siRNA, RelB siRNA or AllStars negative control siRNA (Neg siRNA) for 48 h. Total cellular mRNA and protein were recovered and p65 and RelB mRNA and protein were measured by qPCR (expressed as percentage of non-transfected (NT) control cells after normalization by βactin) and Western blot analysis, respectively. We noted 75% to 80% reduction of mRNA levels and 90–95% reduction of protein levels (as depicted on representative Western blots) of both targeted genes. qPCR results are expressed as mean ± SEM of 4–5 independent experiments. ***p<0.01.
A. Representative Western blot analysis using a phospho-RelB (P-RelB) specific antibody demonstrates significantly greater phosphorylation of RelB in RPTEC cultures 15 min following HGF (50 ng/mL), as compared to TNFα (100 U/mL) treatment. βactin immunoblotting was used to correct for loading and allow semi-quantitative evaluation of the data by densitometry. Results of corrected densitometry, presented as percentage of control non-stimulated (NS) cells are expressed as mean ± SEM of 3 independent experiments. *p<0.05 compared to NS cells B. RPTEC were transfected with RelB silencing RNA (RelB siRNA) 48 h, then stimulated with HGF (50 ng/mL) or TNFα (100 U/mL) for 1h, and A20 mRNA was evaluated by qPCR. Results indicate that RelB knockdown does not affect HGF-induced upregulation of A20 mRNA. Non-transfected (NT) and AllStars negative control siRNA (Neg siRNA) transfected cells were used as controls. Graphs shown represent the statistical analyses of relative A20 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 3–5 independent experiments. ***p<0.001 compared to NS cells within each siRNA treatment group. Pre-incubation with 10 μM of the PI3K inhibitor LY294002 for 30 min, prior to treatment with 50 ng/mL of HGF for 15 min (C) or 100U/mL of TNFα for 30 min (D) significantly decreased phosphorylation of RelB, as shown by Western blot analysis using a phospho-RelB antibody. Immunoblotting with βactin antibody was used to correct for loading and allow semi-quantitative evaluation of the data by densitometry. Results of corrected densitometry, presented as percentage of NS cells are depicted in the graph, and expressed as mean ± SEM of 3–4 independent experiments. *p<0.05; **p<0.01; ***p<0.001
Representative Western blot of RPTEC treated with HGF (50 ng/mL) show IKKα/β phosphorylation 5 min following HGF treatment. Phosphorylation of IKKα/β was abrogated when RPTEC were pre-incubated with LY294002. Immunoblotting with βactin antibody served as loading control. Results shown are representative of 3 independent experiments.
A. HGF (50 ng/mL) or TNFα (100 U/mL) treatment for 1 h significantly upregulate A1 mRNA levels in RPTEC, within of treatment, as measured by qPCR. Graphs shown represent the statistical analyses of relative A1 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 3 independent experiments. *p<0.05; ***p<0.001 compared to non-stimulated (NS) cells. B. This translates into a substantial increase in A1 protein levels 6h after treatment with either HGF or TNFα, as demonstrated by Western blot analysis using an anti-human A1 antibody. Immunoblotting with a GAPDH antibody served as loading control. C. HGF-induced A1 protein upregulation shows a time dependent curve that peaks 6 h, and is still sustained 24 h following addition of 50ng/mL of HGF, as shown by Western blot analysis. Immunoblotting with anti-GAPDH antibody served as loading control. Results are representative of 3 independent experiments
RPTEC were either non-tranfected (NT) or transfected with A20 silencing RNA (A20 siRNA) or AllStars negative control siRNA (Neg siRNA) for 24 h. Cells were then pre-treated with HGF (50 ng/mL) for 4h followed by TNFα (100 U/mL) for 3h, and total RNA was extracted. A20 mRNA levels were measured by qPCR, normalized by βactin. Results show a total (100% inhibition) of HGF-induced and a significant 50% reduction of TNFα-induced A20 mRNA levels in A20 siRNA transfected RPTEC. Results are expressed as mean ± SEM of 4–6 independent experiments. *p<0.05, **p<0.01 when comparing RPTEC with corresponding values in NT, and δp<0.05 when comparing with corresponding values in AllStars siRNA transfected RPTEC.
RPTEC were transfected with A20 silencing RNA (A20 siRNA) or AllStars negative control siRNA (Neg siRNA). Twenty-four h later, cells were stimulated with TNFα (100 U/mL) for 3h. RNA was extracted and MCP-1 and ICAM-1 mRNA levels were determined by qPCR. Our results indicate that A20 silencing significantly increases basal, and TNFα-induced upregulation of MCP-1 and ICAM-1 mRNA. Graphs shown represent the statistical analyses of relative MCP-1 and ICAM-1 mRNA levels after normalization by βactin. Results are expressed as mean ± SEM of 5–7 independent experiments. *p<0.05, **p< 0.01 and ***p< 0.001.
HCAEC and HCASMC were purchased from Lonza, cultured according to the manufacturer’s recommendations, and used between passages 4–8. Western blots of A. HCAEC treated with HGF (25 ng/mL) for 4 and 6 h or B. HCASMC treated with HGF (50 ng/mL) for 3 and 6 h. Cell lysates were run on PAGE, immunoblotted with anti-human A20 antibody and re-probed with anti-GAPDH or anti-βactin antibody to confirm equal loading. Results demonstrate significant upregulation of A20 in EC but not SMC. Data shown are representative of 3 independent experiments. C. Relative A20 mRNA levels measured by qPCR in HCASMC cultures stimulated for 1 h with HGF (25 to 200 ng/mL) or TNFα (100 U/mL), used as control, showed that lack of A20 upregulation in these cells in response to HGF was likely transcriptional. Indeed, we did not detect any increase in A20 mRNA levels after HGF. Histograms represent the statistical analyzes of relative mRNA levels after normalization with the house keeping gene βactin. Results are expressed as mean ± SEM of 2 independent experiments.