

Abstract
Our group has previously identified elevated levels of nonapoptotic active caspase 3 (CASP3) accompanied by increased prosurvival, antiapoptotic signaling in the pregnant mouse uterus during late gestation. We speculated that increased antiapoptotic signaling desensitized the pregnant uterine myocyte to the apoptotic action of uterine CASP3. This current study examines the mechanism by which the pregnant myocyte gains resistance to the apoptotic effects of increased uterine CASP3. Using both primary human pregnant fundal myometrial cultures and the telomerase-immortalized human uterine myocyte cell line (hTERT) as our model systems, uterine myocytes were exposed to UV irradiation and Fas ligand to stimulate both the intrinsic and extrinsic apoptotic pathways. Stimulation of either the intrinsic or extrinsic apoptotic pathways resulted in elevated levels of uterine myocyte CASP3. However, apoptotic cell death was restricted to CASP3 activated by intrinsic stimulation via UV light. In contrast Fas ligand-mediated CASP3 activation was accompanied by increased antiapoptotic signaling mimicking our in vivo observations in the pregnant mouse uterus. Using small interfering RNA to inhibit antiapoptotic signaling, we determined the ability of the human uterine myocyte to resist apoptotic cell death in the absence of the prosurvival, antiapoptotic signaling. Accordingly, suppression of antiapoptotic signaling specifically mediated by myeloid cell leukemia sequence 1 was sufficient to sensitize the uterine myocyte to undergo apoptotic cell death. These data demonstrate that elevated myeloid cell leukemia sequence 1 levels are sufficient to confer apoptotic resistance on the human uterine myocyte despite highly elevated levels of active CASP3.
The signaling that supports uterine quiescence during pregnancy is incompletely understood and is of high clinical significance given the morbidity, mortality, and cost associated with preterm birth. Our group has previously identified high levels of uterine cleaved caspase 3 (CASP3), which we propose moderates uterine quiescence during pregnancy (1). Uterine myocyte CASP3 is regulated by progesterone, which potentiates its levels during gestation (1). It is clear, however, that despite elevated levels of uterine CASP3 during pregnancy, there is minimal evidence of apoptotic cell death (2). The presence of nonapoptotic CASP3 activity has also been identified in other tissues including human cardiac myocytes (3), pregnant rat uterus (4), and fetal membranes (5). In addition, there is evidence from our work in pregnant myometrium that CASP3 targets the contractile apparatus and may lead to impaired contractility (1). Disruption of myocyte contractile architecture in response to cleaved CASP3 has also previously been shown in cardiac (6) (7), skeletal (8), and smooth muscle (9). We have proposed that this may be a mechanism by which active CASP3 acts as a tocolytic to maintain uterine quiescence.
We have recently identified that the pregnant mouse uterine myocyte escapes apoptotic cell death while harnessing the tocolytic action of CASP3 by hosting an antiapoptotic response involving up-regulation members of the Bcl-2 and IAP families (2). Myeloid cell leukemia sequence 1 (MCL1), an antiapoptotic protein in the Bcl-2 family, is an early protector against apoptotic cell death. MCL1 is believed to inhibit cell death through interactions with proapoptotic Bcl-2 family members (10) Previous studies have shown MCL1 to be at the apex of apoptotic signaling and that down-regulation of MCL1 is required for the initiation of caspase activation (11). MCL1 is a critical regulator of apoptosis in many cell types such as the pancreatic β-cell in which down-regulation of MCL1 contributes to the onset of apoptosis (12, 13). On the other hand, overexpression of MCL1 blocks cycloheximide-induced apoptosis (13). Additionally down-regulation of MCL1 with small interfering RNA (siRNA) has been shown to sensitize cells to apoptosis via Fas ligand (FasL) (14), TNF-related apoptosis-inducing ligand (TRAIL) (15), chemotherapeutic agents (16), and radiation therapy (17). In this current study we have observed that despite the onset of CASP3 activation in the human uterine myocyte, the persistence of MCL1 renders the myocyte resistant to apoptotic cell death. We used the human myometrial hTERT cell line (18) and pregnant primary myometrial fundal cells exposed to the intrinsic apoptotic stimulus UV or extrinsic stimulus FasL to elicit human myometrial CASP3 activation. Examination of cellular levels of cleaved CASP3 and cleavage of the nuclear enzyme poly (ADP-ribose) polymerase (PARP) was used to assess activation of apoptotic caspase action (19) along with the assessment of apoptotic indices by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL). MCL1 was confirmed as a critical factor in conferring resistance to CASP3-mediated apoptotic cell death in the pregnant human uterine myocyte.
Results
CASP3 activation in hTERT myometrial cells increases in response to FasL and UV in a dose-dependent manner
hTERT cells were exposed to increased levels of extrinsic (FasL) and intrinsic (UV) apoptotic stimuli in a dose-dependent manner. As shown in Fig. 1 both stimuli have the capacity to activate uterine myocyte cytoplasmic CASP3. FasL-mediated CASP3 activation was barely detectable at 5 ng/ml and reached maximal levels of activation upon exposure to 50 and 100 ng/ml FasL. hTERT cells intrinsically stimulated with increasing intensity of UV light also demonstrate a dose-dependent increase in the appearance of active capase-3 with a maximal response at 500 mJ/cm2.
Fig. 1.

FasL activation of CASP3 is accompanied by elevated levels of the antiapoptotic protein MCL1. Cytoplasmic extracts from hTERT cells were analyzed for levels of active CASP3 and MCL1 by immunoblotting after exposure to FasL (0, 1, 5, 10, 50, and 100 ng/ml) for 24 h after UV exposure (0, 10, 100, and 500 mJ/cm2). PDI was used as our loading control. These data are representative of three independent experiments (n = 3).
MCL1 levels are augmented by FasL and eradicated by UV light in hTERT uterine myocytes
MCL1 levels were examined in hTERT cells exposed to both extrinsic (FasL) and intrinsic (UV light) apoptotic stimuli. As is shown in Fig. 1, MCL1 was found to be highly expressed in the cytoplasm at baseline in the hTERT cell. Unexpectedly increasing concentrations of FasL (0, 1, 5, 10, 50, and 100 ng/ml) resulted in elevated MCL1 levels observed to increase in a dose-dependent manner, reaching maximal levels upon exposure to 50 and 100 ng/ml FasL. In contrast and consistent with previously reported findings (11), UV exposure decreased MCL1 to undetectable levels in a dose-dependent manner with 100 mJ/cm2 being sufficient to eradicate MCL1 levels entirely.
Increased CASP3 activation in hTERT cells in a time course response to FasL and UV light
hTERT cells were exposed to extrinsic (100 ng/ml FasL) and intrinsic (100 mJ/cm2 UV light) apoptotic stimuli in a time course experiment from 0–48 h. As shown in Fig. 2 we demonstrate the appearance of active CASP3 induced in the FasL-stimulated hTERT cells. FasL-mediated CASP3 levels were barely detectable at 1 h, reached maximal levels at 24 h, and declined to minimal levels after 48 h. Similarly, hTERT cells intrinsically stimulated with UV demonstrate the appearance of cleaved active capase-3. UV exposure successfully elicited an active CASP3 response, which was maximal at 48 h after exposure. In data not shown, there is further increased active CASP3 at 72 h after exposure to UV but with poor cell viability and limited protein concentrations.
Fig. 2.

FasL and UV activation of CASP3 with increasing duration of exposure. Cytoplasmic extracts from hTERT cells were analyzed for active CASP3 by immunoblotting after exposure to FasL (100 ng/ml) and UV light (100 mJ/cm2) at 0, 1, 12, 24, and 48 h. PDI was used as our loading control. These data are representative of three independent experiments (n = 3).
hTERT MCL1 levels are modified in a time course response to FasL and UV light
Endogenous MCL1 levels were examined in hTERT cells exposed to both extrinsic (100 ng/ml FasL) and intrinsic (100 mJ/cm2 UV light) apoptotic stimuli in a time course experiment from 0–48 h (Fig. 3). As expected, uterine myocyte MCL1 levels were ablated in response to UV exposure within 12 h. However, in contrast, uterine myocyte MCL1 levels do not decline but remain elevated throughout the 48-h exposure to FasL.
Fig. 3.

Loss of MCL1 after UV exposure. Cytoplasmic extracts from hTERT cells were analyzed for MCL1 by immunoblotting after exposure to FasL (100 ng/ml) and UV light (100 mJ/cm2) at 0, 1, 12, 24, and 48 h. PDI was used as our loading control. These data are representative of three independent experiments (n = 3).
PARP is cleaved upon exposure to UV light but not FasL in the hTERT cell
To determine whether both the extrinsic and intrinsic activated CASP3 in the uterine myocyte had the same apoptotic potential in the presence and in the absence of MCL1, respectively, we examined the marker of apoptotic action of CASP3, i.e. cleaved PARP. Nuclear levels of cleaved PARP (89 kDa) were examined in the hTERT cells in response to FasL and UV exposure. Barely detectable levels of cleaved PARP that remained close to baseline across the time course were observed in the FasL-treated hTERT cell. In contrast, in response to UV exposure, cleaved PARP was detectable in the hTERT cell at robust levels within 12 h with maximal levels of PARP cleavage occurring after 48 h as is shown in Fig. 4.
Fig. 4.

PARP cleavage is demonstrated with UV exposure but not FasL. Nuclear extracts from hTERT cells were analyzed for cleaved PARP by immunoblotting after exposure to FasL and UV light at 0, 1, 12, 24, and 48 h. NCOA3 was used as our loading control. These data are representative of three independent experiments (n = 3).
Fragmentation of uterine myocyte genomic DNA is isolated to the UV light-treated hTERT cells
To further examine CASP3-mediated apoptotic activity, we examined the fragmentation pattern of uterine myocyte genomic DNA in hTERT cells exposed to both intrinsic (100 mJ/cm2 UV light) and extrinsic (100 ng/ml FasL) apoptotic stimuli in a time course experiment from 0–48 h. In accordance with what we observed in Figs. 2, 3 and 4, hTERT cells exposed to FasL that contain active CASP3 and elevated MCL1 levels and lack cleaved PARP continue to retain intact genomic DNA. In contrast, the uterine myocytes exposed to UV light, the intrinsic apoptotic stimuli, demonstrate decreased MCL1 levels and elevated cleaved PARP, resulting in fragmented genomic DNA (Fig. 5). These data suggest that in the absence of MCL1, CASP3 has the capacity to act in an apoptotic manner; however, in the presence of MCL1, uterine myocyte CASP3 is rendered resistant to CASP3–3 mediated apoptotic cell death.
Fig. 5.

UV- but not FasL-induced DNA fragmentation in CASP3-positive hTERT cells. Genomic DNA isolated from hTERT cells was analyzed for CASP3-mediated fragmentation after exposure to FasL and UV light at 0, 24, and 48 h. Apoptotic U937 cells-fragmented DNA is used as a positive control. These data are representative of three independent experiments (n = 3).
TUNEL staining of the hTERT cells confirms CASP3-mediated apoptotic cell death in the UV light-treated, MCL1-negative uterine myocytes
To confirm apoptotic action in hTERT cells in response to activated caspase 3, mediated by exposure to UV and FasL, a TUNEL assay was performed. Figure 6 demonstrates that despite widespread staining for cleaved CASP3 in FasL and UV light-treated hTERT cells, there is no evidence for increased apoptosis as measured by TUNEL in response to FasL exposure. In contrast, an increase in the number of TUNEL-positive cells is clearly limited to those cells exposed to UV light, which are also immunopositive for active CASP3.
Fig. 6.

Increased TUNEL staining was detected in UV- but not FasL-treated CASP3-positive hTERT cells. hTERT myometrial cells were examined 24 h after UV and FasL exposure and analyzed by fluorescent microscopy after TUNEL staining (green) to detect the presence of fragmented nuclear DNA. Cells were also stained for cleaved CASP3 (red), and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Vehicle-treated hTERT cells were used as a negative control. These data are representative of three independent experiments (n = 3).
CASP3 and MCL1 levels are modified in human primary myometrial cultures in a time course response to FasL and UV light
Using primary cultures of human uterine myocytes isolated from fundal myometrium of pregnant laboring women, we observed over a 24-h period an activation pattern of uterine myocyte CASP3 in the presence of both the extrinsic and intrinsic apoptotic stimuli FasL and UV light (Fig. 7). In an almost identical pattern to the hTERT cells (Fig. 3) we also observed that endogenous MCL1 levels were maintained in response to FasL treatment whereas after UV light exposure (12 and 24 h), MCL1 levels were ablated in the primary cultures of pregnant human uterine myocytes.
Fig. 7.
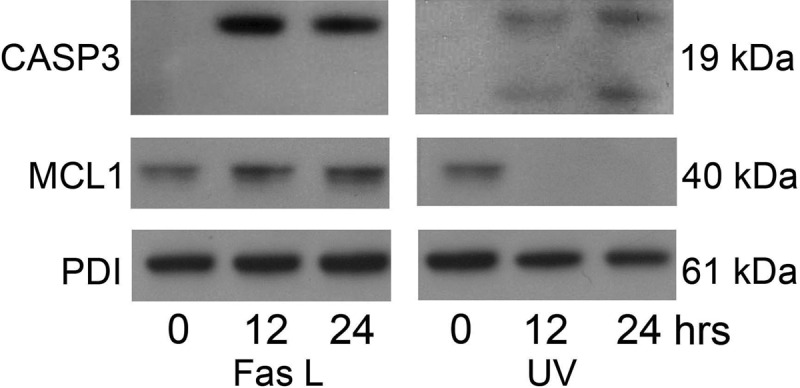
Pregnant primary fundal myometrial cells demonstrate active CASP3 and MCL1 in response to FasL and UV light. Cytoplasmic extracts from pregnant primary fundal myometrial cells were analyzed for active CASP3 and MCL1 by immunoblotting after exposure to FasL (100 ng/ml) and UV light (100 mJ/cm2) at 0, 1, 12, 24, and 48 h. PDI was used as our loading control. These data are representative of three independent experiments (n = 3).
PARP cleavage is limited to human primary myometrial cultures exposed to UV light
To determine whether both the extrinsic (100 ng/ml FasL) and intrinsically (100 mJ/cm2 UV light) activated CASP3 detectable in the primary uterine myocyte had similar apoptotic potential in the presence or the absence of MCL1 (Fig. 7), we examined the marker of apoptotic action of CASP3, cleaved PARP, in a time course experiment from 0–24 h. Figure 8 demonstrates barely detectable levels of PARP cleavage in the FasL-treated primary uterine myocyte with levels remaining close to baseline across the time course. In contrast, cleaved PARP was readily detectable in response to treatment with UV light, in a time-dependent manner with maximal levels of PARP cleavage occurring after 24 h exposure.
Fig. 8.
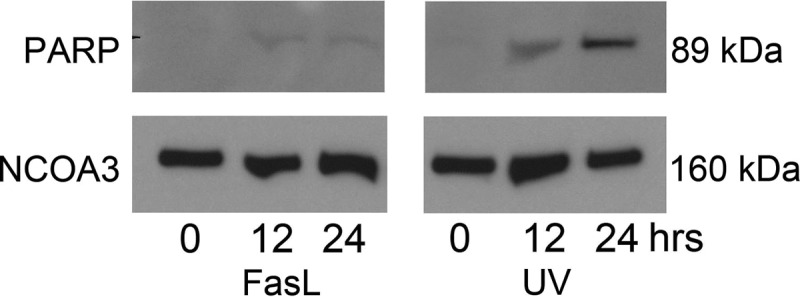
Pregnant primary fundal myometrial cells demonstrate PARP cleavage in response to UV exposure but not FasL. Nuclear extracts from pregnant primary myometrial cells were analyzed for cleaved PARP by immunoblotting after exposure to FasL and UV light at 0, 1, 12, 24, and 48 h. NCOA3 was used as our loading control. These data are representative of three independent experiments (n = 3).
TUNEL staining of the primary myometrial cultures confirms resistance to CASP3-mediated apoptotic cell death in the FasL-treated MCL1-positive uterine myocyte
To determine whether apoptosis occurs in the primary fundal myometrial cells in response to 100 mJ/cm2 UV light and 100 ng/ml FasL, a TUNEL assay and CASP3 immunofluorescent staining were performed (Fig. 9). Despite widespread elevated cleaved CASP3 observed in FasL-treated primary myometrial cells, there was no evidence for increased apoptosis as indicated by the lack of increased numbers of TUNEL-positive cells. In contrast, increased numbers of caspase and TUNEL-positive cells were observed in response to UV exposure.
Fig. 9.

Increased TUNEL staining was detected in UV- but not FasL-treated CASP3-positive pregnant primary fundal myometrial cells. Pregnant primary fundal myometrial cells were treated by UV and Fas for 24 h and analyzed by fluorescent microscopy after TUNEL staining (green) to detect the presence of cleaved nuclear DNA. Cells were also immunostained for cleaved CASP3 (red), and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Vehicle-treated hTERT cells were used as a negative control. These data are representative of three independent experiments (n = 3).
Elimination of MCL1 sensitizes hTERT cells to a FasL-mediated increase in apoptotic potential
hTERT cells were transfected with a validated siRNA for MCL1. Figure 10 demonstrates that MCL1 siRNA transfection of hTERT cells results in efficient knockdown of MCL1 protein levels at 24 h. In the absence of an apoptotic stimuli, MCL1 knockdown alone is insufficient to cause CASP3 activation. FasL treatment induced CASP3 activation whereas knockdown of MCL1 had no affect on the level of FasL-induced CASP3. However, augmented PARP cleavage was observed when MCL1 was ablated despite similar levels of active capase-3 in control cells treated with FasL. These data indicate that human uterine myocytes in the presence of MCL1 are resistant to FasL-stimulated, CASP3-mediated apoptotic cell death. However, in the absence of MCL1, the human uterine myocyte is no longer resistant to FasL-mediated, CASP3-induced apoptotic cell death as indicted by increased targeting and cleavage of the DNA repair enzyme, PARP.
Fig. 10.
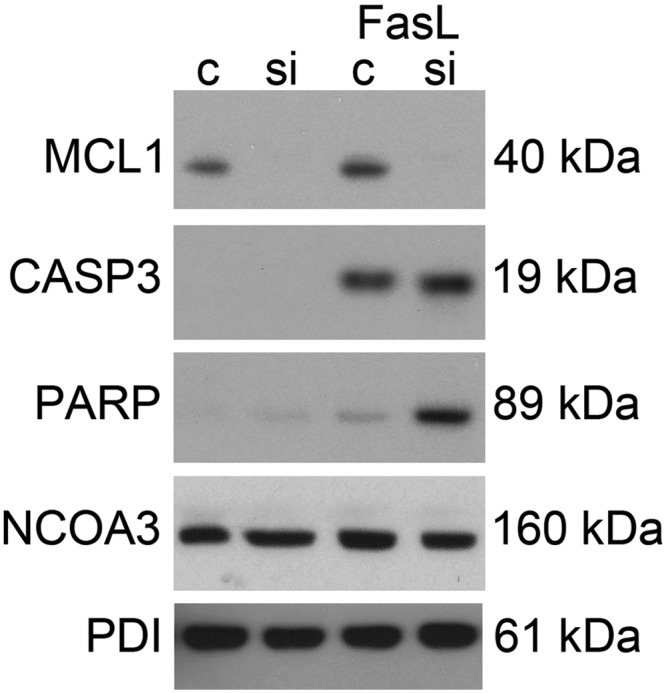
Ablation of myometrial MCL1 levels induced FasL-mediated CASP3 and PARP cleavage. hTERT cells were transfected with MCL1 siRNA for 24 h and treated for an additional 12 h with FasL. Cytoplasmic extracts were analyzed for MCL1 protein levels by immunoblot for the following experimental conditions: scrambled siRNA control, MCL1 siRNA, FasL plus scrambled siRNA, and FasL plus MCL1 siRNA. Cytoplasmic extracts from the same conditions were analyzed for active CASP3. Nuclear extracts from the same conditions were analyzed for cleaved PARP. PDI and NCOA3 were used as loading controls. These data are representative of three independent experiments (n = 3).
Elimination of MCL1 sensitizes hTERT cells to FasL-mediated apoptosis
TUNEL staining was performed on hTERT control and MCL1 siRNA-transfected human myometrial cells in the absence and presence of FasL. Consistent with our observations in Fig. 10, despite elevated CASP3 levels, we find no increase in the number of cells positive for TUNEL with FasL treatment. MCL1 suppression alone was also not sufficient to elicit an apoptotic response as detectable by TUNEL. However, FasL treatment in conjunction with MCL1 knockdown results in myocyte apoptosis as is evidenced by the elevated number of cells positive for both active CASP3 and TUNEL staining (Fig. 11).
Fig. 11.

Ablation of myometrial MCL1 levels induced FasL-mediated TUNEL-positive staining in CASP3-positive hTERT cells. hTERT cells were transfected with MCL1 siRNA and treated with FasL for 12 h. Scrambled siRNA control, FasL plus scrambled siRNA, MCL1 siRNA, and FasL plus MCL1 siRNA were analyzed by fluorescent microscopy after TUNEL staining (green) to detect the cleavage of nuclear DNA. Cells were also stained for cleaved CASP3 (red), and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). These data are representative of three independent experiments (n = 3).
Apoptotic protein array demonstrates reduced antiapoptotic signaling in the FasL-treated, MCL1-negative, uterine myocyte
Using a human apoptosis proteome profile array, we attempted to determine whether there were factors other than MCL1 or MCL1-dependent factors that were modified in the FasL-treated human uterine myocytes that helped to maintain the nonapoptotic CASP3 profile. We also speculated that we would identify either a surge in proapoptotic factors or a decline in antiapoptotic factors in response to MCL1 ablation. We did not see a significant surge in proapoptotic factors between FasL-treated cells in the absence or presence of MCL1 (data not shown). However, we did observe a robust increase in catalase (CAT), a reactive-oxygen species scavenger in response to FasL exposure. However, upon knockdown of MCL1, the hTERT cell fails to elevate CAT levels in response to FasL (Fig. 12).
Fig. 12.

Elevation of antiapoptotic CAT levels in response to FasL were abolished upon MCL1 ablation. A, hTERT cytoplasmic extracts were analyzed by apoptosis proteome array for the following experimental conditions: scrambled siRNA control, MCL1 siRNA, FasL plus scrambled siRNA, and FasL plus MCL1 siRNA. Duplicate dots for pro-CASP3, CASP3, and CAT are represented. B, MCL1, CASP3, and CAT levels were confirmed by immunoblot analysis in hTERT cytoplasmic extracts from scrambled siRNA control, MCL1 siRNA, FasL plus scrambled siRNA, and FasL plus MCL1 siRNA-treated cells. PDI is used as a protein loading control. These data are representative of three independent experiments (n = 3).
Knockdown of CAT fails to promote a proapoptotic response to FasL in the uterine myocyte
hTERT cells were transfected with validated siRNA for human CAT and examined for their CASP3-mediated apoptotic action in response to FasL. As can be observed in Fig. 13, CAT levels are increased in response to FasL exposure. CAT siRNA transfection decreased basal and FasL-treated CAT levels. In the absence of CAT, FasL-induced active CASP3 or apoptotic CASP action, as indicated by the lack of PARP cleavage, did not increase, nor was there a compensatory elevation of MCL1 levels.
Fig. 13.

Ablation of hTERT CAT levels did not induce an apoptotic response to FasL treatment. hTERT cells transfected with CAT siRNA for 24 h were treated for an additional 12 h with FasL. Cytoplasmic extracts were analyzed for CAT protein levels by immunoblot for the following experimental conditions: scrambled siRNA control, FasL plus scrambled siRNA, CAT siRNA, and FasL plus CAT siRNA. Cytoplasmic extracts from the same conditions were analyzed for active CASP3 and MCL1. Nuclear extracts from the same conditions were analyzed for cleaved PARP. PDI and NCOA3 are displayed as protein loading controls. These data are representative of three independent experiments (n = 3).
Uterine myocyte CAT is a target for apoptotic CASP3 action
Uterine myocyte CAT levels were examined in hTERT cells exposed to 100 mJ/cm2 UV light. As can be observed in Fig. 14, a decline in full-length CAT levels can be seen from 0–48 h, which corresponds to an accumulation of CASP3-cleaved CAT fragments reaching maximal levels at 48 h. It has previously been indicated that CAT is a target for apoptotic CASP3 action (20). These data indicate that the decline in CAT levels observed in the MCL1-negative, FasL-treated human uterine myocyte (Fig. 12) may be a result of the apoptotic actions of active CASP3 induced upon MCL1 ablation, which targets and cleaves the FasL-induced CAT, resulting in decreased detection of the full-length form.
Fig. 14.
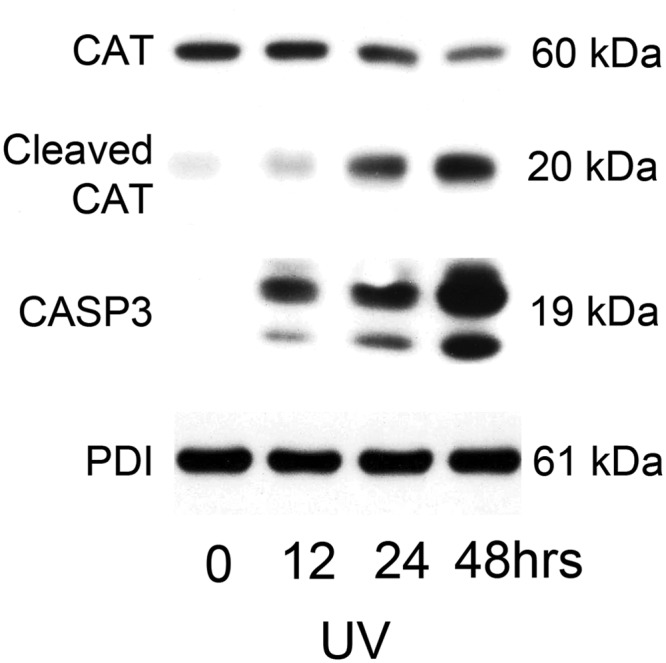
Myometrial CAT is a target for apoptotic CASP3 action. Cytoplasmic extracts from hTERT cells exposed to UV light (100 mJ/cm2) at 0, 1, 12, 24, and 48 h were analyzed by immunoblot for CAT, cleaved CAT, and active CASP3. PDI was used as our loading control. These data are representative of three independent experiments (n = 3).
Discussion
Activation of CASP3 is normally associated with apoptotic cell death (21). However, previous in vivo analysis from our group identified that elevated uterine CASP3 levels did not correlate with apoptotic cell death in the pregnant mouse uterus (2). We have previously proposed that CASP3 plays a nonapoptotic role in regulating uterine quiescence in a progesterone- and progesterone receptor-regulated manner (1). Increased antiapoptotic signaling was also detected in the pregnant mouse uterus concurrent with CASP3 activation, which we proposed maintained the pregnant uterine myocytes in a nonapoptotic state despite the presence of active CASP3. In this current study we attempt to define whether the pregnant human uterine myocyte also has the capacity to avoid CASP3-mediated apoptotic cell death by promoting an antiapoptotic response. This study attempts to define the factors that render human uterine myocyte CASP3 activity nonlethal.
Using human uterine myocytes isolated from term fundal myometrium or from a telomerase-immortalized population of human uterine myocytes (hTERT) (18), we examined the response of the human uterine myocyte to activation of CASP3. Induction of the classic extrinsic (FasL) and intrinsic (UV) apoptotic signaling pathways resulted in robust CASP3 activation in both the hTERT cell and primary cultures (Figs. 1, 2, and 7). However, there was a distinct difference between how the human uterine myocyte responded to CASP3 activation mediated via FasL vs. UV light. UV light-mediated CASP3 activation resulted in apoptotic cell death as is evidenced by increased PARP cleavage (Figs. 4 and 8), DNA laddering (Fig. 5), and TUNEL-positive staining (Figs. 6 and 9). However, FasL-mediated CASP3 activation in both the hTERT and primary cell cultures was not associated with the onset of apoptotic cell death as indicated by the lack of both PARP cleavage (Figs. 4 and 8) and DNA laddering (Fig. 5) and negative TUNEL staining (Figs. 6 and 9).
A dramatic difference in antiapoptotic signaling was also observed between uterine myocytes activated via FasL and UV light. The antiapoptotic factor MCL1 was unexpectedly up-regulated and/or maintained in response to FasL exposure (Figs. 1, 3, and 7), whereas MCL1 levels were dramatically down-regulated in response to UV light (Figs. 1, 3, and 7). MCL1 accumulation in the presence of increasing CASP3 activation was unexpected because MCL1 is primarily thought to be an apical antiapoptotic mediator blocking the initiation of caspase activation (11). However, it seems in the pregnant human uterine myocyte, MCL1 is differentially regulated by the intrinsic and extrinsic pathways of apoptosis. Although the mechanism by which MCL1 levels are up-regulated in response to FasL in the uterine mycoyte is unknown, this antiapoptotic, prosurvival response to an apoptotic stimuli has been documented previously in cancer cells. TRAIL activates CASP3 via extrinsic cell signaling in a similar manner to FasL (22). However, cancer cells have been shown to acquire resistance to TRAIL-activated apoptotic cell death through the up-regulation of the antiapoptotic factor MCL1 (23). This occurs even after apoptotic-mediated mitochondrial outer membrane permeabilization. Up-regulation of MCL1 has been shown to have the capacity to rescue CASP3-positive cells from apoptotic cell death (24). Similarly, the cytotoxic antitumor CASP3 activity of proteasomal inhibitors is also limited by the accumulation and persistence of MCL1 (25). In addition, in response to proteasome inhibitor exposure, MCL1 levels were found to persist in cells that had already undergone CASP3 activation. Similarly, our observations indicate that CASP3 activation in the pregnant uterine myocyte can occur despite the presence of MCL1, and the persistence or accumulation of MCL1 renders the CASP3-positive cell resistant to apoptotic cell death.
Nonapoptotic functions of CASP3 activity have previously been identified in the failing rabbit and rat myocardium (26, 27). Similarly, in the failing human heart it has been shown that cardiomyocytes displaying cytochrome c release, CASP3 activation, and reduced contractile ability surprisingly demonstrate normal nuclei and remain unaffected by the apoptotic process (28). In both the pregnant rat uterus and human fetal membranes, high levels of activated CASP3 in the second trimester of pregnancy were demonstrated with no evidence of wide-scale apoptosis (4, 5). Nonlethal active CASP3 has also been described in the central nervous system, where more than 90% of the active CASP3-positive cells lacked apoptotic markers such as TUNEL and were associated with the differentiation of glial cells rather than cell death (29–31).
We have proposed that the lethal action of active CASP3 in both immortalized and primary pregnant human uterine myocytes is limited as a result of sustained antiapoptotic signaling mediated through MCL1. Accordingly, ablation of uterine myocyte MCL1, either by UV light (Figs. 1, 3, and 7) or by MCL1 siRNA (Fig. 10), confer increased susceptibility to FasL-mediated CASP3 apoptotic cell death as is indicated by increased PARP cleavage and elevated TUNEL-positive staining (Figs. 10 and 11). To further define the manner by which antiapoptotic MCL1 promotes resistance to CASP3-mediated apoptotic cell death, we analyzed a protein array directed against pro- and antiapoptotic mediators using CASP3-positive hTERT myometrial cell extracts in the absence or presence of MCL1 (Fig. 12). We hypothesized that diminished MCL1 levels may reveal increased proapoptotic signaling in response to FasL. We speculated that elevated uterine MCL1 levels may suppress proapoptotic signaling in response to FasL. We considered that because ablation of MCL1 resulted in the onset of apoptotic cell death, it was likely that there were antiapoptotic MCL1-dependent events that assist MCL1 in the suppression of active CASP3-apoptotic action in the pregnant human uterine myocyte.
Uterine myocyte CAT, a reactive oxygen species scavenger, was found to be the sole factor that was uniquely increased in response to FasL but unable to host that response upon ablation of MCL1 (Fig. 12). CAT has previous been demonstrated to act in an antiapoptotic manner by protecting the cell against cellular damage by oxidizing toxins (32, 33). Our data suggest that upon ablation of MCL1, the CAT response was also disabled (Fig. 12). Therefore, these data initially suggested that CAT was an MCL1-sensitive/regulated factor that assisted MCL1 in rendering uterine active CASP3 nonlethal. However, siRNA-mediated inhibition of uterine myocyte CAT failed to initiate an increased proapoptotic response or a compensatory antiapoptotic response (Fig. 13). This was evidenced by the lack of PARP cleavage and unchanged MCL1 levels in response to FasL-mediated CASP3 activation upon diminished CAT levels (Fig. 13). Therefore, we suggest that CAT may assist in limiting the levels of potentially damaging reactive oxygen species released in response to FasL stimulation, but its action is not responsible for maintaining human pregnant uterine myocytes in a nonapoptotic state despite elevated levels of active CASP3. Upon further investigation it became clear that although FasL treatment robustly elevated human uterine myocyte antiapoptotic CAT activity, the ablation of CAT in response to diminished MCL1 levels was likely an indirect result of CAT being a direct target for increased apoptotic CASP3 activity (20). Increased apoptotic CASP3 activity mediated by UV light has the capacity, as is shown in Fig. 14, to cleave and diminish the levels of full-length myometrial CAT.
Taken together, these data indicate that the human pregnant uterine myocyte hosts a prosurvival, antiapoptotic response to avoid CASP3-mediated cell death. MCL1 suppression alone was not sufficient to elicit apoptosis; however, in conjunction with an apoptotic stimuli, diminished MCL1 levels could precipitate cell death in the face of compensatory antiapoptotic signaling. The human myometrial cell hosts a complex and potentially redundant antiapoptotic response to insults such as MCL1 suppression and FasL stimulation, which may contribute to its ability to tolerate active CASP3 during pregnancy. We have previously demonstrated similar antiapoptotic signaling in the face of CASP3 activation in vivo in the pregnant mouse uterine gestational series representing the latter half of gestation. These current in vitro studies suggest that, similar to pregnant mouse uterus, the pregnant human uterine myocyte has the capacity to harness the tocolytic potential of uterine CASP3 while avoiding apoptotic cell death by hosting an antiapoptotic response. We hypothesize that the pregnant uterine myocyte protects itself against the elevated levels of potentially apoptotic CASP3 required to maintain the pregnant uterus in a quiescent state during late gestation. Isolating MCL1 as a critical regulator of nonapoptotic action of uterine CASP3 during human pregnancy may potentially define useful and novel tocolytic agents to assist in the prevention of premature uterine contractions and the onset of preterm birth.
Materials and Methods
hTERT cell culture
The hTERT cell line was derived from premenopausal nonpregnant uterine tissue as previously described (18). Cells were cultured in monolayer in T75 flasks in DMEM/F12 (Invitrogen, Carlsbad, CA) with 10% fetal bovine serum (FBS; Invitrogen) and antibiotic/antimycotic (10,000 U/ml; Invitrogen) at 37 C in 95% air with 5% CO2. For each experiment, hTERT cells were seeded at 1.5–2 × 106 cells per 10-cm2 tissue culture dish, 225,000 cells per well of a six-well plate or 75,000 cells per well of four-well chamber slide in phenol red-free DMEM/F12 with 10% charcoal-stripped FBS for 12 h and serum starved for 12 h before treatment with either FasL or UV light in phenol red-free DMEM/F12 containing 0.5% charcoal-stripped FBS. Each experiment was carried out in triplicate (n = 3).
Primary pregnant fundal myometrial cell culture
Fundal uterine biopsy tissues were obtained from three pregnant women undergoing scheduled cesarean hysterectomy for placenta accreta at 36 wk immediately after delivery. Informed consent was obtained in writing from each subject before surgery using protocols approved by the Institutional Review Board of the University of Pittsburgh. Primary culture of human myometrial smooth muscle cells was performed using a previously described technique (34). Briefly, blood vessels were dissected from each of the biopsies, and the myometrial smooth muscle tissues were minced in ice-cold Hank's buffered salt solution. Tissues were then centrifuged at 3000 rpm for 3 min at 4 C and washed twice with Hank's buffered salt solution. Tissues were then enzymatically digested in DMEM/F12 media containing 10% FBS and antibiotics with 1 mg/ml collagenase type I (Worthington Biochemical Corp., Lakewood, NJ) and 1 mg/ml collagenase type II (Invitrogen) for 2 h in a humidified incubator at 37 C. Tissues were then triturated 20 times with a Pasteur pipette and filtered using a coarse screen. Tissue particles were sequentially passed through 100-μm, 70-μm, and 40-μm filters. Cells were then centrifuged at 3000 rpm for 3 min and washed twice before plating in 10-cm2 tissue culture dishes. Media was changed at least twice daily until 80% confluent at which time cells were trypsinized and seeded into six-well tissue culture dishes and four-well chamber slides for subsequent experiments. Before treatment with UV light or FasL, cells were serum starved for 12 h and treated in phenol red-free DMEM-F12 containing 0.5% FBS. Each experiment was carried out in triplicate (n = 3).
UV light source and exposure
Before treatment with UV light, hTERT and primary culture uterine myocytes were serum starved for 12 h and maintained in phenol red-free DMEM-F12 containing 0.5% FBS. UVC (emission peak at 254 nm) exposure via the HL-2000 HybriLinker (UVP, LLC, Upland, CA) was carried out in 10-cm2 tissue culture dish containing 6 ml media, or in four-chamber Lab-Tek chamber slides (Labtek, Scotts Valley, CA) containing 0.5 ml media. In the initial dose-response experiments hTERT cells were exposed to 10, 100, and 500 mJ/cm2 UV light and placed back into a humidified tissue culture incubator at 37 C for 16 h. In the time course experiments, after 100 mJ/cm2 exposure, cells were placed back into a humidified tissue culture incubator at 37 C for 1, 12, 24, and 48 h. Each experiment was carried out in triplicate (n = 3).
FasL treatment of hTERT and primary cultures of uterine myocytes
Before treatment with FasL, cells were serum starved for 12 h and maintained in phenol red-free DMEM-F12 containing 0.5% FBS. Cells were treated with recombinant human FasL (R&D Systems, Minneapolis, MN) at 0, 1, 5, 10, 50, and 100 ng/ml in the presence of 10 mg/ml of a cross-linking antibody (MAB2020, R&D Systems) for 16 h. For time-course experiments, hTERT and primary myometrial cells were treated with 100 ng/ml FasL and 10 mg/ml of the cross-linking antibody for 0, 1, 12, 24, and 48 h. Each experiment was carried out in triplicate (n = 3).
Reverse transfection of hTERT cells with MCL1 and CAT siRNA
Silencer Select siRNA specific for MCL1 (siRNA ID s8583, control sense 5′-CCAGUAUACUUCUUAGAAATT-3′ and test antisense 5′-UUUCUAAGAAGUAUACUGGGA-3′) were obtained from Ambion (Austin, TX). Silencer select siRNA specific for CAT (siRNA ID s2445, control sense 5′-GACUGACCAGGGCAUCAAAtt-3′ and test antisense 5′-UUUGAUGCCCUGGUCAGUCtt-3′) were obtained from Ambion. Per vendor protocol, 5 ml of Lipofectamine transfection reagent (Invitrogen, Carlsbad, CA) per well of a six-well plate was diluted in OptiMEM media without phenol red (Invitrogen) and incubated at room temperature for 5 min. Lipofectamine and MCL1 siRNA transfection complexes were formed for a final concentration of 5 nm siRNA at room temperature for 15 min. Lipofectamine in OptiMEM media was used as a control for each experiment. Lipofectamine alone or transfection complexes were aliquoted into each well. hTERT cells were trypsinized and diluted in OptiMEM media containing 10% charcoal-stripped FBS without antibiotics. Cells were overlaid in each well for a final volume of 2.5 ml and incubated at 37 C for 24 h with transfection complexes or Lipofectamine alone. At 24 h after transfection, baseline (time zero) protein extracts were obtained. Media were then replaced with DMEM/F12 without phenol red containing 0.5% charcoal-stripped FBS with and without FasL/antipolyhistidine cross-linking antibody. Each experiment was carried out in triplicate (n = 3).
Subcellular fractionation
Isolation of cytoplasmic and nuclear extracts from cultured cells were performed as described previously (35, 36). Briefly, cells grown in monolayer were washed with phosphate-buffered solution, scraped and centrifuged at 3000 × g for 1 min at 4 C. The pellet was homogenized in ice-cold NE1 buffer containing 10 mm HEPES (pH 7.5), 10 mm MgCl2, 5 mm KCl, 0.1% Triton X-100, and 1× protease/phosphatase inhibitor cocktail (Roche, Mannheim, Germany) with a 23-gauge needle. The homogenate was centrifuged at 3000 × g for 10 min at 4 C, and the supernatant was retained as cytoplasmic fraction. The pellet was washed in NE1 and then resuspended in ice-cold NE2 buffer containing 25% glycerol, 20 mm HEPES (pH 7.9), 500 mm NaCl, 1.5 mm MgCl2, and 0.2 mm EDTA (pH 8.0) and was incubated on ice for 30 min, vortexed every 5 min, and centrifuged at 10,600 × g for 10 min at 4 C. The supernatant was retained as the nuclear fraction. Subcellular protein concentrations were determined in triplicate by bicinchoninic acid protein assay (Promega Corp., Madison, WI).
Immunoblotting
Equal amounts of cytoplasmic and nuclear proteins, quantified by colorimetric bicinchoninic acid protein assay, were separated in gradient polyacrylamide gels (Invitrogen) and transferred onto Hybond-P (Amersham Pharmacia, Piscataway, NJ). Blots were probed using primary antibodies against cleaved CASP3 (1:750 dilution; no. 9664; Cell Signaling Technology, Danvers, MA), cleaved PARP 1:750 dilution; no. 9541; Cell Signaling Technology), MCL1 (1:1000 dilution; no. Sc-819; Santa Cruz Biotechnology, Inc., Santa Cruz, CA), CAT (1:500 dilution; ab1877; AbCam, Cambridge MA), disulfide isomerase (PDI) (1:2000 dilution; no. 2446; Cell Signaling Technology), and nuclear receptor coactivator 3 (NCOA3) (1:2000 dilution; no. PA1–845, Pierce Chemical Co., Rockford, IL).
Primary antibodies were diluted in 5% milk-1× Tris-buffered saline-Tween 20 buffer, followed by incubation with horseradish peroxidase-conjugated secondary antibodies diluted in 5% milk-1× Tris-buffered saline-Tween 20 buffer. Immunoreactive bands were visualized using an enhanced chemiluminescence detection system (Amersham Pharmacia). NIH ImageJ was used to identify immunoreactive protein band intensity. The relative OD was obtained by normalizing immunoreactive cytoplasmic protein band intensity to PDI, which we have found to be maintained at constant levels under the experimental conditions used across this study. Nuclear band intensity was normalized to NCOA3, a steroid-receptor coactivator localized to the nucleus, which we have found to be maintained at constant levels under the experimental conditions used across this study.
TUNEL and immunofluorescent CASP3 staining
hTERT and primary uterine myocytes were grown on four-well chamber slides. Cells were plated at 75,000 per well of four-well chamber slide in phenol red-free DMEM/F12 with 10% charcoal stripped FBS for 12 h and serum starved for 12 h before treatment with either FasL or UV light in phenol red-free DMEM/F12. Cells were fixed with 4% paraformaldehyde for 25 min at room temperature and stored in 1× PBS at 4 C. Cells were permeabilized with 0.2% Triton-X in PBS and stained with a fluorometric TUNEL assay (Promega, no. G3250). Subsequent immunofluorescent staining for cleaved CASP3 (Cell Signaling Technology, no. 9664) at 1:200 in PBS for 2 h at room temperature was performed with Texas Red antirabbit secondary secondary antibody (Jackson Laboratories, Newmarket Suffolk, UK) at 1 h at room temperature in PBS. Each experiment was carried out in triplicate (n = 3).
DNA fragmentation assay
hTERT cells were cultured as described above and treated with FasL or UV light for 12 and 24 h. Genomic DNA was extracted from cells using an apoptotic DNA ladder assay kit (Roche). DNA was incubated with 100 μg/ml of ribonuclease A at room temperature for 30 min. Equal amounts of DNA were separated on 1% agarose gel containing ethidium bromide and photographed under UV light. The positive control apoptotic DNA ladder was an U937 cell extract provided by Roche. Each experiment was carried out in triplicate (n = 3).
Apoptosis proteome array
siRNA transfection for MCL1 with subsequent treatment with FasL 100 ng/ml for 12 h was performed, and cytoplasmic protein was extracted. For the scrambled control, MCL1 siRNA alone, FasL alone, and MCL1 siRNA with FasL, 250 mg of protein for each condition, was incubated with an array membrane seeded with antibodies against 35 pro- and apoptotic proteins (R&D, no. ARY009, Minneapolis, MN) per manufacturer's instructions. Immunoreactive dots were visualized using an enhanced chemiluminescence detection system (Amersham Pharmacia). Each experiment was carried out in triplicate (n = 3).
Acknowledgments
We thank Professor Stanley N. Caritis and Professor Hygraiv Simhan (Department of Obstetrics and Gynecology, University of Pittsburgh) for providing us with human uterine tissue used in this study.
This work was supported by grants from the March of Dimes (21-FY2008-555) and the National Institute of Child Health and Human Development (1RO1HD065011-01A1).
Disclosure Summary: A.S.-F., J.M., A.S., S.N.C., H.S., P.J., and J.C.C. have nothing to disclose.
Footnotes
- CASP3
- Caspase 3
- CAT
- catalase
- FasL
- Fas ligand
- FBS
- fetal bovine serum
- MCL1
- myeloid cell leukemia sequence 1
- NCOA3
- nuclear receptor coactivator 3
- PARP
- polymerase
- PDI
- disulfide isomerase
- siRNA
- small interfering RNA
- TRAIL
- TNF-related apoptosis-inducing ligand
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling.
References
- 1. Jeyasuria P, Wetzel J, Bradley M, Subedi K, Condon JC. 2009. Progesterone-regulated caspase 3 action in the mouse may play a role in uterine quiescence during pregnancy through fragmentation of uterine myocyte contractile proteins. Biol Reprod 80:928–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jeyasuria P, Subedi K, Suresh A, Condon JC. 2011. Elevated levels of uterine anti-apoptotic signaling may activate NFKB and potentially confer resistance to caspase 3-mediated apoptotic cell death during pregnancy in mice. Biol Reprod 85:417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haider N, Narula N, Narula J. 2002. Apoptosis in heart failure represents programmed cell survival, not death, of cardiomyocytes and likelihood of reverse remodeling. J Cardiac Fail 8 (Suppl 6):S512–S517 [DOI] [PubMed] [Google Scholar]
- 4. Shynlova O, Oldenhof A, Dorogin A, Xu Q, Mu J, Nashman N, Lye SJ. 2006. Myometrial apoptosis: activation of the caspase cascade in the pregnant rat myometrium at midgestation. Biol Reprod 74:839–849 [DOI] [PubMed] [Google Scholar]
- 5. Runić R, Lockwood CJ, LaChapelle L, Dipasquale B, Demopoulos RI, Kumar A, Guller S. 1998. Apoptosis and Fas expression in human fetal membranes. J Clin Endocrinol Metab 83:660–666 [DOI] [PubMed] [Google Scholar]
- 6. Communal C, Sumandea M, de Tombe P, Narula J, Solaro RJ, Hajjar RJ. 2002. Functional consequences of caspase activation in cardiac myocytes. Proc Natl Acad Sci USA 99:6252–6256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD, Dal Bello B, Semigran MJ, Bielsa-Masdeu A, Dec GW, Israels S, Ballester M, Virmani R, Saxena S, Kharbanda S. 1999. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc Natl Acad Sci USA 96:8144–8149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Supinski GS, Callahan LA. 2006. Caspase activation contributes to endotoxin-induced diaphragm weakness. J Appl Physiol 100:1770–1777 [DOI] [PubMed] [Google Scholar]
- 9. Hong SK, Son H, Kim SW, Oh SJ, Choi H. 2005. Effect of glycine on recovery of bladder smooth muscle contractility after acute urinary retention in rats. BJU Int 96:1403–1408 [DOI] [PubMed] [Google Scholar]
- 10. Youle RJ, Strasser A. 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9:47–59 [DOI] [PubMed] [Google Scholar]
- 11. Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. 2003. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev 17:1475–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Allagnat F, Cunha D, Moore F, Vanderwinden JM, Eizirik DL, Cardozo AK. 2011. Mcl-1 downregulation by pro-inflammatory cytokines and palmitate is an early event contributing to β-cell apoptosis. Cell Death Differ 18:328–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adams KW, Cooper GM. 2007. Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J Biol Chem 282:6192–6200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chetoui N, Sylla K, Gagnon-Houde JV, Alcaide-Loridan C, Charron D, Al-Daccak R, Aoudjit F. 2008. Down-regulation of mcl-1 by small interfering RNA sensitizes resistant melanoma cells to fas-mediated apoptosis. Mol Cancer Res 6:42–52 [DOI] [PubMed] [Google Scholar]
- 15. Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ, Kaufmann SH, Gores GJ. 2004. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res 64:3517–3524 [DOI] [PubMed] [Google Scholar]
- 16. Schulze-Bergkamen H, Fleischer B, Schuchmann M, Weber A, Weinmann A, Krammer PH, Galle PR. 2006. Suppression of Mcl-1 via RNA interference sensitizes human hepatocellular carcinoma cells towards apoptosis induction. BMC Cancer 6:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guoan X, Hanning W, Kaiyun C, Hao L. 2010. Adenovirus-mediated siRNA targeting Mcl-1 gene increases radiosensitivity of pancreatic carcinoma cells in vitro and in vivo. Surgery 147:553–561 [DOI] [PubMed] [Google Scholar]
- 18. Condon J, Yin S, Mayhew B, Word RA, Wright WE, Shay JW, Rainey WE. 2002. Telomerase immortalization of human myometrial cells. Biol Reprod 67:506–514 [DOI] [PubMed] [Google Scholar]
- 19. Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. 1994. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371:346–347 [DOI] [PubMed] [Google Scholar]
- 20. Iwai K, Kondo T, Watanabe M, Yabu T, Kitano T, Taguchi Y, Umehara H, Takahashi A, Uchiyama T, Okazaki T. 2003. Ceramide increases oxidative damage due to inhibition of catalase by caspase-3-dependent proteolysis in HL-60 cell apoptosis. J Biol Chem 278:9813–9822 [DOI] [PubMed] [Google Scholar]
- 21. Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. 1993. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 β-converting enzyme. Cell 75:641–652 [DOI] [PubMed] [Google Scholar]
- 22. Srivastava RK. 2001. TRAIL/Apo-2L: mechanisms and clinical applications in cancer. Neoplasia 3:535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W, Jin Z, Liu YY, Dicker DT, Chiao PJ, Flaherty KT, Smith CD, El-Deiry WS. 2007. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell 12:66–80 [DOI] [PubMed] [Google Scholar]
- 24. Son JK, Varadarajan S, Bratton SB. 2010. TRAIL-activated stress kinases suppress apoptosis through transcriptional upregulation of MCL-1. Cell Death Diff 17:1288–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nencioni A, Hua F, Dillon CP, Yokoo R, Scheiermann C, Cardone MH, Barbieri E, Rocco I, Garuti A, Wesselborg S, Belka C, Brossart P, Patrone F, Ballestrero A. 2005. Evidence for a protective role of Mcl-1 in proteasome inhibitor-induced apoptosis. Blood 105:3255–3262 [DOI] [PubMed] [Google Scholar]
- 26. Laugwitz KL, Moretti A, Weig HJ, Gillitzer A, Pinkernell K, Ott T, Pragst I, Städele C, Seyfarth M, Schömig A, Ungerer M. 2001. Blocking caspase-activated apoptosis improves contractility in failing myocardium. Hum Gene Ther 12:2051–2063 [DOI] [PubMed] [Google Scholar]
- 27. Ruetten H, Badorff C, Ihling C, Zeiher AM, Dimmeler S. 2001. Inhibition of caspase-3 improves contractile recovery of stunned myocardium, independent of apoptosis-inhibitory effects. J Am Coll Cardiol 38:2063–2070 [DOI] [PubMed] [Google Scholar]
- 28. Narula J, Haider N, Arbustini E, Chandrashekhar Y. 2006. Mechanisms of disease: apoptosis in heart failure–seeing hope in death. Nat Clin Pract Cardiovasc Med 3:681–688 [DOI] [PubMed] [Google Scholar]
- 29. Oomman S, Finckbone V, Dertien J, Attridge J, Henne W, Medina M, Mansouri B, Singh H, Strahlendorf H, Strahlendorf J. 2004. Active caspase-3 expression during postnatal development of rat cerebellum is not systematically or consistently associated with apoptosis. J Comp Neurol 476:154–173 [DOI] [PubMed] [Google Scholar]
- 30. Oomman S, Strahlendorf H, Finckbone V, Strahlendorf J. 2005. Non-lethal active caspase-3 expression in Bergmann glia of postnatal rat cerebellum. Brain Res Dev Brain Res 160:130–145 [DOI] [PubMed] [Google Scholar]
- 31. Finckbone V, Oomman SK, Strahlendorf HK, Strahlendorf JC. 2009. Regional differences in the temporal expression of non-apoptotic caspase-3-positive bergmann glial cells in the developing rat cerebellum. Front Neuroanat 3:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bechtel W, Bauer G. 2009. Catalase protects tumor cells from apoptosis induction by intercellular ROS signaling. Anticancer Res 29:4541–4557 [PubMed] [Google Scholar]
- 33. Nishikawa T, Nishikawa S, Akiyama N, Natori S. 2004. Correlation between the catalase level in tumor cells and their sensitivity to N-β-alanyl-5-S-glutathionyl-3,4-dihydroxyphenylalanine (5-S-GAD). J Biochem 135:465–469 [DOI] [PubMed] [Google Scholar]
- 34. Tribe RM, Moriarty P, Poston L. 2000. Calcium homeostatic pathways change with gestation in human myometrium. Biol Reprod 63:748–755 [DOI] [PubMed] [Google Scholar]
- 35. Condon JC, Hardy DB, Kovaric K, Mendelson CR. 2006. Up-regulation of the progesterone receptor (PR)-C isoform in laboring myometrium by activation of nuclear factor-κB may contribute to the onset of labor through inhibition of PR function. Mol Endocrinol 20:764–775 [DOI] [PubMed] [Google Scholar]
- 36. Blough E, Dineen B, Esser K. 1999. Extraction of nuclear proteins from striated muscle tissue. Biotechniques 26:202–204 [DOI] [PubMed] [Google Scholar]