

Abstract
In the title compound, C6H10N2O2·H2O, the imidazole ring is essentially planar, with a maximum deviation of 0.012 (2) Å. In the crystal, molecules are connected via N—H⋯O and O—H⋯O hydrogen bonds, forming a supramolecular tape along the a axis.
Related literature
For details and applications of hydantoins, see: El-Deeb et al. (2010 ▶); Rajic et al. (2006 ▶); Carmi et al. (2006 ▶); Sergent et al., (2008 ▶); Yu et al. (2004 ▶). For related structues, see: Delgado et al. (2007 ▶); Ciechanowicz-Rutkowska et al. (1994 ▶). For the synthetic procedure, see: Abdel-Aziz (2007 ▶). For a description of the Cambridge Structural Database, see: Allen (2002 ▶).
Experimental
Crystal data
C6H10N2O2·H2O
M r = 160.18
Orthorhombic,
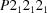
a = 6.2688 (3) Å
b = 9.2387 (4) Å
c = 14.8280 (7) Å
V = 858.77 (7) Å3
Z = 4
Cu Kα radiation
μ = 0.84 mm−1
T = 296 K
0.90 × 0.21 × 0.16 mm
Data collection
Bruker SMART APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.518, T max = 0.879
5702 measured reflections
1497 independent reflections
1378 reflections with I > 2σ(I)
R int = 0.027
Refinement
R[F 2 > 2σ(F 2)] = 0.037
wR(F 2) = 0.098
S = 1.09
1497 reflections
117 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.12 e Å−3
Δρmin = −0.18 e Å−3
Absolute structure: Flack (1983 ▶), with 592 Friedel pairs
Flack parameter: 0.2 (3)
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812002838/is5056sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812002838/is5056Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812002838/is5056Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N1⋯O2i | 0.81 (2) | 2.12 (2) | 2.927 (2) | 174.0 (19) |
| N2—H1N2⋯O1Wii | 0.87 (3) | 1.88 (3) | 2.751 (2) | 173 (2) |
| O1W—H1W1⋯O1 | 0.82 (4) | 1.95 (4) | 2.767 (2) | 173 (3) |
| O1W—H2W2⋯O1iii | 0.86 (4) | 1.98 (4) | 2.839 (2) | 171 (4) |
Symmetry codes: (i) 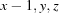 ; (ii)
; (ii) 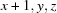 ; (iii)
; (iii) 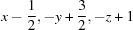 .
.
Acknowledgments
AAA, ASE and AMA thank Universiti Sains Malaysia and King Saud University for supporting this study. MH and HKF thank the Malaysian Government and Universiti Sains Malaysia for Research University Grant No. 1001/PFIZIK/811160. MH also thanks Universiti Sains Malaysia for a postdoctoral research fellowship.
supplementary crystallographic information
Comment
Hydantoins (imidazolidine-2,4-dione) are important classes of compounds which have long attracted attention, owing to their remarkable biological and pharmacological properties, such as antitumor activity, antiviral activity, insulinotropic properties and EGFR inhibitors (El-Deeb et al., 2010; Rajic et al., 2006; Carmi et al., 2006; Sergent et al., 2008). The crystal structures of (S)-5-Benzylimidazolidine-2,4-dione monohydrate (Delgado et al., 2007) and diphenylhydantoin derivatives (Ciechanowicz-Rutkowska et al., 1994) have been reported in the literature. The title compound was successfully obtained in an optical active form without racemization by dehydrative cyclization in one-pot reaction of L-valine and urea in the presence of DPPOX as catalyst (Abdel-Aziz, 2007).
The asymmetric unit contains one (S)-5-isopropylimidazolidine-2,4-dione molecule and one water molecule as shown in Fig. 1. The imidazole (N1,N2/C1–C3) ring is essentially planar, with maximum deviations of 0.012 (2) Å for atom C3. The N1—C1—O1 [128.32 (19)°] angle is greater than the N2—C1—O1 [124.05 (17)°] angle. This difference is also observed in the hydantoin molecule (Yu et al., 2004) and 50 other hydantoin derivatives reported in the Cambridge Structural Database (Version 5.28; Allen, 2002) with both unsubstituted NH groups and sp3-hybridization at C3. In the crystal structure (Fig. 2), the molecules are connected via N—H···O and O—H···O hydrogen bonds (Table 1), forming a supramolecular tape along the a axis.
Experimental
The DPPOX (1.5 equiv) was added to the equimolar solution of L-valine and urea in MeCN in addition to Et3N (1.5 equiv) and the mixture was stirred at 50°C for 60 min. After removal of the solvent, the residue was taken up in organic solvent EtOAc, and washed successively with HCl aq and NaHCO3 aq. Evaporation of the dried organic solvent gave the title compound. The colourless single-crystals suitable for X-ray analysis was obtained by recrystallization from ethanol (m.p. 145–147 °C; yield: 95%).
Refinement
Atoms H1N1, H1N2, H1W1 and H2W2 were located in a difference Fourier map and refined freely [N—H = 0.80 (2)–0.87 (3) Å; O—H = 0.82 (4)–0.87 (4) Å]. The remaining H atoms were positioned geometrically [C—H = 0.96 or 0.98 Å] and were refined using a riding model, with Uiso(H) = 1.2 or 1.5Ueq(C). A rotating group model was applied to the methyl groups. Even though there is sufficient anomalous dispersion to find the absolute configuration as the compound crystallize out in a chiral space group and Cu radiation was used, this was unsuccessful as the crystal is a inversion twin [BASF ratio of 0.8 (3):0.2 (3)].
Figures
Fig. 1.

The asymmetric unit of the title compound, showing 50% probability displacement ellipsoids. The O—H···O hydrogen bond is shown by a dashed line.
Fig. 2.

A partial packing view of the title compound. The hydrogen bonds are shown by dashed lines.
Crystal data
| C6H10N2O2·H2O | F(000) = 344 |
| Mr = 160.18 | Dx = 1.239 Mg m−3 |
| Orthorhombic, P212121 | Cu Kα radiation, λ = 1.54178 Å |
| Hall symbol: P 2ac 2ab | Cell parameters from 2281 reflections |
| a = 6.2688 (3) Å | θ = 5.6–65.9° |
| b = 9.2387 (4) Å | µ = 0.84 mm−1 |
| c = 14.8280 (7) Å | T = 296 K |
| V = 858.77 (7) Å3 | Needle, colourless |
| Z = 4 | 0.90 × 0.21 × 0.16 mm |
Data collection
| Bruker SMART APEXII CCD area-detector diffractometer | 1497 independent reflections |
| Radiation source: fine-focus sealed tube | 1378 reflections with I > 2σ(I) |
| graphite | Rint = 0.027 |
| φ and ω scans | θmax = 67.3°, θmin = 5.6° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −7→5 |
| Tmin = 0.518, Tmax = 0.879 | k = −11→10 |
| 5702 measured reflections | l = −17→17 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.037 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.098 | w = 1/[σ2(Fo2) + (0.0552P)2 + 0.0813P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.09 | (Δ/σ)max = 0.001 |
| 1497 reflections | Δρmax = 0.12 e Å−3 |
| 117 parameters | Δρmin = −0.18 e Å−3 |
| 0 restraints | Absolute structure: Flack (1983), 592 Friedel pairs |
| Primary atom site location: structure-invariant direct methods | Flack parameter: 0.2 (3) |
Special details
| Experimental. 1H NMR (DMSO–d6): 10.54 (s, 1H, NH), 7.87 (s, 1H, NH), 3.89 (s, 1H), 2.01–1.97 (m, 1H), 0.94–0.92 (d, 3H, J = 7.0 Hz), 0.80–0.78 (d, 3H, J = 6.5 Hz). 13C NMR (DMSO–d6): 175.87, 158.28, 63.22, 30.01, 18.93, 16.31. |
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.6583 (2) | 0.56794 (15) | 0.46328 (11) | 0.0679 (5) | |
| O2 | 1.2150 (2) | 0.27203 (15) | 0.39487 (11) | 0.0590 (4) | |
| N1 | 0.6703 (3) | 0.34173 (17) | 0.39871 (11) | 0.0472 (4) | |
| N2 | 0.9719 (2) | 0.44507 (16) | 0.43611 (11) | 0.0503 (4) | |
| C1 | 0.7524 (3) | 0.46070 (19) | 0.43523 (13) | 0.0481 (4) | |
| C2 | 1.0337 (3) | 0.31526 (19) | 0.40226 (13) | 0.0440 (4) | |
| C3 | 0.8324 (3) | 0.23514 (18) | 0.37618 (12) | 0.0436 (4) | |
| H3A | 0.8156 | 0.1507 | 0.4156 | 0.052* | |
| C4 | 0.8317 (3) | 0.1851 (2) | 0.27789 (14) | 0.0534 (5) | |
| H4A | 0.9533 | 0.1196 | 0.2701 | 0.064* | |
| C5 | 0.6312 (4) | 0.0986 (3) | 0.25796 (19) | 0.0828 (8) | |
| H5A | 0.6202 | 0.0197 | 0.2998 | 0.124* | |
| H5B | 0.6381 | 0.0613 | 0.1976 | 0.124* | |
| H5C | 0.5086 | 0.1602 | 0.2639 | 0.124* | |
| C6 | 0.8624 (5) | 0.3102 (3) | 0.21256 (16) | 0.0810 (7) | |
| H6A | 0.9926 | 0.3600 | 0.2266 | 0.122* | |
| H6B | 0.7446 | 0.3760 | 0.2177 | 0.122* | |
| H6C | 0.8694 | 0.2736 | 0.1520 | 0.122* | |
| H1N1 | 0.543 (4) | 0.329 (2) | 0.3981 (13) | 0.045 (5)* | |
| H1N2 | 1.059 (5) | 0.509 (3) | 0.4591 (17) | 0.078 (8)* | |
| O1W | 0.2489 (3) | 0.63212 (17) | 0.52044 (13) | 0.0665 (4) | |
| H1W1 | 0.374 (6) | 0.617 (3) | 0.507 (2) | 0.096 (11)* | |
| H2W2 | 0.214 (6) | 0.721 (4) | 0.531 (3) | 0.122 (13)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0438 (9) | 0.0566 (8) | 0.1033 (12) | 0.0068 (6) | −0.0017 (7) | −0.0302 (8) |
| O2 | 0.0292 (8) | 0.0607 (8) | 0.0871 (11) | 0.0039 (6) | −0.0030 (6) | −0.0086 (7) |
| N1 | 0.0246 (8) | 0.0507 (8) | 0.0662 (10) | −0.0020 (6) | −0.0013 (6) | −0.0127 (7) |
| N2 | 0.0309 (9) | 0.0475 (8) | 0.0725 (11) | −0.0035 (6) | −0.0045 (7) | −0.0147 (8) |
| C1 | 0.0372 (10) | 0.0474 (9) | 0.0597 (12) | 0.0028 (8) | −0.0024 (8) | −0.0079 (8) |
| C2 | 0.0312 (9) | 0.0468 (9) | 0.0541 (10) | −0.0018 (7) | −0.0011 (7) | −0.0001 (8) |
| C3 | 0.0323 (9) | 0.0394 (8) | 0.0591 (10) | −0.0025 (6) | 0.0003 (7) | −0.0028 (7) |
| C4 | 0.0443 (11) | 0.0491 (9) | 0.0667 (12) | −0.0016 (8) | 0.0028 (8) | −0.0161 (8) |
| C5 | 0.0645 (16) | 0.0895 (17) | 0.0945 (18) | −0.0206 (13) | −0.0049 (14) | −0.0376 (14) |
| C6 | 0.098 (2) | 0.0838 (16) | 0.0617 (14) | −0.0079 (15) | 0.0041 (13) | −0.0044 (12) |
| O1W | 0.0438 (10) | 0.0557 (8) | 0.1000 (12) | −0.0042 (7) | −0.0067 (8) | −0.0203 (8) |
Geometric parameters (Å, °)
| O1—C1 | 1.226 (2) | C4—C5 | 1.518 (3) |
| O2—C2 | 1.210 (2) | C4—C6 | 1.520 (3) |
| N1—C1 | 1.329 (2) | C4—H4A | 0.9800 |
| N1—C3 | 1.454 (2) | C5—H5A | 0.9600 |
| N1—H1N1 | 0.80 (2) | C5—H5B | 0.9600 |
| N2—C2 | 1.357 (2) | C5—H5C | 0.9600 |
| N2—C1 | 1.383 (3) | C6—H6A | 0.9600 |
| N2—H1N2 | 0.87 (3) | C6—H6B | 0.9600 |
| C2—C3 | 1.513 (2) | C6—H6C | 0.9600 |
| C3—C4 | 1.529 (3) | O1W—H1W1 | 0.82 (4) |
| C3—H3A | 0.9800 | O1W—H2W2 | 0.87 (4) |
| C1—N1—C3 | 112.51 (15) | C5—C4—C3 | 110.30 (17) |
| C1—N1—H1N1 | 120.6 (15) | C6—C4—C3 | 112.16 (16) |
| C3—N1—H1N1 | 126.2 (15) | C5—C4—H4A | 107.2 |
| C2—N2—C1 | 111.89 (15) | C6—C4—H4A | 107.2 |
| C2—N2—H1N2 | 124.4 (19) | C3—C4—H4A | 107.2 |
| C1—N2—H1N2 | 123.6 (19) | C4—C5—H5A | 109.5 |
| O1—C1—N1 | 128.32 (19) | C4—C5—H5B | 109.5 |
| O1—C1—N2 | 124.05 (17) | H5A—C5—H5B | 109.5 |
| N1—C1—N2 | 107.63 (16) | C4—C5—H5C | 109.5 |
| O2—C2—N2 | 126.42 (17) | H5A—C5—H5C | 109.5 |
| O2—C2—C3 | 126.79 (16) | H5B—C5—H5C | 109.5 |
| N2—C2—C3 | 106.79 (15) | C4—C6—H6A | 109.5 |
| N1—C3—C2 | 101.12 (13) | C4—C6—H6B | 109.5 |
| N1—C3—C4 | 114.95 (15) | H6A—C6—H6B | 109.5 |
| C2—C3—C4 | 113.19 (15) | C4—C6—H6C | 109.5 |
| N1—C3—H3A | 109.1 | H6A—C6—H6C | 109.5 |
| C2—C3—H3A | 109.1 | H6B—C6—H6C | 109.5 |
| C4—C3—H3A | 109.1 | H1W1—O1W—H2W2 | 116 (3) |
| C5—C4—C6 | 112.4 (2) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N1···O2i | 0.81 (2) | 2.12 (2) | 2.927 (2) | 174.0 (19) |
| N2—H1N2···O1Wii | 0.87 (3) | 1.88 (3) | 2.751 (2) | 173 (2) |
| O1W—H1W1···O1 | 0.82 (4) | 1.95 (4) | 2.767 (2) | 173 (3) |
| O1W—H2W2···O1iii | 0.86 (4) | 1.98 (4) | 2.839 (2) | 171 (4) |
Symmetry codes: (i) x−1, y, z; (ii) x+1, y, z; (iii) x−1/2, −y+3/2, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IS5056).
References
- Abdel-Aziz, A. A.-M. (2007). Eur. J. Med. Chem. 42, 614–626. [DOI] [PubMed]
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Carmi, C., Cavazzoni, A., Zuliani, V., Lodola, A., Bordi, F., Plazzi, P. V., Alfieri, R. R., Petronini, P. G. & Mor, M. (2006). Bioorg. Med. Chem. Lett. 16, 4021–4025. [DOI] [PubMed]
- Ciechanowicz-Rutkowska, M., Kieć-Kononowicz, K., Howard, S. T., Lieberman, H. & Hursthouse, M. B. (1994). Acta Cryst. B50, 86–96. [DOI] [PubMed]
- Delgado, G. E., Mora, A. J., Uzcátegui, J., Bahsas, A. & Briceño, A. (2007). Acta Cryst. C63, o448-o450. [DOI] [PubMed]
- El-Deeb, I. M., Bayoumi, S. M., El-Sherbeny, M. A. & Abdel-Aziz, A. A.-M. (2010). Eur. J. Med. Chem. 45, 2516–2530. [DOI] [PubMed]
- Flack, H. D. (1983). Acta Cryst. A39, 876–881.
- Rajic, Z., Zorc, B., Raic-Malic, S., Ester, K., Kralj, M., Pavelic, K., Balzarini, J., De Clercq, E. & Mintas, M. (2006). Molecules, 11, 837–848. [DOI] [PMC free article] [PubMed]
- Sergent, D., Wang, Q., Sasaki, N. A. & Ouazzani, J. (2008). Bioorg. Med. Chem. Lett. 18, 4332–4335. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Yu, F.-L., Schwalbe, C. H. & Watkin, D. J. (2004). Acta Cryst. C60, o714–o717. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536812002838/is5056sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812002838/is5056Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812002838/is5056Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report