

Abstract
Cancer research has been devoted toward an understanding of the molecular regulation and functional significance of cell-cycle regulators in the pathogenesis and development of cancers. Cyclin-dependent Kinase 2-associated Protein 1 (CDK2AP1) is one such cell-cycle regulator, originally identified as a growth suppressor and a prognostic marker for human oral/head and neck cancers. Functional importance and the molecular mechanism of CDK2AP1-mediated cell-cycle regulation have been documented over the years. Recent progress has shown that CDK2AP1 is a competency factor in embryonic stem cell differentiation. Deletion of CDK2AP1 leads to early embryonic lethality, potentially through altered differentiation capability of embryonic stem cells. More intriguingly, CDK2AP1 exerts its effect on stem cell maintenance/differentiation through epigenetic regulation. Cancer cells and stem cells share common cellular characteristics, most prominently in maintaining high proliferative potential through an unconventional cell-cycle regulatory mechanism. Cross-talk between cellular processes and molecular signaling pathways is frequent in any biological system. Currently, it remains largely elusive how cell-cycle regulation is mechanistically linked to epigenetic control. Understanding the molecular mechanism underlying CDK2AP1-mediated cell-cycle regulation and epigenetic control will set an example for establishing a novel and effective molecular link between these two important regulatory mechanisms.
Keywords: Cyclin-dependent kinase 2-associated protein 1 (CDK2AP1), cell cycle, epigenetic regulation, oral/head and neck cancer, embryonic stem cells, cancer stem cells
Introduction
Recent progress in cancer research has unveiled many important cellular processes potentially related to the pathogenesis of cancers. Despite significant advances in understanding the biology of cancer development, we still need to unravel cellular and molecular mechanisms that will be essential to the invention of clinical strategies for the prevention, treatment, and cure of cancers. It is widely accepted that cancer is a disease of cellular differentiation. Two of the major alterations of differentiation are seen phenotypically as uncontrolled proliferation of cells and/or deregulation of programmed cell death (Hanahan and Weinberg, 2011). Altered proliferation and apoptosis have been significantly correlated to the early pathogenesis of many cancers. As predicted, the most prominent feature of cellular/molecular alterations in cancers is related to cell-cycle regulatory machinery. Molecular mechanisms governing these cellular processes have been extensively studied, and the functional consequences of their deregulation have been demonstrated. However, cellular and molecular heterogeneity in cancers makes it difficult to establish a generalized mechanism that can be utilized for understanding cancer development, and in designing therapeutic strategies for a cure.
Recently, the concept of cancer stem cells has begun to emerge (Visvader and Lindeman, 2008). Cancer cells and stem cells share many key biological properties. A cardinal trait possessed by both cell types is the extensive proliferative potential essential for embryogenesis and tumor development. In this review, we will examine the dual regulatory roles of CDK2AP1 in cell-cycle regulation and in epigenetic control, linking these two important cellular and molecular processes in cancer development and stem cell maintenance. The resemblance in molecular mechanisms between these two biological models and the utility of understanding cell-cycle and epigenetic regulation with a stem cell model will be discussed toward advancing our knowledge in cancer research, in view of the genesis of cancer stem cells.
CDK2AP1 in Cancers
The hallmark of cancer is rapid and uncontrolled growth. In head and neck cancers, several key cell-cycle regulatory molecules have been implicated in its pathogenesis. An essential component in cell-cycle transition is the cyclin-CDK complex and Retinoblastoma Protein (RB). Upon RB phosphorylation by the cyclin/CDK complexes on RB, there is a release of E2F, subsequently allowing E2F to transcribe the necessary components for the cell to continue through the G1/S transition. Specifically, RB function is mediated by cyclin E/CDK2 activity through E2F transcriptionally regulating cyclin E. In contrast, CDK4 and CDK6 act upstream of RB, and they inhibit RB function by phosphorylation (Hinds et al., 1992, 1994; Sherr, 1993). Loss of RB function has mainly been implicated in retinoblastomas; however, both down- and up-regulation of RB function has been observed in head and neck cancer, conferring a greater degree of malignancy and aggressiveness, dependent upon cellular context (Pavelic et al., 1996; Regezi et al., 1999). Down-regulation of RB function obviously allows the cell cycle to remain unchecked and leads to continual cell division and cell proliferation; however, up-regulation may also be detrimental, leading to a decrease in pro-apoptotic signals that are triggered during the cell cycle. In either case, changes in the RB pathway alter cell-cycle transition and allow for greater cancer cell survival. Alteration of these cyclins and CDKs has been observed in head and neck cancers as well.
Nevertheless, there are negative regulators of CDK activity that are an integral part of the cell cycle and the RB pathway. These inhibitors belong to either the CIP/KIP family or the INK family, and function by arresting the cell cycle in the G1 phase. Members of the CIP/KIP family include proteins such as p21WAF1, p27KIP-1, and p57, and members of the INK family include proteins such as p16INK4a, p18INK4C, p19INK4d, and p14ARF (Sherr and Roberts, 1999). For example, p21WAF1 functions as a cell-cycle regulator by binding to and inhibiting complex formation of CDK2 and CDK4 with their respective cyclins, thereby down-regulating CDK activity. p21WAF1 as a CDK2 inhibitor specifically disrupts the RB pathway, with subsequent effects on down-regulation of E2F activity, leading to cell-cycle inhibition. However, these molecules may have multiple roles—for example, p21WAF1 also functions as an inhibitor of DNA synthesis by competing for a binding partner to DNA polymerase δ (Abbas and Dutta, 2009). Down-regulation of these inhibitors has been implicated in oral/head and neck cancer, such as reduction of p27KIP-1 or loss of p16INK4a function (Timmermann et al., 1997; Schoelch et al., 1999). However, context may be important for these regulators as well, since p21WAF1 has been shown to have both tumor-suppressive and oncogenic properties in oral cancers (Abbas and Dutta, 2009), the tumor-suppressive effects coming from the obvious inhibition of the cell cycle and tight regulation by p53. Nevertheless, cellular senescence by p21WAF1 can also lead to the inhibition of apoptosis, since an active cell cycle is necessary for the proper apoptotic signals, similar to the up-regulation of RB discussed above (Abbas and Dutta, 2009), thus allowing head and neck cancer (HNC) another mechanism of altering CDK activity and abrogating proper cell-cycle control. Alteration in cell-cycle control is one of the mechanisms involved in promoting a cancer phenotype. As discussed above, cell-cycle alteration can be accomplished by mutating or altering levels of various molecules in this pathway. Recent studies have implicated a new molecule, CDK2AP1, which is now coming to the forefront as a key cell-cycle regulator that is altered in head and neck cancer (Zolochevska and Figueiredo, 2009a,b, 2010, 2011; Zheng et al., 2011).
Human CDK2AP1 is a highly conserved, ubiquitously expressed gene located on chromosome 12q24 and is a 115-aa nuclear polypeptide that is down-regulated in ~70% of oral cancers (Tsuji et al., 1998; Shintani et al., 2001). Murine Cdk2ap1, with only 3 amino acid deviations from the human CDK2AP1, is located at chromosome 5 (Kim et al., 2001, 2005). In vitro studies have unveiled cellular roles of CDK2AP1. It functions as an S-phase regulator through 2 important cellular partners: CDK2 and DNA polymerase-alpha/primase (Matsuo et al., 2000). CDK2AP1 has also been shown to play a role in TGF-β-induced growth arrest, cisplatin-induced genotoxicity, and apoptosis (Kohno et al., 2002; Hu et al., 2004; Kim et al., 2005). CDK2AP1 has been shown to form a homodimer, which is the active form for its growth-suppressing effect (Buajeeb et al., 2004; Kim et al., 2005). We have shown that Cdk2ap1 expression in a xenograft mouse model of head and neck cancer in vivo inhibits tumor growth by reducing proliferation and increasing apoptotic indices (Figueiredo et al., 2005).
Modulation of TGF-β and downstream effector molecules have been shown to alter cell proliferation, survival, and differentiation (Massagué, 2008; Padua and Massagué, 2009). A majority of oral carcinomas exhibit reduced expression of TGF-β receptor II (TGFβRII), and only a few tumors show mutations in the members of the TGF-β pathway (Prime et al., 2004). Numerous studies have shown that most primary oral squamous cell carcinomas retain TGF-β signaling, which diminishes as the disease progresses from carcinoma to metastasis (Meng et al., 2011). During this transition, there is a significant decrease in TGFβRII expression, resulting from down-regulation of gene expression without detectable mutations (Paterson et al., 2001). The significance of CDK2AP1 in TGF-β signaling has been examined in normal and malignant keratinocytes (Fig. 1). TGF-β1 treatment of normal human keratinocytes increased cellular levels of CDK2AP1 mRNA and protein (Hu et al., 2004). Analysis of the data suggests that SMAD induced by TGF-β1 binds at the proximal promoter of the CDK2AP1 gene. A significant correlative expression of TGF-β receptor II (TGFβRII) and CDK2AP1 has been found in human oral squamous cell carcinoma (OSCC) tissues (Peng et al., 2006). There was a loss of expression of CDK2AP1 and p21WAF1 in OSCC. A decrease in TGFβRII expression had a statistically significant correlation with decreased CDK2AP1 and p27KIP-1 expression. Further in vitro studies showed that there is a correlative decrease in TGFβRII, CDK2AP1, SMAD2, and pSMAD2/3 in human OSCC lines. It has also been found that OSCC lines resistant to TGF-β1 failed to induce pSMAD2/3 and expression of CDK2AP1 (Peng et al., 2006). These results provide evidence of a link between the disrupted TGF-β-SMAD signaling pathway and loss of the cell-cycle-inhibitory proteins CDK2AP1, p21WAF1, and p27KIP-1 in OSCC, which may lead to resistance of the TGF-β1 growth-inhibitory effect in OSCC.
Figure 1.
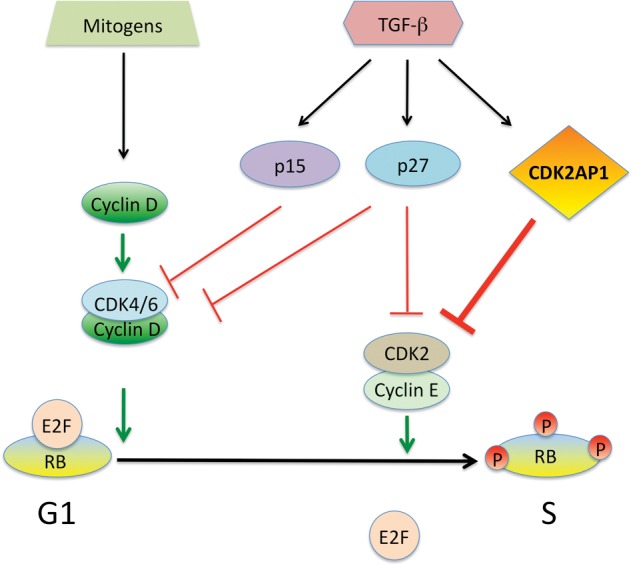
CDK2AP1 in cell-cycle regulation. Studies have shown that TGF-β induces CDK2AP1, and this blocks CDK2 from allowing the cells to pass the G1/S checkpoint. Normally, cyclin D and cyclin E, in conjunction with their respective CDKs, will allow hyperphosphorylation of RB releasing E2F and progression through the cell cycle. CDK2AP1 in this context blocks CDK2’s activities in RB phosphorylation, thereby inhibiting the cell cycle. (Adapted from Hu et al., 2004.)
The importance of CDK2AP1 is also being recognized in cancers outside of the head and neck. In prostate cancer (PCa), CDK2 inhibitors have been associated with a decrease in PCa cell growth. PCa cells naturally decrease CDK2AP1 expression to confer survival. To understand the mechanisms behind this down-regulation, investigators forced PCa cells to overexpress CDK2AP1, which led to cell-cycle arrest, apoptosis, and reduced invasion capacity (Zolochevska and Figueiredo, 2009a). Also, CDK2AP1 enhanced demethylation at the Androgen Receptor (AR) promoter, subsequently leading to an increase in AR expression and enhanced AR transcriptional activity (Zolochevska and Figueiredo, 2009a). Analysis of these data suggests that CDK2AP1 may restore a tumor-suppressive effect of the AR by modulation of specific transcriptional functions involved in the AR pathway. Furthermore, CDK2AP1 was shown to reduce chemotaxis to osteoblasts and reduce osteoblastic proliferation (Zolochevska and Figueiredo, 2011). Also altered were signaling pathways involved in prostate cancer/bone cross-talk, suggesting a role of CDK2AP1 in altering the bone microenvironment in favor of a benign state (Zolochevska and Figueiredo, 2011).
In gastric cancer, CDK2AP1 was negatively correlated with stage and invasiveness (Choi et al., 2009). Patients with CDK2AP1 present in their gastric tissue samples had a better life expectancy, while p53 status made no difference (Choi et al., 2009). In another study, CDK2AP1 was shown to play a role in epithelial-to-mesenchymal transition (EMT), determining both cell fate and differentiation (Tsuji et al., 2008). This was determined in the context of hamster pouch carcinoma cells, where CDK2AP1 induction was growth-inhibitory yet increased EMT. This is contrary to our overall understanding of CDK2AP1 as a tumor suppressor. Therefore, cellular context is important in determination of the role of CDK2AP1 as a tumor suppressor or an oncogene.
The role of CDK2AP1 has been delineated in colon cancer. Most colorectal cancers are characterized by alterations in the length of their DNA repeat sequences, known as microsatellite instability. This instability has been considered to alter growth-regulatory elements, allowing for the progression of a cancerous phenotype. Microsatellite-unstable colorectal cancer lines were studied, and an inverse relationship between microsatellite stability and CDK2AP1 status was found, suggesting that CDK2AP1 helped keep these regions of DNA stable (Yuan et al., 2003). Analysis of the data suggests that CDK2AP1 has a tumor-suppressor function in colon epithelial cells. CDK2AP1 has been shown to be a key molecule in the cell cycle and may function as a growth suppressor in various cancer settings. It will be essential to assess how CDK2AP1 functions in a developmental context to determine how it is involved in cell-cycle regulation and differentiation. It is in this light that we have recently unraveled a dual role of CDK2AP1 functions in stem cells and in epigenetic control.
CDK2AP1 in ESC Differentiation
Embryonic stem cells (ESCs) possess an unlimited potential for self-renewal and the capacity to differentiate into 3 primary germ layers. It is not surprising that ESCs should have a cell-cycle profile that is significantly different from that of somatic cells, since ESCs have a unique biological function of balancing self-renewal and differentiation. Previous studies have revealed that mouse embryonic stem cells (mESCs) possess (1) a distinctive cell-cycle profile, (2) a rapid rate of cell division, (3) a lack of the RB pathway, (4) different cyclin-CDK levels, and (5) elevated cell-cycle-independent CDK activity (Savatier et al., 1994; Stead et al., 2002). The distinct cell-cycle profile of mESCs includes a truncated G1 phase and a relatively longer S phase (Faast et al., 2004). Cell-cycle time of mESCs is approximately 8-11 hrs, which is only half to a third of the cell-cycle time of somatic cells (Stead et al., 2002). The prominent feature of cell-cycle regulation in mESCs is the lack of the RB pathway involvement. Hypo-phosphorylated RB is virtually undetectable during the truncated G1 phase in mESCs (Savatier et al., 1994). Moreover, mESCs show a low level of cyclin D and CDK4-associated kinase activity, but show constitutive cyclin E/CDK2 kinase activity (Savatier et al., 1996). During differentiation, there is a robust expression of D-type cyclins and CDK4-associated kinase activity, along with a reduction of cyclin E/CDK2 kinase activity, resulting in G1/S control by the RB pathway (Savatier et al., 1996). Furthermore, mESCs show that individual cyclins stay activated throughout the cell cycle and do not oscillate, and CDK activity is elevated significantly with the absence of CDK-inhibitory molecules (Savatier et al., 1994; Stead et al., 2002; Faast et al., 2004). From these findings, it is suggested that the cell cycle plays a major role in ESC self-renewal and differentiation.
During the normal cell cycle, CDK2AP1 functions as an inhibitor of G1/S transition through down-regulation of CDK2 (Fig. 1) (Matsuo et al., 2000). mESCs with disrupted expression of Cdk2ap1 showed an altered cell-cycle profile along with increased CDK2 activity, increased proliferation, loss of stem cell differentiation potential, and an altered pRB phosphorylation (Kim et al., 2005). Cdk2ap1 KO mESCs showed no significant change in G1 population (2-3% alteration), but there was a significant increase in the S phase (up to 13.3%, p < 0.0001) and a decrease in the G2/M phase (up to 90%, p < 0.0003) (Kim et al., 2005). Compared with WT cells, Cdk2ap1 KO mESCs showed a two-fold increase in proliferation (Kim et al., 2005). Perhaps more interestingly, Cdk2ap1 KO mESCs resulted in abrogation of leukemia-inhibitory factor (LIF) withdrawal-induced differentiation and showed a dramatic increase in the phosphorylated form of RB (Kim et al., 2009a). Furthermore, teratoma formation analysis showed that tumors from Cdk2ap1 KO mESCs contained only mesoderm lineage tissues and failed to differentiate properly into all 3 lineages (Kim et al., 2009b). Based on these findings, we conclude that CDK2AP1 plays a major role in mESCs differentiation. These findings also explain, in part, the lethality of Cdk2ap1 KO mice during embryonic development.
Epigenetic Control and Cross-talk with the Cell Cycle
Epigenetics has become a growing field in the scientific community in recent years. Several groups are studying how cells alter expression and phenotype without altering the DNA sequence itself but modifying the DNA with methylation or accessibility due to chromatin structure via histone modification (Egger et al., 2004; Jiang et al., 2004). DNA methylation is mainly associated with a down-regulation in gene expression (Tate and Bird, 1993; Newell-Price et al., 2000; Jaenisch and Bird, 2003). Histones that are involved in the overall chromatin structure can be acetylated to allow for a more open structure, or have various degrees of methylation signifying various heterochromatic states and transcriptional status (Bradbury, 1992; Brownell and Allis, 1996). Alteration of these epigenetic marks by various proteins that modify histones as well as alter DNA methylation have been shown to cause cells to both differentiate and de-differentiate, based on cellular context and the types of modifications involved. Acetylation on histones brings a negative charge to histones, which are positively charged (Allfrey et al., 1964). This, in turn, decreases the interaction between the N termini of histones and the negatively charged phosphate groups of DNA. The weakened interaction relaxes the condensed chromatin into a more relaxed structure, which may lead to greater levels of gene transcription (Wolffe and Pruss, 1996). The process of acetylation and deacetylation is catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively. HATs are known to modulate cell-cycle progression through p300/CBP (transcriptional co-activators with the HAT domain) and P300/CBP-associated factor (PCAF) (Nakatani et al., 1996). HDACs have also been implicated in the regulation of the cell cycle (Kouzarides, 1998). However, it is not totally clear how these epigenetic modifying proteins function as a whole, and, depending upon context, the same enzyme can be functioning either in pluripotency or differentiation; one such example is with histone deacetylating enzymes, where inhibition in one context can lead to stem cell renewal, and inhibition in another context can lead to mesenchymal stem cell differentiation (Cho et al., 2005; Dovey et al., 2010). All the mechanisms involved in epigenetic regulation and controlling cell fate are not known, yet there is a growing body of evidence suggesting that cell-cycle regulators may play a partial role in controlling epigenetic status.
RB is well-known as a cell-cycle regulator and tumor suppressor. However, RB has also been shown to interact with several proteins involved with heterochromatin structure, including HP1, Suv39h1, DNMT1, and the SWI/SNF chromatin remodeling complex (Siddiqui et al., 2007). Knocking out RB led to changes in levels of histone modifications as well as heterochromatin protein dynamics. Similarly, change in p53 status has been shown to alter DNA methyltransferase levels. In p53 knockout mice, DNMT levels have been shown to increase in the thymus and in the liver, which, in turn, led to an increased level of global methylation in these organs (Park et al., 2005). However, in specific regions, there was hypomethylation instead of hypermethylation, suggesting that p53 was involved not only in overall DNA methylation changes, but also in maintenance of specific DNA methylation patterns (Park et al., 2005). In another more molecular study, p53 was mutated and unable to suppress DNMT1 expression, leading to an overall increase in methylation and specific hypermethylation of p16INK4A (Guo et al., 2008). This led to a subsequent decrease in p16INK4A gene expression and potential alteration of the cell cycle (Guo et al., 2008). Analysis of these data together suggests that there is a growing link between cell-cycle molecules and epigenetic control, either by direct interaction between epigenetic regulators and cell-cycle regulators, or by cell-cycle regulators altering the expression of epigenetic regulators. Similar to the above-mentioned findings, CDK2AP1 (originally thought to be only a cell-cycle molecule) is now known to be involved in epigenetics, which will be discussed further below.
CDK2AP1 in Epigenetic Regulation
From fertilization to gastrulation, ESCs are challenged by an extensive array of signals which include: (1) gene expression changes at stage-specific embryonic development, (2) secreted small molecules and growth factors, (3) appropriate cellular responses, and (4) communication between cells and the surrounding environment (Koledova et al., 2010). One possible mechanism by which ESCs can process an overwhelming number of different signals is through epigenetics. DNA methylation, genomic imprinting, and bivalent chromatin allow many important tissue-specific genes to be primed for expression but ‘held back’ for an appropriate development time by either DNA methylation and/or histone modification (Spivakov and Fisher, 2007). When a critical time is reached to turn a gene either on or off, DNA methylation regulatory enzymes such as DNMTs and/or nucleosome remodeling complexes such as NuRD can rapidly modify DNA and/or histone to respond to appropriate developmental cues (Tong et al., 1998; Muller and Leutz, 2001; Becker and Horz, 2002). Epigenetic mechanisms are essential for determining stem cell properties by regulating gene expression without altering DNA sequence. As ESCs differentiate, pluripotency-associated factors become silenced, and tissue- and lineage-specific genes become activated. Expression of Oct4, for example, is under epigenetic control. Oct4 expression is down-regulated as mESCs differentiate by increased promoter methylation. An alteration of DNA methylation and histone modification by 5-aza-2-deoxycytidine or trichostatin A (TSA) at the Oct4 promoter lead to failed stem cell differentiation, which suggests that epigenetics plays an intricate role in stem cell regulation (Tsuji-Takayama et al., 2004).
Recent studies linked CDK2AP1 to epigenetic regulation and stem cell biology. CDK2AP1 has been identified as a core subunit of the Nucleosome Remodeling and Histone Deacetylation complex (NuRD) (Fig. 2) associated with Methyl CpG Binding Domain proteins 2 and 3 (MBD2 and MBD3) (Le Guezennec et al., 2006; Spruijt et al., 2010). NuRD-mediated gene-silencing has a crucial function in the lineage commitment of mESCs, since this process allows cells to exit pluripotency (Kaji et al., 2006). It has been reported that mESCs lacking MBD3 (a component of NuRD) display a profound defect in differentiation that results in persistent self-renewal (Kaji et al., 2006), a phenotype that is remarkably similar to Cdk2ap1-nulled mESCs. These reports suggest functional collaboration of MBD3 and CDK2AP1 in the NuRD complex toward regulation of ESC differentiation via epigenetic mechanisms. The epigenetic role of CDK2AP1 in mESCs is further explained in Deshpande et al. (2009). It was found that CDK2AP1 promoted Oct4 promoter methylation during mESC differentiation by physically interacting with MBD3, and that deletion of Cdk2ap1 resulted in retention of Oct4 expression (Fig. 3) (Deshpande et al., 2009). Although further study is needed before we can understand the full extent of CDK2AP1-mediated epigenetic regulation in stem cell self-renewal and differentiation, it is clear that the epigenetic regulatory role of CDK2AP1 may have global genome-wide implications beyond one, yet important, master switch, OCT4. Interestingly, a recent study showed that CDK1 and CDK2 phosphorylated Enhancer of Zeste Homolog 2 (EZH2) that led to a decrease in EZH2 methyltransferase activity, resulting in inhibition of cancer cell invasion (Wei et al., 2011). This suggests that there may be a signaling link between CDKs and EZH2, which epigenetically controls osteogenic differentiation of mesenchymal stem cells (Wei et al., 2011). It will be intriguing to see if CDK2AP1-mediated epigenetic control has any functional correlation to its cell-cycle-regulatory role (Kim et al., 2005).
Figure 2.
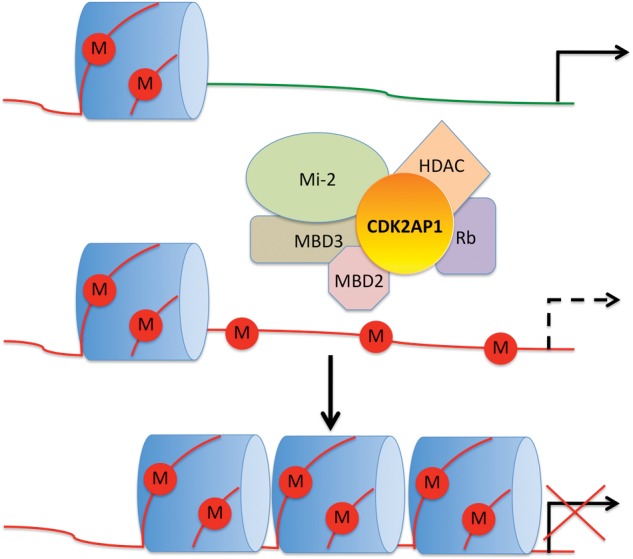
CDK2AP1 in the nucleosome remodeling complex. A hypothetical model can be established describing how CDK2AP1 may be involved in altering transcription globally in the cell. In this model, a target gene is normally transcribed (top); however, this transcription may shut down due to the incorporation of methylation marks on CpG sites (middle). The establishment of these marks is uncertain, but may involve the function of various DNMTs. CDK2AP1’s interaction with members of the nucleosome remodeling complex (NuRD), including MBD2 and 3, HDAC1 and 2, and RB, may alter the chromatin landscape (middle and bottom), subsequently leading to a closed chromatin structure and down-regulation of transcription (bottom).
Figure 3.

Schematic for the role of CDK2AP1 as the epigenetic regulator of Oct4. A hypothetical model can be established where CDK2AP1 is a core component of the NuRD complex, and it has the ability to down-regulate Oct4 expression by binding to methylated CpG islands (filled circle = methylated CpG site; open circle = unmethylated CpG site) during mESC differentiation. In undifferentiated mESCs, the NuRD complex is in its inactive state, the promoter of Oct4 is hypomethylated, and Oct4 expression remains high (left). Upon LIF removal, mESCs differentiate into embryoid bodies (EBs), leading to Oct4 promoter hypermethylation, an active NuRD complex, and Oct4 down-regulation (right). The role of CDK2AP1 as an epigenetic regulator may not be limited to Oct4, but could play a global role in both cancer signaling pathways and the pluripotency of stem cells. (Adapted from Deshpande et al., 2009.)
Summary
In this review, we have examined several lines of evidence demonstrating the significance of cell-cycle regulatory mechanisms in cancer development as well as in stem cell maintenance and differentiation. It is generally considered that cancer cells are quite different from stem cells, since they are generated from fully differentiated cells. However, two important cellular and molecular similarities between cancer cells and stem cells provide scientific justification for how studying stem cell models can be beneficial in understanding cancer biology. First, cancer cells share many molecular pathways that are important in the maintenance and differentiation of stem cells. Second, evidence supports that genesis of cancer cells may involve a de-differentiation process, which eventually causes terminally differentiated cellular phenotypes to revert to a stem-cell-like state. This involves reactivation or inactivation of key molecular and cellular pathways that resemble those in stem cells. In addition, the emerging concept of cancer stem cells provides us with sufficient justification to use stem cells as a proper experimental model to understand the mechanism of the genesis and development of cancers. As reviewed in this article, CDK2AP1 is involved in both cancer development and stem cell fate. Moreover, CDK2AP1 is emerging as one of the key molecules involved in the interplay between cell-cycle regulation and epigenetic regulation. CDK2AP1 may potentially be regulating molecular networks, rendering mutual intra-regulatory influences. It is intriguing that a given cell-cycle regulator, such as CDK2AP1, has a dual role in cell-cycle and epigenetic control.
Footnotes
This work was supported by grants from NIH-NIDCR (R01 DE014857 to D.T.W.W.; F30 DE020003 to J.J.K.; T32 DE07269 to O.K.) and CIRM Basic Biology Award II (RB2-01562 to Y.K.).
The authors declare no potential conflicts of interests with respect to the authorship and/or publication of this article.
References
- Abbas T, Dutta A. (2009). p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9:400-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey VG, Faulkner R, Mirsky AE. (1964). Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci USA 51:786-794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker PB, Horz W. (2002). ATP-dependent nucleosome remodeling. Annu Rev Biochem 71:247-273 [DOI] [PubMed] [Google Scholar]
- Bradbury EM. (1992). Reversible histone modifications and the chromosome cell cycle. Bioessays 14:9-16 [DOI] [PubMed] [Google Scholar]
- Brownell JE, Allis CD. (1996). Special HATs for special occasions: linking histone acetylation to chromatin assembly and gene activation. Curr Opin Genet Dev 6:176-184 [DOI] [PubMed] [Google Scholar]
- Buajeeb W, Zhang X, Ohyama H, Han D, Surarit R, Kim Y, et al. (2004). Interaction of the CDK2-associated protein-1, p12(DOC-1/CDK2AP1), with its homolog, p14(DOC-1R). Biochem Biophys Res Commun 315:998-1003 [DOI] [PubMed] [Google Scholar]
- Cho HH, Park HT, Kim YJ, Bae YC, Suh KT, Jung JS. (2005). Induction of osteogenic differentiation of human mesenchymal stem cells by histone deacetylase inhibitors. J Cell Biochem 96:533-542 [DOI] [PubMed] [Google Scholar]
- Choi MG, Sohn TS, Park SB, Paik YH, Noh JH, Kim KM, et al. (2009). Decreased expression of p12 is associated with more advanced tumor invasion in human gastric cancer tissues. Eur Surg Res 42:223-229 [DOI] [PubMed] [Google Scholar]
- Deshpande AM, Dai YS, Kim Y, Kim J, Kimlin L, Gao K, et al. (2009). Cdk2ap1 is required for epigenetic silencing of Oct4 during murine embryonic stem cell differentiation. J Biol Chem 284:6043-6047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovey OM, Foster CT, Cowley SM. (2010). Histone deacetylase 1 (HDAC1), but not HDAC2, controls embryonic stem cell differentiation. Proc Natl Acad Sci USA 107:8242-8247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger G, Liang G, Aparicio A, Jones PA. (2004). Epigenetics in human disease and prospects for epigenetic therapy. Nature 429:457-463 [DOI] [PubMed] [Google Scholar]
- Faast R, White J, Cartwright P, Crocker L, Sarcevic B, Dalton S. (2004). Cdk6-cyclin D3 activity in murine ES cells is resistant to inhibition by p16(INK4a). Oncogene 23:491-502 [DOI] [PubMed] [Google Scholar]
- Figueiredo ML, Kim Y, St John MA, Wong DT. (2005). p12CDK2-AP1 gene therapy strategy inhibits tumor growth in an in vivo mouse model of head and neck cancer. Clin Cancer Res 11:3939-3948 [DOI] [PubMed] [Google Scholar]
- Guo Z, Tsai MH, Shiao YH, Chen LH, Wei ML, Lv X, et al. (2008). DNA (cytosine-5)-methyltransferase 1 as a mediator of mutant p53-determined p16(ink4A) down-regulation. J Biomed Sci 15:163-168 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. (2011). Hallmarks of cancer: the next generation. Cell 144:646-674 [DOI] [PubMed] [Google Scholar]
- Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. (1992). Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell 70:993-1006 [DOI] [PubMed] [Google Scholar]
- Hinds PW, Dowdy SF, Eaton EN, Arnold A, Weinberg RA. (1994). Function of a human cyclin gene as an oncogene. Proc Natl Acad Sci USA 91:709-713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MG, Hu GF, Kim Y, Tsuji T, McBride J, Hinds P, et al. (2004). Role of p12(CDK2-AP1) in transforming growth factor-beta1-mediated growth suppression. Cancer Res 64:490-499 [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33(Suppl):245S-254S [DOI] [PubMed] [Google Scholar]
- Jiang YH, Bressler J, Beaudet AL. (2004). Epigenetics and human disease. Annu Rev Genomics Hum Genet 5:479-510 [DOI] [PubMed] [Google Scholar]
- Kaji K, Caballero IM, MacLeod R, Nichols J, Wilson VA, Hendrich B. (2006). The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat Cell Biol 8:285-292 [DOI] [PubMed] [Google Scholar]
- Kim Y, Tsuji T, Elovic A, Shintani S, Mihara M, Salih E, et al. (2001). Murine doc-1 cDNA cloning, sequencing and expression in normal adult tissues. Int J Oral Biol 2001:87-91 [Google Scholar]
- Kim Y, Ohyama H, Patel V, Figueiredo M, Wong DT. (2005). Mutation of Cys105 inhibits dimerization of p12CDK2-AP1 and its growth suppressor effect. J Biol Chem 280:23273-23279 [DOI] [PubMed] [Google Scholar]
- Kim Y, Deshpande A, Dai Y, Kim JJ, Lindgren A, Conway A, et al. (2009a). Cyclin-dependent kinase 2-associating protein 1 commits murine embryonic stem cell differentiation through retinoblastoma protein regulation. J Biol Chem 284:23405-23414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, McBride J, Kimlin L, Pae EK, Deshpande A, Wong DT. (2009b). Targeted inactivation of p12, CDK2 associating protein 1, leads to early embryonic lethality. PLoS One 4:e4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno Y, Patel V, Kim Y, Tsuji T, Chin BR, Sun M, et al. (2002). Apoptosis, proliferation and p12(doc-1) profiles in normal, dysplastic and malignant squamous epithelium of the Syrian hamster cheek pouch model. Oral Oncol 38:274-280 [DOI] [PubMed] [Google Scholar]
- Koledova Z, Kramer A, Kafkova LR, Divoky V. (2010). Cell-cycle regulation in embryonic stem cells: centrosomal decisions on self-renewal. Stem Cells Dev 19:1663-1678 [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (1998). Transcription factors and cell differentiation. 20th Annual Symposium, Burnham Institute, La Jolla, CA, USA, 22 May 1998 Biochim Biophys Acta 1378:R59-R61 [DOI] [PubMed] [Google Scholar]
- Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E, et al. (2006). MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol 26:843-851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J. (2008). TGFbeta in cancer. Cell 134:215-230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo K, Shintani S, Tsuji T, Nagata E, Lerman M, McBride J, et al. (2000). p12(DOC-1), a growth suppressor, associates with DNA polymerase alpha/primase. FASEB J 14:1318-1324 [DOI] [PubMed] [Google Scholar]
- Meng W, Xia Q, Wu L, Chen S, He X, Zhang L, et al. (2011). Downregulation of TGF-beta receptor types II and III in oral squamous cell carcinoma and oral carcinoma-associated fibroblasts. BMC Cancer 11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller C, Leutz A. (2001). Chromatin remodeling in development and differentiation. Curr Opin Genet Dev 11:167-174 [DOI] [PubMed] [Google Scholar]
- Nakatani Y, Bagby S, Ikura M. (1996). The histone folds in transcription factor TFIID. J Biol Chem 271:6575-6578 [DOI] [PubMed] [Google Scholar]
- Newell-Price J, Clark AJ, King P. (2000). DNA methylation and silencing of gene expression. Trends Endocrinol Metab 11:142-148 [DOI] [PubMed] [Google Scholar]
- Padua D, Massagué J. (2009). Roles of TGFbeta in metastasis. Cell Res 19:89-102 [DOI] [PubMed] [Google Scholar]
- Park IY, Sohn BH, Choo JH, Joe CO, Seong JK, Lee YI, et al. (2005). Deregulation of DNA methyltransferases and loss of parental methylation at the insulin-like growth factor II (Igf2)/H19 loci in p53 knockout mice prior to tumor development. J Cell Biochem 94:585-596 [DOI] [PubMed] [Google Scholar]
- Paterson IC, Matthews JB, Huntley S, Robinson CM, Fahey M, Parkinson EK, et al. (2001). Decreased expression of TGF-beta cell surface receptors during progression of human oral squamous cell carcinoma. J Pathol 193:458-467 [DOI] [PubMed] [Google Scholar]
- Pavelic ZP, Lasmar M, Pavelic L, Sorensen C, Stambrook PJ, Zimmermann N, et al. (1996). Absence of retinoblastoma gene product in human primary oral cavity carcinomas. Eur J Cancer B Oral Oncol 32(B):347-351 [DOI] [PubMed] [Google Scholar]
- Peng H, Shintani S, Kim Y, Wong DT. (2006). Loss of p12CDK2-AP1 expression in human oral squamous cell carcinoma with disrupted transforming growth factor-beta-Smad signaling pathway. Neoplasia 8:1028-1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prime SS, Davies M, Pring M, Paterson IC. (2004). The role of TGF-beta in epithelial malignancy and its relevance to the pathogenesis of oral cancer (part II). Crit Rev Oral Biol Med 15:337-347 [DOI] [PubMed] [Google Scholar]
- Regezi JA, Dekker NP, McMillan A, Ramirez-Amador V, Meneses-Garcia A, Ruiz-Godoy Rivera LM, et al. (1999). p53, p21, Rb, and MDM2 proteins in tongue carcinoma from patients < 35 versus > 75 years. Oral Oncol 35:379-383 [DOI] [PubMed] [Google Scholar]
- Savatier P, Huang S, Szekely L, Wiman KG, Samarut J. (1994). Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene 9:809-818 [PubMed] [Google Scholar]
- Savatier P, Lapillonne H, van Grunsven LA, Rudkin BB, Samarut J. (1996). Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. Oncogene 12:309-322 [PubMed] [Google Scholar]
- Schoelch ML, Regezi JA, Dekker NP, Ng IO, McMillan A, Ziober BL, et al. (1999). Cell cycle proteins and the development of oral squamous cell carcinoma. Oral Oncol 35:333-342 [DOI] [PubMed] [Google Scholar]
- Sherr CJ. (1993). Mammalian G1 cyclins. Cell 73:1059-1065 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. (1999). CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13:1501-1512 [DOI] [PubMed] [Google Scholar]
- Shintani S, Mihara M, Terakado N, Nakahara Y, Matsumura T, Kohno Y, et al. (2001). Reduction of p12DOC-1 expression is a negative prognostic indicator in patients with surgically resected oral squamous cell carcinoma. Clin Cancer Res 7:2776-2782 [PubMed] [Google Scholar]
- Siddiqui H, Fox SR, Gunawardena RW, Knudsen ES. (2007). Loss of RB compromises specific heterochromatin modifications and modulates HP1alpha dynamics. J Cell Physiol 211:131-137 [DOI] [PubMed] [Google Scholar]
- Spivakov M, Fisher AG. (2007). Epigenetic signatures of stem-cell identity. Nat Rev Genet 8:263-271 [DOI] [PubMed] [Google Scholar]
- Spruijt CG, Bartels SJ, Brinkman AB, Tjeertes JV, Poser I, Stunnenberg HG, et al. (2010). CDK2AP1/DOC-1 is a bona fide subunit of the Mi-2/NuRD complex. Mol Biosyst 6:1700-1706 [DOI] [PubMed] [Google Scholar]
- Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, et al. (2002). Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene 21:8320-8333 [DOI] [PubMed] [Google Scholar]
- Tate PH, Bird AP. (1993). Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev 3:226-231 [DOI] [PubMed] [Google Scholar]
- Timmermann S, Hinds PW, Munger K. (1997). Elevated activity of cyclin-dependent kinase 6 in human squamous cell carcinoma lines. Cell Growth Differ 8:361-370 [PubMed] [Google Scholar]
- Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. (1998). Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 395:917-921 [DOI] [PubMed] [Google Scholar]
- Tsuji T, Duh FM, Latif F, Popescu NC, Zimonjic DB, McBride J, et al. (1998). Cloning, mapping, expression, function, and mutation analyses of the human ortholog of the hamster putative tumor suppressor gene Doc-1. J Biol Chem 273:6704-6709 [DOI] [PubMed] [Google Scholar]
- Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A, et al. (2008). Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res 68:10377-10386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji-Takayama K, Inoue T, Ijiri Y, Otani T, Motoda R, Nakamura S, et al. (2004). Demethylating agent, 5-azacytidine, reverses differentiation of embryonic stem cells. Biochem Biophys Res Commun 323:86-90 [DOI] [PubMed] [Google Scholar]
- Visvader JE, Lindeman GJ. (2008). Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8:755-768 [DOI] [PubMed] [Google Scholar]
- Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, et al. (2011). CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol 13:87-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolffe AP, Pruss D. (1996). Targeting chromatin disruption: transcription regulators that acetylate histones. Cell 84:817-819 [DOI] [PubMed] [Google Scholar]
- Yuan Z, Sotsky Kent T, Weber TK. (2003). Differential expression of DOC-1 in microsatellite-unstable human colorectal cancer. Oncogene 22:6304-6310 [DOI] [PubMed] [Google Scholar]
- Zheng J, Xue H, Wang T, Jiang Y, Liu B, Li J, et al. (2011). miR-21 downregulates the tumor suppressor P12 CDK2AP1 and stimulates cell proliferation and invasion. J Cell Biochem 112:872-880 [DOI] [PubMed] [Google Scholar]
- Zolochevska O, Figueiredo ML. (2009a). Cell cycle regulator cdk2ap1 inhibits prostate cancer cell growth and modifies androgen-responsive pathway function. Prostate 69:1586-1597 [DOI] [PubMed] [Google Scholar]
- Zolochevska O, Figueiredo ML. (2009b). Expression of cell cycle regulator cdk2ap1 suppresses tumor cell phenotype by non-cell-autonomous mechanisms. Oral Oncol 45(9):e106-e112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolochevska O, Figueiredo ML. (2010). Novel tumor growth inhibition mechanism by cell cycle regulator cdk2ap1 involves antiangiogenesis modulation. Microvasc Res 80:324-331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolochevska O, Figueiredo ML. (2011). Cell-cycle regulators cdk2ap1 and bicalutamide suppress malignant biological interactions between prostate cancer and bone cells. Prostate 71:353-367 [DOI] [PubMed] [Google Scholar]