

Abstract
The mechanisms underlying the effects of long-term deep brain stimulation of the subthalamic nucleus (STN DBS) as a therapy for Parkinson’s disease (PD) remain poorly understood. The present study examined whether functionally effective, long-term STN DBS modulates glial cell line-derived neurotrophic factor (GDNF) and/or brain-derived neurotrophic factor (BDNF) in both unlesioned and unilateral 6-hydroxydopamine lesioned rats. Lesioned rats that received two weeks of continuous unilateral STN DBS exhibited significant improvements in parkinsonian motor behaviors in tests of forelimb akinesia and rearing activity. Unilateral STN DBS did not increase GDNF in the nigrostriatal system, primary motor cortex (M1), or hippocampus of unlesioned rats. In contrast, unilateral STN DBS increased BDNF protein 2–3 fold bilaterally in the nigrostriatal system with the location (substantia nigra vs. striatum) dependent upon lesion status. Further, BDNF protein was bilaterally increased in M1 cortex by as much as 2 fold regardless of lesion status. STN DBS did not impact cortical regions that receive less input from the STN. STN DBS also was associated with bilateral increases in BDNF mRNA in the substantia nigra (SN) and internal globus pallidus (GPi). The increase observed in GPi was completely blocked by pretreatment with 5-Methyl-10,11-dihydro-5 H-dibenzo[a,d]cyclohepten-5,10-imine (MK-801), suggesting that the activation of N-methyl-D-aspartate (NMDA) receptors was involved in this phenomenon. The upregulation of BDNF associated with long term STN DBS suggest that this therapy may exert pronounced and underappreciated effects on plasticity in the basal ganglia circuitry that may play a role in the symptomatic effects of this therapy as well as support the neuroprotective effect of stimulation documented in this rat model.
Keywords: Deep brain stimulation, Parkinson’s disease, basal ganglia, trophic factors
INTRODUCTION
Over 40,000 patients have received high frequency electrical stimulation of the subthalamic nucleus (STN DBS) to treat the motor symptoms of Parkinson’s disease (PD) as of 2007 [1]. Pre-clinical studies demonstrate that STN DBS can provide neuroprotection for the dopaminergic neurons of the substantia nigra (SN) in both the 6-hydroxydopamine (6-OHDA) rodent model and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) non-human primate model of PD [2–6]. It also has been shown that electrical stimulation of the dopaminergic neurons of the ventral tegmental area (VTA) increases brain-derived neurotrophic factor (BDNF) mRNA and protein in its primary target area, the prefrontal cortex [7]. GDNF and BDNF are known to be potent trophic factors that are important for the survival of the dopaminergic neurons in the SN [8]. However, to date no investigation of the effect of STN DBS on trophic factors within the basal ganglia and STN target structures has been conducted.
The STN serves as an important modulating influence within the basal ganglia circuitry, providing glutamatergic projections to the cortex, globus pal-lidus externa (GPe) (globus pallidus [GP] in rats), globus pallidus interna (GPi or entopeduncular nucleus [EP] in rats), substantia nigra pars reticulate and pars compacta (SNpr and SNpc), and striatum [9–14]. Evidence that STN DBS has the potential to impact the trophic environment, particularly BDNF levels, comes from our understanding of the impact of stimulation on hippocampal neurons and the ability of glutamate and γ-aminobutyric acid (GABA) release to upregulate BDNF. High-frequency stimulation of hippocampal glutamatergic synapses induces the presynaptic and postsynaptic release of BDNF [15–17], and in the basal ganglia, increased glutamatergic transmission can increase BDNF mRNA in the SN [18]. Additionally, STN DBS has been shown to increase GABA levels in STN target structures [19–23], and pharmacological activation of hippocampal GABA(B) receptors induces BDNF release [24]. Collectively, these findings suggest that STN DBS has the potential to alter BDNF protein and mRNA levels in STN target structures.
We hypothesized that STN DBS would increase BDNF, but not GDNF, in STN target structures. We examined this issue using our recently described rat model of STN DBS [6, 25]. In the current study we initially investigated the functional impact of long term STN DBS on parkinsonian motor behaviors in the cylinder task. Next, we examined the effects of STN DBS on GDNF protein in the nigrostriatal system, M1 cortex, and hippocampus of unlesioned rats. We subsequently measured BDNF protein levels within the STN and its target structures in both intact and 6-hydroxydopamine (6-OHDA) lesioned rats. We then measured BDNF mRNA and protein levels within the M1 cortex, GPe, GPi, SN, hippocampus and the STN itself. Finally, to assess whether increases in BDNF gene expression associated with STN DBS may be linked to glutamate transmission we examined the effects of NMDA receptor blockade using MK-801.
MATERIALS AND METHODS
Animals
A total of 109 male, Sprague Dawley rats (Harlan, 200–250 g) received unilateral STN stimulators with or without unilateral intrastriatal 6-OHDA. Twenty-eight rats were ultimately excluded from the final analyses due to either lesion failure (<20% reduction in contralateral forepaw use), failure to complete the entire 2-weeks stimulation interval (cable connection malfunction), or inappropriate stimulator placement outside the STN upon postmortem analysis. After exclusions, there remained 50 animals with unilateral STN stimulator implantation and no 6-OHDA, 31 animals with unilateral intrastriatal 6-hydroxydopamine (6-OHDA) injections in addition to STN stimulator implantation, and 4 additional animals that served as naïve controls for BDNF mRNA levels. All animals were given food and water ad libitum, and housed in reverse light-dark cycle conditions in the University of Cincinnati Vontz Center vivarium, which is fully AAALAC approved.
Experimental overview
STN DBS in unlesioned rats
All rats (n = 50) were implanted unilaterally with stimulating electrodes into the STN. Rats were allowed to recover from surgery for 2 weeks. Rats were then randomly divided into ACTIVE (n = 22) or INACTIVE (n = 28) stimulation groups. Rats in the ACTIVE group had their stimulators connected to an external stimulation source and received STN stimulation continuously for two weeks. Rats in the INACTIVE group received no stimulation during the identical two-week interval. One subgroup of animals was implanted with osmotic pumps in the interscapular space (Model 2002, Azlet Osmotic Pumps, Cupertino, CA) 10 days post-electrode implantation to allow for the continuous subcutaneous delivery of MK-801 or saline (ACTIVE MK-801 n = 3, ACTIVE saline n = 2, INACTIVE MK-801 n = 6, INACTIVE saline n = 4), These rats were then allowed to recover for 4 more days before beginning stimulation (2 weeks post electrode implantation). At the end of the two weeks of stimulation rats were sacrificed within 1–6 hours of the cessation of stimulation. Brains were processed for ELISA and/or qPCR analysis.
STN DBS in 6-OHDA lesioned rats
All rats (n = 31) were assessed for forelimb akinesia and rearing activity utilizing the cylinder task prior to 6-OHDA lesion surgery. The following day rats were unilaterally lesioned via injections of 6-OHDA into the striatum and implanted unilaterally (ipsilateral to 6-OHDA) with stimulating electrodes into the STN during the same surgical session. Following a two-week recovery period, all rats were reassessed for the degree of contralateral forelimb akinesia and rearing via the cylinder task and were divided into ACTIVE (n = 20) or INACTIVE (n = 11) stimulation groups. Rats in the ACTIVE group had their stimulators connected to an external stimulation source and received STN stimulation continuously for two weeks. Rats in the INACTIVE group received no stimulation during the two-week interval. At the end of the two weeks a subset of rats were reassessed for forelimb akinesia (ACTIVE n = 8, INACTIVE n = 7) and rearing activity (ACTIVE n = 12, INACTIVE n = 5) via the cylinder task both on and after 24 hours off of stimulation. All lesioned rats were sacrificed 1–24 hours after the cessation of stimulation and brains processed for ELISA analysis.
Intrastriatal 6-OHDA injections
Rats were unilaterally injected with 6-OHDA as described previously [6]. Briefly, rats were injected in two sites in the striatum (AP +1.6 mm, ML +2.4 mm, DV −4.2 mm and AP −0.2 mm, ML +2.6 mm, DV −7.0 mm relative to bregma) with 6-OHDA (MP Biomedicals, Solon, OH; 5 μg/μl 6-OHDA in 0.02% ascorbic acid, 0.9% saline solution, injection rate 0.5 μl/minute, 2 μl per site).
Electrode implantation
All rats were implanted unilaterally with stimulating electrodes. Electrode implantation occurred immediately following intrastriatal 6-OHDA injection in rats in the STN DBS in 6-OHDA Lesioned Rats experiment. A bipolar concentric microelectrode (Plastics One, Roanoke VA) was lowered to the dorsal border of the STN (AP −3.4, L +2.5, and DV −7.7 from bregma) as identified by previous experiments in which extracellular-guided recording was utilized [6]. The electrode was then fixed in place using dental acrylic and bone screws. The dorsal border of the STN was chosen as the implantation location in order to minimize any damage to the STN resulting from electrode implantation. Post-operative pain relief was provided via subcutaneous injections of buprenorphine (0.05 mL/100 g BW).
Osmotic pump implantation and MK-801 delivery
Osmotic pumps (Model 2002, Alzet Osmotic Pumps, Cupertino, CA) filled with either 7.5 mg/ml (+)-MK-801 hydrogen maleate (MK-801) (Sigma, St. Louis, MO) in 0.9% saline or 0.9% saline were implanted subcutaneously to the interscapular space in a subset of anesthetized, unlesioned animals. This concentration and osmotic pump configuration allowed for the continuous subcutaneous delivery of a total of 0.3 mg/kg/day MK-801 for the entire two-week stimulation interval. This osmotic pump concentration was chosen based on previous reports [26]. Pumps were examined at the time of sacrifice to confirm that the contents had been delivered.
Cylinder testing
Non-drugged, spontaneous use of the forepaws and rearing activity was measured in rats as originally described by Schallert [27] and using standard procedures in our laboratory [6]. Briefly, during the dark cycle rats were placed in a clear Plexiglas cylinder and videotaped for 5 minutes or 20-weight bearing paw placements on the side of the cylinder, whichever came first. Videotapes were analyzed by a rater blinded to treatment. The number of times the rat reared (both forepaws raised off the platform) and used its left, right, or both paws for weight bearing on the side of the cylinder was determined and recorded. Data for forepaw use is reported as the percentage of contralateral (to 6-OHDA injection) forelimb use: [(contralateral+1/2 both) / (ipsilateral+contralateral+both)] × 100 as described previously [6, 27]. Data for rearing activity is reported as the total number of rears/minute. A 20% reduction in contralateral forelimb use two weeks after 6-OHDA injection (ACTIVE n = 8, INACTIVE n = 7) was utilized to confirm striatal lesion prior to enrollment in either the ACTIVE stimulation or INACTIVE stimulation groups. Animals were included in the rearing analysis if they exhibited a 20% reduction in rearing two weeks after 6-OHDA (ACTIVE n = 12, INACTIVE n = 5).
Long-term stimulation
Rats assigned to the ACTIVE group were stimulated in a freely moving set-up as described previously [6]. The stimulus signal was supplied by an Accupulser Signal Generator (World Precision Instruments, Sarasota, FL) via a battery-powered Constant Current Bipolar Stimulus Isolator (World Precision Instruments, Sarasota, FL). Rats were stimulated at frequency of 130 Hz, 60 μs pulse width, and an intensity of 30–50 μA, which was set below the threshold of contralateral forepaw and orofacial dyskinesia, thereby preventing problems with feeding or locomotion during the stimulus interval. Stimulation commenced two-weeks following 6-OHDA/electrode implantation surgery and was active twenty-four hours/day for a two-week period. Animals with INACTIVE electrodes were housed individually in standard shoebox cages for the identical two-week interval.
Sacrifice
After cessation of stimulation rats were deeply anesthetized (60 mg/kg, pentobarbital, i.p.) and perfused intracardially with heparinized saline at 37°C followed by ice-cold saline in order to remove endogenous circulating growth factors from blood vessels [28, 29]. The brains were immediately removed, flash frozen in 3-methyl butane, and stored at −80°C. This same perfusion and storage procedure was also used for the naïve rats.
Microdissections
Brains were placed in a −20°C cryostat for approximately one hour. The brains were then sliced into 1–2 mm slabs using chilled (−20°C) single edge razor blades and an aluminum brain blocker. The brain slabs were dissected on a cold plate set at −12°C (ThermoElectric Cooling America Corp, Chicago, IL). All microdissection instruments and the cold plate were wiped down with RNase Away (Invitrogen, Carlsbad, CA) to prevent contamination from RNase and DNA. The rat equivalent of the GPi (entopeduncular nucleus), GPe, STN, SN, hippocampus, M1 cortex, M2 cortex, and parietal cortex were dissected out of the frozen brain slabs using either irridectomy scissors or a small tissue punch. The cortical regions were delineated as per Degos et al. [9], and small tissue punches were taken from the middle of each cortical region to ensure precision. Slabs containing the STN were examined for visual verification of electrode placement prior to dissection of this nucleus. For animals in which the STN was not used for ELISA or PCR, this STN slab was then immediately immersed in 10% phosphate buffered formalin for subsequent electrode placement analysis. Each dissected brain structure was placed in a separate pre-frozen RNase free microcentrifuge tube and immediately placed back on dry ice. All samples were stored at −80°C until time of assay.
STN brain slab processing
The microdissected coronal slab containing the STN from each rat was analyzed for appropriate stimulator placement. STN slabs were stored in 10% phosphate buffered formalin for a minimum of 1 week and then transferred to 30% sucrose in 0.1 M PO4 buffer. For sectioning, slabs were frozen on dry ice and sectioned at 80 μm thickness using a sliding microtome. Every section throughout the STN was stained using the Kluver-Barrera technique according to previously described methods [63]. Briefly, mounted sections were washed in xylene then ETOH and then stained with Luxol Fast Blue (0.1 g Solvent Blue 38, Sigma, St. Louis, MO, 95% ETOH, and 10% Glacial Acetic Acid) for 15 minutes. The sections were then washed again with ETOH and distilled water and then differentiated in 0.05% Lithium Carbonate, ethanol, and water. Sections were then stained with Cresyl Violet (0.1%, Harleco, Lawrence, KS) for 5 minutes. Sections were then dehydrated to xylene and coverslipped with Cytoseal. Stimulating electrode placement was considered appropriately targeted to the STN if the tip of the stimulating electrode was observed to be within or at the dorsal border of the STN within any of the sections examined (see Fig. 1). Rats in which electrode placement was determined to be outside of the STN were excluded from analysis. Our laboratory has previously documented that neither electrode implantation nor stimulation under identical conditions impacts STN cell survival [6].
Fig. 1.
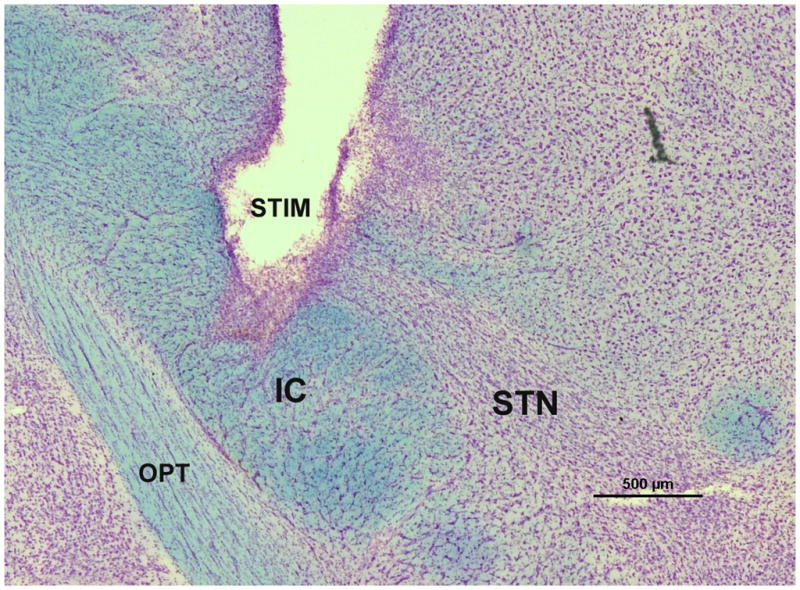
STN stimulator placement. A representative micrograph showing stimulating electrode placement in a coronal section through the rat STN stained via the Kluver-Barrera method. The luxol fast blue component of Kluver-Barrera stains the fiber tracts and the cresyl violet component stains the cell bodies. Therefore, this staining makes the dense cellular swath of the STN immediately dorsal to the internal capsule easily identifiable. Electrode placement was considered to be appropriately targeted to the STN if the tip was within or at the border of the STN within any of the sections examined. STIM = stimulator, IC = internal capsule, OPT = optic tract, STN = subthalamic nucleus. Scale bar = 500μm.
Protein assay
In order to determine total protein levels, tissue was homogenized in T-PER (Pierce, Rockford, IL) using a 300 V/T Ultrasonic Homogenizer (BioLogics, Manassas, VA) and a portion of each sample for ELISA was added to 2% SDS solution and then added to a BD Falcon 96-well Microtest plate (Fisher, Morris Plains, NJ) along with a BSA standard curve (Pierce, Rockford, IL). CuSO4 (4%) was then added to the samples and the plate was incubated at 37°C for 20 minutes and then read at 590 nm on a spectrophotometer.
ELISA
The ELISA reaction was completed (in triplicate) using Immunolon 4 HBX or Costar 96 well plates (Fisher, Morris Plains, NJ) according to the ELISA manufacturer’s instructions (GDNF or BDNF Emax ImmunoAssay Systems Kit, Promega, Madison, WI). The samples were then read at 450 nm within 30 minutes using a spectrophotometer with unknown values determined through interpolation against the GDNF or BDNF standard curve. Each structure was run individually with both ACTIVE and INACTIVE animals on the same plate. Results are expressed as GDNF or BDNF pg/mg of protein. Data was normalized to the percent of INACTIVE rats control. The control for the ACTIVE left hemisphere (stimulator and 6-OHDA hemisphere) was the mean GDNF or BDNF pg/mg protein value of the INACTIVE left hemisphere and the control for the ACTIVE right hemisphere was the mean GDNF or BDNF pg/mg protein value of the INACTIVE right hemisphere.
RNA isolation
RNA was isolated using the QIAshredder (Qiagen, Valencia, CA) and RNeasy Plus Mini kit (Qiagen, Valencia, CA). The Qiagen protocol for purification of total RNA from animal tissue was used with the addition of an extra wash with 500 μl buffer RPE to remove residual salts.
BDNF qPCR
RNA from the tissue was then converted to cDNA via a two-step process using random hexamers and MultiScribe Reverse Transcriptase (ABI, Carlsbad, CA). The RNA was assumed to be converted 100% to cDNA. The PCR reactions were carried out with 1X TaqMan Universal MasterMix (ABI, Carlsbad, CA), and the TaqMan gene expression assay kit for BDNF (Rn02531967_s1, ABI, Carlsbad, CA). This kit includes 900 nM of each manufactured primer, and 900 nM of a BDNF MGB-florescent probe. A total of 20 ng of cDNA was added to each 50 μl reaction mixture. Additional reactions containing RNA only (no cDNA) were amplified to ensure no genomic contamination. qPCR reactions to control for cDNA quantities were run using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an endogenous control (Rn99999916_s1, ABI, Carlsbad, CA). The qPCR reactions were run on an ABI 7500 real-time thermo-cycler using the following method: Step 1: incubation at 50°C for 2 min. Step 2: incubation at 95°C for 10 min. Step 3: denaturation at 95°C for 15 s followed by annealing-elongation at 60°C for 1 min followed by data collection. Step 3 was cycled 40 times for the qPCR run. Cycle thresholds were chosen during the linear phase of amplification using the AutoCT function. Structures were run individually and all plates contained structures from both ACTIVE and INACTIVE animals as well as at least 1 naïve animal, which were used as the controls. Analysis was first carried out using the 2−ΔCT method [64]. The 2−ΔCT values for the naïve animals were then averaged and used as the control for the percent control conversion.
Statistical analysis
One-way, two-way, or two-way repeated measures analysis of variance (RM-ANOVA) followed by a Holm-Sidak or Tukeys post-hoc test were used to determine significant differences in behavior, BDNF protein levels and BDNF gene expression levels. SigmaStat 3.0 software (Systat Software, San Jose, CA) and SPSS (Chicago, IL) was used for all statistical analyses and the level of statistical significance was set at 0.05. All results are expressed as the mean ± the standard error of the mean (SEM).
RESULTS
Impact of STN DBS on Parkinsonian motor behaviors
In 6-OHDA lesioned animals, all rats exhibited reductions in both rearing activity and contralateral forepaw use compared to prelesion behaviors during spontaneous exploration in a Plexiglas cylinder (cylinder task). Overall there was a significant difference between the ACTIVE and INACTIVE groups across time in both contralateral forepaw use and rearing activity (forepaw F(3,13) = 20.915, p ≤ 0.001; rearing F(3,15) = 17.348, p ≤ 0.001). Rats that received two weeks of continuous active STN stimulation (ACTIVE) exhibited significant increases in both rearing activity and contralateral forepaw use compared to rats that received STN stimulating electrodes that were never activated (INACTIVE control group, p < 0.05). This difference was not apparent 24 hours after the cessation of stimulation (p > 0.05). These results indicate that our long-term STN stimulation parameters provide functional improvement in the parkinsonian motor behaviors induced by intrastriatal 6-OHDA. These results are depicted in Fig. 2.
Fig. 2.

Long term STN DBS reverses 6-OHDA induced deficits in forepaw akinesia and rearing activity. A–H) Spontaneous rearing and forepaw use in the cylinder test. Rats received either no stimulation (A–D, n = 7) or 2 weeks of STN DBS (E–H, n = 12). Rats were videotaped and analyzed for the number of spontaneous hind leg rears and number of weight bearing forepaw placements (red plusses) on the cylinder wall before 6-OHDA (A, E, Pre), 2 weeks following intrastriatal 6-OHDA (B, F, Post-lesion), during stimulation on day 14 (C, G, On Stim), and 24 hours after the cessation of stimulation (D, H, Off Stim). I, J) Quantification of rearing activity (I) and forepaw placement (J). All rats exhibited a significant decrease in rears/minute and contralateral forepaw use as a result of 6-OHDA lesioning. Rats stimulated continuously for a two-week period (red triangles) exhibited significantly more rears/minute and significantly increased use of their contralateral forepaw (*, p < 0.05) compared to rats that received no stimulation. Twenty-four hours after cessation of stimulation, stimulation-associated improvement was no longer apparent. Values represent the mean for each group at each time point ± SEM.
Impact of STN DBS on GDNF protein levels in intact animals
There was no significant difference in GDNF protein levels in the SN, striatum, M1 cortex, or hippocampus between the ACTIVE and INACTIVE groups (p > 0.05). Nigral levels of GDNF were 294.85 ± 10.27 pg/mg protein in ACTIVE rats and 220.33 ± 20.43 pg/mg in INACTIVE rats. Striatal levels of GDNF were 81.36 ± 2.52 pg/mg in ACTIVE rats and 87.06 ± 6.61 pg/mg in INACTIVE rats. M1 levels of GDNF were 106.69 ± 4.34 pg/mg in ACTIVE rats and 98.69 ± 7.72 in INACTIVE rats. Hippocampal levels of GDNF were 65.12 ± 10.65 pg/mg in ACTIVE rats and 55.33 ± 12.71 pg/mg in INACTIVE rats. These results are illustrated in Fig. 3.
Fig. 3.

Long Term STN DBS does not increase GDNF protein in unlesioned animals. Long term STN DBS does not increase GDNF protein in the striatum (A) substantia nigra (B) M1 cortex (C) or hippocampus (D) of intact rats. Values expressed as the mean percent of control ± SEM for each group. The control for the ACTIVE left hemisphere (stimulator and 6-OHDA hemisphere) was the mean GDNF pg/mg protein value of the INACTIVE left hemisphere and the control for the ACTIVE right hemisphere was the mean GDNF pg/mg protein value of the INACTIVE right hemisphere.
Impact of STN DBS on BDNF protein levels in the nigrostriatal system
BDNF protein levels were significantly increased in both striatal hemispheres of unlesioned ACTIVE rats as compared to the striatum of unlesioned INACTIVE rats (F(1,8) = 11.674, p = 0.005). ACTIVE rats possessed 115.80 ± 20.69 pg/mg BDNF in the unlesioned striatum compared to 35.93 ± 6.60 pg/mg BDNF in unlesioned INACTIVE rats. There was no significant difference (p > 0.05) in BDNF protein levels between the ACTIVE and INACTIVE groups in the SN of unlesioned animals. Nigral levels of BDNF were 114.19 ± 21.97 pg/mg in ACTIVE rats and 82.89 ± 18.61 pg/mg in INACTIVE rats. These results indicate that STN DBS induces a significant upregulation of striatal BDNF in rats with an intact nigrostriatal system.
STN DBS also was associated with significant increases in BDNF in the nigrostriatal system of rats lesioned with intrastriatal 6-OHDA, however the location of this BDNF protein upregulation differed from the location in unlesioned rats. Specifically, there was a significant bilateral increase in BDNF protein in the SN of the ACTIVE group in lesioned rats as compared to the INACTIVE group of lesioned rats (F(1,17) = 6.733, p = 0.019). ACTIVE rats possessed 29.85 ± 3.24 pg/mg BDNF in the lesioned nigra compared to 12.87 ± 1.66 pg/mg BDNF in lesioned INACTIVE rats. No significant difference in striatal BDNF levels was observed between INACTIVE and ACTIVE lesioned rats (p > 0.05). Striatal levels of BDNF were 8.15 ± 1.93 pg/mg in ACTIVE rats and 3.81 ± 1.69 pg/mg in INACTIVE rats and many of the samples were at or slightly below the minimum sensitivity of the assay. Collectively, these results indicate that STN DBS induces a significant bilateral increase in BDNF protein levels in the nigrostriatal system and that the location of this upregulation is dependent upon lesion status. The effects of STN DBS on BDNF levels in the nigrostriatal system are illustrated in Fig. 4.
Fig. 4.

Long Term STN DBS increases BDNF protein in the nigrostriatal system. No increase in nigral BDNF protein was seen with ACTIVE stimulation in intact rats (n = 4) (A). In 6-OHDA lesioned animals BDNF protein is bilaterally increased in the substantia nigra of rats receiving ACTIVE stimulation (n = 12, white bars) as compared to INACTIVE stimulator controls (n = 6, black bars) (B, *p < 0.05). BDNF protein was bilaterally increased in the striatum of rats receiving ACTIVE stimulation (n = 4, white bars) as compared to INACTIVE stimulator controls (n = 4, black bars) in intact rats (C, *p < 0.05), but was not increased in the striatum of 6-OHDA lesioned rats (ACTIVE n = 12, INACTIVE n = 6) (D). Values expressed as the mean percent of control ± SEM for each group. The control for the ACTIVE left hemisphere (stimulator and 6-OHDA hemisphere) was the mean BDNF pg/mg protein value of the INACTIVE left hemisphere and the control for the ACTIVE right hemisphere was the mean BDNF pg/mg protein value of the INACTIVE right hemisphere.
Impact of STN DBS on BDNF protein levels in the cortex
We also examined BDNF levels in cortical areas that receive varying degrees of innervation from the STN, including the M1, M2, and parietal cortices [9, 12]. In the M2 and parietal cortices, structures that receive sparse STN innervation, there were no significant differences in BDNF protein levels between the ACTIVE and INACTIVE groups in lesioned animals (p > 0.05). BDNF levels in the M2 and parietal cortex of ACTIVE rats were 5.78 ± 0.92 and 3.10 ± 0.15 pg/mg, respectively. BDNF levels in the M2 and parietal cortex of INACTIVE rats were 5.95 ± 0.78 and 3.83 ± 0.44 pg/mg, respectively. However, in the M1 cortex, which receives the preponderance of innervation from the STN [9], we found a significant bilateral increase in BDNF protein in the ACTIVE as compared to the INACTIVE group in both unlesioned (F(1,26)=5.935, p = .019) and lesioned (F(1,18)=5.611, p = .031) rats. BDNF levels in the M1 cortex of ACTIVE unlesioned and lesioned rats were 11.25 ± 1.47 and 17.0 ± 3.15 pg/mg, respectively whereas BDNF levels in the M1 cortex of INACTIVE unlesioned and lesioned rats were 7.15 ± 0.62 and 7.48 ± 1.37, respectively. These results indicate that STN DBS is associated with a significant bilateral increase in BDNF in the M1 cortex in both unlesioned and 6-OHDA lesioned rats. These results are illustrated in Fig. 5.
Fig. 5.

Long term STN DBS increases BDNF protein in the M1 cortex. Long term STN DBS does not increase BDNF protein in the parietal or M2 cortex of 6-OHDA lesioned rats (ACTIVE n = 5, INACTIVE n = 4) (A–B). Long term STN DBS bilaterally increases BDNF protein (*p < 0.05) in the M1 cortex of rats receiving ACTIVE stimulation (white bars) as compared to INACTIVE stimulator controls (black bars) in both 6-OHDA lesioned (ACTIVE n = 12, INACTIVE n = 7) and intact rats (ACTIVE n = 8, INACTIVE n = 10) (C–D). Values expressed as the mean percent of control ± SEM for each group. The control for the ACTIVE left hemisphere (stimulator and 6-OHDA hemisphere) was the mean BDNF pg/mg protein value of the INACTIVE left hemisphere and the control for the ACTIVE right hemisphere was the mean BDNF pg/mg protein value of the INACTIVE right hemisphere.
Impact of STN DBS on BDNF protein levels in the STN, GPe, GPi and hippocampus
In addition, we measured BDNF protein levels in the hippocampi of unlesioned animals in order to determine whether integration into STN circuitry was required for STN DBS to impact BDNF levels. Indeed, we observed no significant difference (p > 0.05) in BDNF protein levels between the ACTIVE and INACTIVE groups in these animals. Hippocampal levels of BDNF were 19.83 ± 1.63 pg/mg in ACTIVE unlesioned rats and 21.82 ± 1.42 pg/mg in INACTIVE unlesioned rats. Lastly, we attempted to measure BDNF protein levels in the GPe and GPi, as well as in the STN itself. However, the BDNF protein levels in these structures were below the detectable limits of the ELISA assay for both the ACTIVE and INACTIVE groups in both unlesioned and lesioned animals.
Impact of STN DBS and MK-801 on BDNF gene expression in STN target structures
STN DBS significantly increased BDNF mRNA in both the SN and GPi in unlesioned rats (SN: F(2,36) = 2.955, p = 0.046; GPI: F(4,14) = 5.941, p = 0.005). Rats in the ACTIVE treatment group displayed an approximate 30% increase in BDNF mRNA in the SN whereas a doubling in GPi BDNF mRNA was observed compared to both INACTIVE and naïve rats. Further, while MK-801 administration had no impact on STN DBS-associated increases in nigral BDNF mRNA (p > 0.05), in the GPi MK-801 completely prevented the STN DBS induced increase in BDNF mRNA. These results are illustrated in Fig. 6A, B. Examination of additional structures including GPe, striatum, M1 cortex, or the STN itself revealed no significant differences in BDNF mRNA levels associated with either STN DBS or MK-801 administration (p > 0.05).
Fig. 6.

The effects of long term STN DBS on BDNF mRNA expression in the GPi and SN. A) Long term STN DBS bilaterally increased BDNF gene expression in the SN of rats receiving ACTIVE stimulation (No MK-801 n = 8, MK-801 n = 5) regardless of MK-801 status (black bars, *p < 0.05) as compared to INACTIVE stimulator control rats (white bars, No MK-801 n = 8, MK-801 n = 10) or naïve rats with no stimulators (n = 4, grey bars). B.) Long-term STN DBS bilaterally increased BDNF gene expression in the GPi of saline treated rats receiving ACTIVE stimulation (n = 2, black bars, *p < 0.05) as compared to INACTIVE stimulator control rats (saline n = 4, MK801 n = 6, white bars) or naïve rats with no stimulators (n = 4, grey bars). Further, ACTIVE stimulation rats treated with MK-801 (n = 3) also expressed significantly lower levels of BDNF mRNA in the GPi compared to ACTIVE saline treated rats (n = 4, black bars, *p < 0.05). Values are expressed as the mean percent of control where the control is the mean 2−ΔCT of the naïve rats.
DISCUSSION
The present experiments characterize the effects of STN DBS on parkinsonian motor behaviors and the expression of GDNF and BDNF within the STN and its target structures in a rat model of PD. Specifically, we found that our STN DBS parameters reversed the 6-OHDA lesion-induced decrease in contralateral forepaw use and rearing activity as measured in the cylinder task. This functional recovery was dependent on active STN DBS since it was not observed after stimulation was turned off. We found that STN DBS did not increase GDNF protein in all structures examined, including the SN, striatum, M1 cortex and hippocampus. However, we observed a STN DBS-associated increase in BDNF protein and mRNA levels in several of the STN target structures examined. Specifically, STN DBS produced a bilateral upregulation in BDNF protein in the nigrostriatal system and the M1 cortex. Further, STN DBS increased BDNF mRNA in both the GPi and the SN. In the GPi, this increase in BDNF mRNA expression was blocked by the administration of the NMDA receptor antagonist MK-801.
Previous work has demonstrated that acute STN DBS can reverse 6-OHDA lesion induced impairments in motor behaviors, including contralateral forepaw akinesia and reduced rearing activity [30, 31] and other motor deficits induced by striatal DA denervation [32, 33]. Here we have demonstrated that 24 hours after the cessation of stimulation both contralateral forepaw use and rearing activity returned to impaired prestimulation performance levels. Forepaw use also has been shown to be enhanced immediately following the cessation of STN DBS and is reversible by MK-801 [31].
In the present study we did not examine the impact of MK-801 on 6-OHDA induced motor impairments. It is tempting to speculate that had we been able to measure BDNF levels during stimulation that more marked increases in BDNF would have been detected. However, given the fact that elevated BDNF levels persisted after behavioral improvements were no longer apparent it is unlikely that the observed improvements in rearing and contralateral forelimb use are mediated by BDNF. Regardless, the finding of improved motor function resulting from STN DBS remains important as it indicates that our long-term STN DBS model is functionally effective, and thus clinically relevant for continued translational research to enhance our understanding of the biological underpinnings of this therapy.
The mechanism whereby long term STN DBS increases BDNF remains to be determined. However, previous in vitro studies provide possible clues. High-frequency stimulation of hippocampal glutamatergic synapses can induce the dendritic release of BDNF [15] with prolonged stimulation leading to axonal release [16]. The results of the present study provide in vivo evidence of stimulation-induced increases in BDNF release, as has previously been observed following stimulation of the VTA [7], perhaps via similar mechanisms as reported in vitro. Alternatively, STN DBS-mediated increases in BDNF could be due to an increased activity level resulting from improved motor functioning in the ACTIVE group. Exercise has been shown to increase BDNF levels in the rat brain [34–36]. However, several differences between our study and the exercise studies decrease the likelihood that exercise is involved in the STN DBS elevations in BDNF. Exercise-induced increases in BDNF result from rigorous wheel or treadmill running [34–37]. Although ACTIVE rats in our study were more physically active than INACTIVE rats, their activity was considerably less than treadmill or wheel running and stimulation did not return their activity to even baseline levels. Further, exercise-induced increases in BDNF consistently demonstrate upregulation in the hippocampus [34–37]. We observed no impact of STN DBS on hippocampal BDNF protein levels. Therefore, it is unlikely that the slight increase in activity associated with long term STN DBS is a key contributor to the increases in BDNF observed in ACTIVE rats.
Unilateral STN DBS produced a significant bilateral increase in BDNF mRNA in the SN. In intact rats the increase in BDNF protein was localized to the striatum whereas in lesioned rats with depleted striatal DA terminals [6] the STN DBS-associated increase in BDNF protein was instead located in the SN. STN DBS did not impact BDNF mRNA levels in either the M1 or the STN. Collectively, our results suggest that STN DBS-associated increases in the nigrostriatal system result from increased SN BDNF mRNA that ultimately enhances BDNF protein levels in either the SN or striatum, depending upon the integrity of the nigrostriatal connections. Increases in BDNF within the nigrostriatal system have the potential to exert a multitude of effects. First, several preclinical studies have demonstrated STN DBS to be neuroprotective to the dopaminergic neurons in the SN [2–6]. STN DBS-induced increases in BDNF may participate in this neuroprotection. Second, BDNF plays an essential role in the maintenance of post-synaptic spine density of the striatal medium spiny neurons (MSNs) [38, 39]. Loss of MSN spines has been demonstrated in both preclinical models and postmortem PD patients [40–43]. Striatal MSN spines are the site of interaction for nigral dopamine neurons and glutamatergic cortical and thalamic neurons [44] and this interaction is necessary for normal basal ganglia functioning and thus normal motor function [45]. It is possible that STN DBS induced BDNF may impact MSN spine density and morphology, thus leading to the improved motor functioning seen with STN DBS [46–49]. Finally, it has been previously demonstrated that BDNF can increase dopamine turnover and/or release in the nigrostriatal system [50–53]. Upregulation of BDNF within the nigrostriatal system may also impact striatal dopaminergic transmission and thus improve parkinsonian motor symptoms.
The primary motor cortex (M1) is central to the execution and planning of movement. Reductions in motor cortex excitability and output have been shown in PD [54–56], which is thought to underlie bradykinesia [57]. STN DBS associated increases in BDNF in the M1 cortex may impact excitability [58] and thereby increase cortical output. Further, BDNF may participate in the decreases in cortico-subcortical oscillatory activity believed to underlie STN DBS associated improvements in tremor [59, 60]. Ultimately, the significance of STN DBS induced increases in M1 BDNF remains to be elucidated.
Our finding that STN DBS upregulates BDNF protein in the M1 cortex but not the M2 or parietal cortex is interesting when viewed in an anatomical context. In the rat the M1 cortex receives the preponderance of STN glutamatergic projections as compared to the M2 and parietal cortices. The M1 also provides a dense reciprocal projection to the STN [9]. Discrete antidromic activation of M1-STN circuitry improves 6-OHDA induced motor deficits [61] suggesting that activation of this same pathway may play a role in the motor improvements associated with STN DBS in patients. Combined with findings that high frequency stimulation of neurons can cause dendritic release of BDNF [15], this raises the possibility that the mechanism of BDNF upregulation in the M1 is antidromic activation of M1-STN circuitry. However, since high frequency activation of neurons can also induce axonal release of BDNF [16], orthodromic activation of the STN-M1 circuitry cannot be ruled out. Further investigation will focus on the source of increased BDNF in the M1 associated with STN DBS.
STN DBS increased BDNF mRNA expression in both the SN and the GPi. STN DBS increases glutamate and GABA in the SN, whereas in the GPi STN DBS has only been associated with increased glutamate [19–21, 23, 62]. Stimulation of NMDA and GABA(B) receptors can lead to BDNF release [15, 16, 24]. Our finding that blockade of NMDA receptors (via MK-801) prevents STN DBS-associated BDNF mRNA upregulation in the GPi but not in the SN suggests that NMDA receptor activation mediates the upregulation of BDNF in the GPi but not the SN. It is possible that the BDNF increase in the SN may be mediated by both NMDA and GABA(B) receptors. The mechanism whereby STN DBS-mediated BDNF mRNA expression is increased in the GPi will be the focus of further experiments.
In summary, the finding that long term STN DBS increases BDNF indicates that this therapy may exert pronounced and underappreciated effects on plasticity in the basal ganglia circuitry, beyond basic inhibition and excitation of neuronal activity. The present study focused on the impact of STN DBS on the trophic factors GDNF and BDNF. It remains to be determined whether long term STN DBS impacts the expression of additional trophic factors. A complete understanding of the impact of STN DBS on the trophic environment and the consequences of this for efficacy and side effects has the potential to influence patient selection and refinement of the therapy itself. It may also lead to the development of less invasive and more efficacious treatments for PD. The ability of stimulation to modulate BDNF expression also opens up the possibility for this therapy to be used to treat other disorders which may involve BDNF, such as Alzheimer’s, Huntington’s, traumatic brain injury, post traumatic stress disorder, or mood disorders. Ultimately, a deeper understanding of this ever-expanding therapeutic approach is essential to optimizing its safety and efficacy.
Acknowledgments
This work was supported by the NS064696 (ASE), the Davis Phinney Foundation for Parkinson’s and the Morris K. Udall Center of Excellence for Parkinson’s Disease Research at Michigan State University NS058830 (TJC).
References
- 1.Pereira EA, Green AL, Nandi D, Aziz TZ. Deep brain stimulation: indications and evidence. Expert Rev Med Devices. 2007;4:591–603. doi: 10.1586/17434440.4.5.591. [DOI] [PubMed] [Google Scholar]
- 2.Maesawa S, Kaneoke Y, Kajita Y, Usui N, Misawa N, Nakayama A, Yoshida J. Long-term stimulation of the subthalamic nucleus in hemiparkinsonian rats: Neuroprotection of dopaminergic neurons. J Neurosurg. 2004;100:679–687. doi: 10.3171/jns.2004.100.4.0679. [DOI] [PubMed] [Google Scholar]
- 3.Temel Y, Visser-Vandewalle V, Kaplan S, Kozan R, Daemen MA, Blokland A, Schmitz C, Steinbusch HW. Protection of nigral cell death by bilateral subthalamic nucleus stimulation. Brain Res. 2006;1120:100–105. doi: 10.1016/j.brainres.2006.08.082. [DOI] [PubMed] [Google Scholar]
- 4.Wallace BA, Ashkan K, Heise CE, Foote KD, Torres N, Mitrofanis J, Benabid AL. Survival of midbrain dopaminergic cells after lesion or deep brain stimulation of the subthalamic nucleus in MPTP-treated monkeys. Brain. 2007;130:2129–2145. doi: 10.1093/brain/awm137. [DOI] [PubMed] [Google Scholar]
- 5.Harnack D, Meissner W, Jira JA, Winter C, Morgenstern R, Kupsch A. Placebo-controlled chronic high-frequency stimulation of the subthalamic nucleus preserves dopaminergic nigral neurons in a rat model of progressive parkinsonism. Exp Neurol. 2008;210:257–260. doi: 10.1016/j.expneurol.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 6.Spieles-Engemann AL, Behbehani MM, Collier TJ, Wohlgenant SL, Steece-Collier K, Paumier K, Daley BF, Gombash S, Madhavan L, Mandybur GT, Lipton JW, Terpstra BT, Sortwell CE. Stimulation of the rat subthalamic nucleus is neuroprotective following significant nigral dopamine neuron loss. Neurobiol Dis. 2010;39:105–115. doi: 10.1016/j.nbd.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedman A, Frankel M, Flaumenhaft Y, Merenlender A, Pinhasov A, Feder Y, Taler M, Gil-Ad I, Abeles M, Yadid G. Programmed acute electrical stimulation of ventral tegmental area alleviates depressive-like behavior. Neuropsychopharmacology. 2009;34:1057–1066. doi: 10.1038/npp.2008.177. [DOI] [PubMed] [Google Scholar]
- 8.Collier TJ, Sortwell CE. Therapeutic potential of nerve growth factors in Parkinson’s disease. Drugs Aging. 1999;14:261–287. doi: 10.2165/00002512-199914040-00003. [DOI] [PubMed] [Google Scholar]
- 9.Degos B, Deniau JM, Le Cam J, Mailly P, Maurice N. Evidence for a direct subthalamo-cortical loop circuit in the rat. Eur J Neurosci. 2008;27:2599–2610. doi: 10.1111/j.1460-9568.2008.06229.x. [DOI] [PubMed] [Google Scholar]
- 10.Jackson A, Crossman AR. Subthalamic nucleus efferent projection to the cerebral cortex. Neuroscience. 1981;6:2367–2377. doi: 10.1016/0306-4522(81)90023-3. [DOI] [PubMed] [Google Scholar]
- 11.Kita H, Chang HT, Kitai ST. The morphology of intracellularly labeled rat subthalamic neurons: a light microscopic analysis. J Comp Neurol. 1983;215:245–257. doi: 10.1002/cne.902150302. [DOI] [PubMed] [Google Scholar]
- 12.Kita H, Kitai ST. Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol. 1987;260:435–452. doi: 10.1002/cne.902600309. [DOI] [PubMed] [Google Scholar]
- 13.Kitai ST, Kita H. In: The Basal Ganglia II-Structure and Function: Current Concepts. Carpenter MB, Jayaraman A, editors. Plenum Press; New York, NY: 2006. pp. 357–373. [Google Scholar]
- 14.Parent A, Hazrati LN. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Brain Res Rev. 1995;20:128–154. doi: 10.1016/0165-0173(94)00008-d. [DOI] [PubMed] [Google Scholar]
- 15.Hartmann M, Heumann R, Lessmann V. Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J. 2001;20:5887–5897. doi: 10.1093/emboj/20.21.5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuda N, Lu H, Fukata Y, Noritake J, Gao H, Mukherjee S, Nemoto T, Fukata M, Poo MM. Differential activity-dependent secretion of brain-derived neurotrophic factor from axon and dendrite. J Neurosci. 2009;29:14185–14198. doi: 10.1523/JNEUROSCI.1863-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagappan G, Zaitsev E, Senatorov VV, Jr, Yang J, Hempstead BL, Lu B. Control of extracellular cleavage of proBDNF by high frequency neuronal activity. Proc Natl Acad Sci U S A. 2009;106:1267–1272. doi: 10.1073/pnas.0807322106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bustos G, Abarca J, Campusano J, Bustos V, Noriega V, Aliaga E. Functional interactions between somatodendritic dopamine release, glutamate receptors and brain-derived neurotrophic factor expression in mesencephalic structures of the brain. Brain Res Brain Res Rev. 2004;47:126–144. doi: 10.1016/j.brainresrev.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 19.Windels F, Bruet N, Poupard A, Urbain N, Chouvet G, Feuerstein C, Savasta M. Effects of high frequency stimulation of subthalamic nucleus on extracellular glutamate and GABA in substantia nigra and globus pallidus in the normal rat. Eur J Neurosci. 2000;12:4141–4146. doi: 10.1046/j.1460-9568.2000.00296.x. [DOI] [PubMed] [Google Scholar]
- 20.Windels F, Bruet N, Poupard A, Feuerstein C, Bertrand A, Savasta M. Influence of the frequency parameter on extracellular glutamate and gamma-aminobutyric acid in substantia nigra and globus pallidus during electrical stimulation of subthalamic nucleus in rats. J Neurosci Res. 2003;72:259–267. doi: 10.1002/jnr.10577. [DOI] [PubMed] [Google Scholar]
- 21.Windels F, Carcenac C, Poupard A, Savasta M. Pallidal origin of GABA release within the substantia nigra pars reticulata during high-frequency stimulation of the subthalamic nucleus. J Neurosci. 2005;25:5079–5086. doi: 10.1523/JNEUROSCI.0360-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bruet N, Windels F, Carcenac C, Feuerstein C, Bertrand A, Poupard A, Savasta M. Neurochemical mechanisms induced by high frequency stimulation of the subthalamic nucleus: increase of extracellular striatal glutamate and GABA in normal and hemiparkinsonian rats. J Neuropathol Exp Neurol. 2003;62:1228–1240. doi: 10.1093/jnen/62.12.1228. [DOI] [PubMed] [Google Scholar]
- 23.Boulet S, Lacombe E, Carcenac C, Feuerstein C, Sgambato-Faure V, Poupard A, Savasta M. Subthalamic stimulation-induced forelimb dyskinesias are linked to an increase in glutamate levels in the substantia nigra pars reticulata. J Neurosci. 2006;26:10768–10776. doi: 10.1523/JNEUROSCI.3065-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fiorentino H, Kuczewski N, Diabira D, Ferrand N, Pangalos MN, Porcher C, Gaiarsa JL. GABA(b) receptor activation triggers BDNF release and promotes the maturation of GABAergic synapses. J Neurosci. 2009;29:11650–11661. doi: 10.1523/JNEUROSCI.3587-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spieles-Engemann AL, Collier TJ, Sortwell CE. A functionally relevant and long-term model of deep brain stimulation of the rat subthalamic nucleus: advantages and considerations. Eur J Neurosci. 2010;32:1092–1099. doi: 10.1111/j.1460-9568.2010.07416.x. [DOI] [PubMed] [Google Scholar]
- 26.Armentero MT, Fancellu R, Nappi G, Bramanti P, Blandini F. Prolonged blockade of NMDA or mGLuR5 glutamate receptors reduces nigrostriatal degeneration while inducing selective metabolic changes in the basal ganglia circuitry in a rodent model of Parkinson’s disease. Neurobiol Dis. 2006;22:1–9. doi: 10.1016/j.nbd.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 27.Schallert T. Behavioral tests for preclinical intervention assessment. NeuroRx. 2006;3:497–504. doi: 10.1016/j.nurx.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Radka SF, Holst PA, Fritsche M, Altar CA. Presence of brain-derived neurotrophic factor in brain and human and rat but not mouse serum detected by a sensitive and specific immunoassay. Brain Res. 1996;709:122–130. doi: 10.1016/0006-8993(95)01321-0. [DOI] [PubMed] [Google Scholar]
- 29.Koprich JB, Campbell NG, Lipton JW. Neonatal 3,4-methylenedioxymethamphetamine (ecstasy) alters dopamine and serotonin neurochemistry and increases brain-derived neurotrophic factor in the forebrain and brainstem of the rat. Brain Res Dev Brain Res. 2003;147:177–182. doi: 10.1016/s0165-3806(03)00219-0. [DOI] [PubMed] [Google Scholar]
- 30.Shi LH, Woodward DJ, Luo F, Anstrom K, Schallert T, Chang JY. High-frequency stimulation of the subthalamic nucleus reverses limb-use asymmetry in rats with unilateral 6-hydroxydopamine lesions. Brain Res. 2004;1013:98–106. doi: 10.1016/j.brainres.2004.03.053. [DOI] [PubMed] [Google Scholar]
- 31.Akita H, Honda Y, Ogata M, Noda K, Saji M. Activation of the NMDA receptor involved in the alleviating after-effect of repeated stimulation of the subthalamic nucleus on motor deficits in hemiparkinsonian rats. Brain Res. 2010;1306:159–167. doi: 10.1016/j.brainres.2009.09.040. [DOI] [PubMed] [Google Scholar]
- 32.Temel Y, Visser-Vandewalle V, Aendekerk B, Rutten B, Tan S, Scholtissen B, Schmitz C, Blokland A, Steinbusch HW. Acute and separate modulation of motor and cognitive performance in parkinsonian rats by bilateral stimulation of the subthalamic nucleus. Exp Neurol. 2005;193:43–52. doi: 10.1016/j.expneurol.2004.12.025. [DOI] [PubMed] [Google Scholar]
- 33.Li XH, Wang JY, Gao G, Chang JY, Woodward DJ, Luo F. High-frequency stimulation of the subthalamic nucleus restores neural and behavioral functions during reaction time task in a rat model of Parkinson’s disease. J Neurosci Res. 2010;88:1510–1521. doi: 10.1002/jnr.22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neeper SA, Gomez-Pinilla F, Choi J, Cotman CW. Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 1996;726:49–56. [PubMed] [Google Scholar]
- 35.Marais L, Stein DJ, Daniels WM. Exercise increases BDNF levels in the striatum and decreases depressive-like behavior in chronically stressed rats. Metab Brain Dis. 2009;24:587–597. doi: 10.1007/s11011-009-9157-2. [DOI] [PubMed] [Google Scholar]
- 36.Wu SY, Wang TF, Yu L, Jen CJ, Chuang JI, Wu FS, Wu CW, Kuo YM. Running exercise protects the substantia nigra dopaminergic neurons against inflammation-induced degeneration via the activation of BDNF signaling pathway. Brain Behav Immun. 2011;25:135–146. doi: 10.1016/j.bbi.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Carro E, Nunez A, Busiguina S, Torres-Aleman I. Circulating insulin-like growth factor i mediates effects of exercise on the brain. J Neurosci. 2000;20:2926–2933. doi: 10.1523/JNEUROSCI.20-08-02926.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saylor AJ, Meredith GE, Vercillo MS, Zahm DS, McGinty JF. BDNF heterozygous mice demonstrate age-related changes in striatal and nigral gene expression. Exp Neurol. 2006;199:362–372. doi: 10.1016/j.expneurol.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 39.Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNeill TH, Brown SA, Rafols JA, Shoulson I. Atrophy of medium spiny I striatal dendrites in advanced Parkinson’s disease. Brain Res. 1988;455:148–152. doi: 10.1016/0006-8993(88)90124-2. [DOI] [PubMed] [Google Scholar]
- 41.Ingham CA, Hood SH, van Maldegem B, Weenink A, Arbuthnott GW. Morphological changes in the rat neostriatum after unilateral 6-hydroxydopamine injections into the nigrostriatal pathway. Exp Brain Res. 1993;93:17–27. doi: 10.1007/BF00227776. [DOI] [PubMed] [Google Scholar]
- 42.Stephens B, Mueller AJ, Shering AF, Hood SH, Taggart P, Arbuthnott GW, Bell JE, Kilford L, Kingsbury AE, Daniel SE, Ingham CA. Evidence of a breakdown of corticostriatal connections in Parkinson’s disease. Neuroscience. 2005;132:741–754. doi: 10.1016/j.neuroscience.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 43.Zaja-Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, Deutch AY, Montine TJ. Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology. 2005;64:545–547. doi: 10.1212/01.WNL.0000150591.33787.A4. [DOI] [PubMed] [Google Scholar]
- 44.Smith Y, Bennett BD, Bolam JP, Parent A, Sadikot AF. Synaptic relationships between dopaminergic afferents and cortical or thalamic input in the sensorimotor territory of the striatum in monkey. J Comp Neurol. 1994;344:1–19. doi: 10.1002/cne.903440102. [DOI] [PubMed] [Google Scholar]
- 45.Obeso JA, Rodríguez-Oroz MC, Benitez-Temino B, Blesa FJ, Guridi J, Marin C, Rodriguez M. Functional organization of the basal ganglia: therapeutic implications for Parkinson’s disease. Mov Disord. 2008;23(Suppl 3):S548–S559. doi: 10.1002/mds.22062. [DOI] [PubMed] [Google Scholar]
- 46.Kumar R, Lozano AM, Kim YJ, Hutchison WD, Sime E, Halket E, Lang AE. Double-blind evaluation of subthalamic nucleus deep brain stimulation in advanced Parkinson’s disease. Neurology. 1998;51:850–855. doi: 10.1212/wnl.51.3.850. [DOI] [PubMed] [Google Scholar]
- 47.Vesper J, Klostermann F, Stockhammer F, Funk T, Brock M. Results of chronic subthalamic nucleus stimulation for Parkinson’s disease: a 1-year follow-up study. Surg Neurol. 2002;57:306–311. doi: 10.1016/s0090-3019(02)00691-2. discussion 311–313. [DOI] [PubMed] [Google Scholar]
- 48.Rodriguez-Oroz MC, Obeso JA, Lang AE, Houeto JL, Pollak P, Rehncrona S, Kulisevsky J, Albanese A, Volkmann J, Hariz MI, Quinn NP, Speelman JD, Guridi J, Zamarbide I, Gironell A, Molet J, Pascual-Sedano B, Pidoux B, Bonnet AM, Agid Y, Xie J, Benabid AL, Lozano AM, Saint-Cyr J, Romito L, Contarino MF, Scerrati M, Fraix V, Van Blercom N. Bilateral deep brain stimulation in Parkinson’s disease: A multicentre study with 4 years follow-up. Brain. 2005;128:2240–2249. doi: 10.1093/brain/awh571. [DOI] [PubMed] [Google Scholar]
- 49.Liang GS, Chou KL, Baltuch GH, Jaggi JL, Loveland-Jones C, Leng L, Maccarone H, Hurtig HI, Colcher A, Stern MB, Kleiner-Fisman G, Simuni T, Siderowf AD. Long-term outcomes of bilateral subthalamic nucleus stimulation in patients with advanced Parkinson’s disease. Stereotact Funct Neurosurg. 2006;84:221–227. doi: 10.1159/000096495. [DOI] [PubMed] [Google Scholar]
- 50.Altar CA, Boylan CB, Jackson C, Hershenson S, Miller J, Wiegand SJ, Lindsay RM, Hyman C. Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc Natl Acad Sci U S A. 1992;89:11347–11351. doi: 10.1073/pnas.89.23.11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Altar CA, Boylan CB, Fritsche M, Jones BE, Jackson C, Wiegand SJ, Lindsay RM, Hyman C. Efficacy of brain-derived neurotrophic factor and neurotrophin-3 on neurochemical and behavioral deficits associated with partial nigrostriatal dopamine lesions. J Neurochem. 1994;63:1021–1032. doi: 10.1046/j.1471-4159.1994.63031021.x. [DOI] [PubMed] [Google Scholar]
- 52.Martin-Iverson MT, Todd KG, Altar CA. Brain-derived neurotrophic factor and neurotrophin-3 activate striatal dopamine and serotonin metabolism and related behaviors: interactions with amphetamine. J Neurosci. 1994;14:1262–1270. doi: 10.1523/JNEUROSCI.14-03-01262.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shults CW, Matthews RT, Altar CA, Hill LR, Langlais PJ. A single intramesencephalic injection of brain-derived neurotrophic factor induces persistent rotational asymmetry in rats. Exp Neurol. 1994;125:183–194. doi: 10.1006/exnr.1994.1023. [DOI] [PubMed] [Google Scholar]
- 54.Albin RL, Young AB, Penney JB. Functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 55.DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- 56.Ridgel AL, Vitek JL, Alberts JL. Forced, not voluntary, exercise improves motor function in Parkinson’s disease patients. Neurorehabil Neural Repair. 2009;23:600–608. doi: 10.1177/1545968308328726. [DOI] [PubMed] [Google Scholar]
- 57.Jenkins IH, Fernandez W, Playford ED, Lees AJ, Frackowiak RS, Passingham RE, Brooks DJ. Impaired activation of the supplementary motor area in Parkinson’s disease is reversed when akinesia is treated with apomorphine. Ann Neurol. 1992;32:749–757. doi: 10.1002/ana.410320608. [DOI] [PubMed] [Google Scholar]
- 58.Kafitz KW, Rose CR, Thoenen H, Konnerth A. Neurotrophin-evoked rapid excitation through TrkB receptors. Nature. 1999;401:918–921. doi: 10.1038/44847. [DOI] [PubMed] [Google Scholar]
- 59.Kuhn AA, Kempf F, Brücke C, Gaynor Doyle L, Martinez-Torres I, Pogosyan A, Trottenberg T, Kupsch A, Schneider GH, Hariz MI, Vandenberghe W, Nuttin B, Brown P. High-frequency stimulation of the subthalamic nucleus suppresses oscillatory beta activity in patients with Parkinson’s disease in parallel with improvement in motor performance. J Neurosci. 2008;28:6165–6173. doi: 10.1523/JNEUROSCI.0282-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Timmermann L, Gross J, Dirks M, Volkmann J, Freund HJ, Schnitzler A. The cerebral oscillatory network of parkinsonian resting tremor. Brain. 2003;126:199–212. doi: 10.1093/brain/awg022. [DOI] [PubMed] [Google Scholar]
- 61.Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stefani A, Fedele E, Galati S, Raiteri M, Pepicelli O, Brusa L, Pierantozzi M, Peppe A, Pisani A, Gattoni G, Hainsworth AH, Bernardi G, Stanzione P, Mazzone P. Deep brain stimulation in Parkinson’s disease patients: biochemical evidence. J Neural Transm Suppl. 2006;70:401–408. doi: 10.1007/978-3-211-45295-0_60. [DOI] [PubMed] [Google Scholar]
- 63.Kluver H, Barrera E. A method for the combined staining of cells and fibers in the nervous system. J Neuropathol Exp Neurol. 1953;12:400–403. doi: 10.1097/00005072-195312040-00008. [DOI] [PubMed] [Google Scholar]
- 64.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]