

Abstract
Lynch syndrome (LS) individuals are predisposed to a variety of cancers, most commonly colorectal, uterine, urinary tract, ovarian, small bowel, stomach and biliary tract cancers. The risk of extracolonic manifestations appears to be highest in MSH2 mutations carriers.
We present a carrier case with a novel MSH2 gene mutation that clearly demonstrates the broad extent of LS phenotypic expression and highlights several important clinical aspects. Current evidence suggests that colorectal tumors from LS patients tend to have better prognoses than their sporadic counterparts, however survival benefits for other cancers encountered in LS are unclear.
In this article we describe a family with a novel protein truncating mutation of c.2388delT in the MSH2 gene, particularly focusing on one individual carrier affected with multiple primary cancers who is surviving 25 years on. Our report of multiple primary tumors occurring in the 12-25 years interval might suggest these patients do not succumb to other extracolonic cancers, provided they are regularly followed-up.
Keywords: Lynch syndrome, MSH2 mutation, increased survival, multiple cancers, HNPCC
Introduction
Lynch syndrome (LS), or hereditary nonpolyposis colorectal cancer (HNPCC) syndrome, is genetically heterogeneous autosomal dominant disease, caused by mutations in one of at least four mismatch repair (MMR) genes, most frequently MLH1 or MSH2, which account for about 50% and 40% of cases respectively [1]. More than 1000 unique mutations were reported in each of these genes http://www.insight-group.org. Truncating mutations are the most frequent cause of deficient MMR function, comprising about 68% and 82% of the types of mutations found in the MLH1 and MSH2 genes respectively [1].
Clinically LS is characterized by a high risk for early-onset colorectal and endometrial cancers and also a moderate risk for urinary tract, ovarian, small bowel, stomach and biliary tract cancers. LS accounts for 2% to 5% of all colorectal cancers [2]. Recognition of LS, leading to molecular screening for MMR genes in a proband, is currently based on the clinico-familial Amsterdam II criteria and/or clinico-pathological Bethesda criteria [3]. Identification of LS causing mutations is important for clinical surveillance in carriers and genetic testing for at risk relatives.
Considerable variability exists in phenotypic expression and cancer risk among different gene mutations carriers. The risk of colon cancer appears to be greater in carriers of MLH1 vs other MMR genes, but the overall risk of all cancers (colonic and extracolonic) combined appears with MSH2 mutations [4]. In MSH2 mutation carriers, relevant genotype-phenotype correlations exist for benign and malignant sebaceous glands tumors (Muir-Torre syndrome) [2].
It is well known that colorectal tumors from LS patients tend to have better prognoses than sporadic colorectal tumors [5] and are associated with distinct histological characteristics such as tumor-infiltrating lymphocytes, mucinous/signet-ring differentiation and/or medullary growth patterns, as well as proximal localization [6]. However, the survival benefits for other cancers in LS are unclear.
There is some inconsistency among cases reported in the literature regarding the involvement of other tumors in LS, such as breast cancer [7], although breast cancer is not classically regarded a LS spectrum tumor [8].
In this article we describe a family with the novel protein truncating mutation of c.2388delT in the MSH2 gene, focusing particularly on one individual carrier affected with multiple primary cancers who is surviving 25 years on.
Case report
Currently 63 year old index female patient (III:4) with a positive colorectal cancer family history was considered having LS after she presented with a colorectal cancer in her 47th year (1995), after she already had been diagnosed with ovarian cancer at the age of 38 (1986) (Figure 1). Her personal medical history revealed a total hysterectomy and bilateral-salpingoophorectomy as a consequence of treatment for ovarian cancer. The patient then conformed to Amsterdam II and Bethesda criteria [3]. For research purposes the patient was enrolled in a LS screening project and found to be a carrier of c.2388delT mutation in the 14 exon of MSH2 gene by direct sequencing. This genetic data was included in a collaborative study report of LS mutation spectrum from the Baltic countries and Poland [9], recently referenced in InSiGHT http://www.insight-group.org, although this mutation and phenotype were not characterized in more detail at the time.
Figure 1.
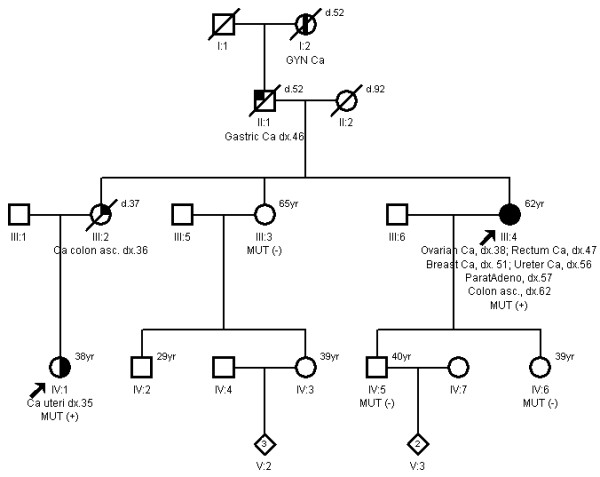
Pedigree of the family with MSH2 c.2388delT mutation.
The patient at the time of diagnosis received standard treatment in accordance with protocols established at the Lithuanian Institute of Oncology (briefly outlined in Table 1). After the rectal cancer diagnosis, she was followed-up by 1-2 yearly colonoscopies and clinical examinations as an outpatient.
Table 1.
The development of multiple primary tumors in patient (individual - index III: 4).
| Site of tumor | Age at diagnosis (years) | Histology | Stage (TNM) | Treatment |
|---|---|---|---|---|
| Ovary (right) | 38 | Mucinous cystadenocarcinoma | I (T1N0M0) | Radical hysterectomy+chemotherapy+ radiotherapy (1986) |
| Rectosigmoid | 47 | Mucinous adenocarcinoma | III (T3N1M0) | Resection of rectosigmoid part with "end-to-end anastomosis"+ chemotherapy |
| Breast (left) | 51 | Ductal adenocarcinoma | II (T2N0M0) | Mastectomy + radiotherapy (4 fields), tamoxifen (20 mg) 3 years |
| Ureter (left) | 56 | Transitional cell ureter carcinoma (G2) | II (T2N0M0) |
Resection of ureter + nephroectomy |
| Adenoma of parathyroid | 57 | Ultrasound biopsy | ||
| Ureteral metastasis in liver (S5-6) | 60 | Transitional cell ureter carcinoma (G2) | IV | Radiofrequency ablation |
| Colon (ascending) | 61 | Adenocarcinoma with partial mucinous differentiation (G2) | II (T3N0M0) | Right hemicolectomy |
All available pathology and medical reports of cancers occurring in this patient were reviewed.
During a prophylactic mammography performed at the age of 51, a tumor (3 × 2.5 cm) was localized in the lower-inner quadrant of the left breast and estrogen receptor positive infiltrative adenocarcinoma stage II (T2N0M0) was diagnosed. The patient underwent a left modified mastectomy, radiotherapy and received Tamoxifen treatment for 3 years. Chemotherapy was postponed due to persistent leucopenia and thrombocytopenia; the patient agreed to receive only one CMF (Cyclophosphamide, Methotrexate and 5-FU) regimen cycle.
After 5 years thereafter, at the age of 56, the patient arrived with complaints of left sided loin-flank pain. An enlarged ureter and left hydronephrosis were found and a moderately differentiated transitional cell carcinoma of the left ureter was confirmed histopathologically after partial resection. A left kidney nephrectomy was performed a few months later, since no kidney function was detected by excretional urography. Four years later an ureter carcinoma metastasis was suspected in the liver by CT scan, confirmed pathologically after liver mass biopsy and treated by percutaneous radiofrequency ablation.
Benign adenoma of the parathyroid gland was diagnosed by ultrasound guided biopsy at the age of 57.
At the age of 62, 1 year and 4 months after the last normal colonoscopy, the patient subsequently developed a stage II adonocarcinoma of the ascending colon (visualized endoscopically as a flat 2 cm lesion) and a right hemicolectomy was performed.
Retrospective immunohistochemical analysis of MMR proteins in the patients colorectal cancer and urinary tract carcinoma showed a consistent loss of expression of MSH2/MSH6 protein complexes.
The niece of the index patient (IV:1, 38 years) was independently referred to the cancer geneticist in 2010 due to a cancer family history and endometrial cancer diagnosis at the age of 35 years (Figure 1), meeting both Amsterdam II and Bethesda criteria. Analysis of mononucleotide BAT25, BAT26 and CAT25 markers [10] showed instability of all of them and immunohistochemistry of MLH1/PMS2 and MSH2/MSH6 proteins indicated the loss of MSH2/MSH6 expression in the endometrial tumor; direct sequencing of the MSH2 gene on ABI 3500 (Applied Biosytems) genetic analyzer revealed the same c.2388delT mutation (p.Thr796ThrfxX15) in exon 14 (Figure 2). This frame shifting mutation is predicted to induce a premature "stop" codon at the downstream 811 amino acid level, which results in truncated MSH2 protein, presumably further degraded by nonsense mediated decay (NMD) [11]. This mutation has not been reported in detail in the literature.
Figure 2.
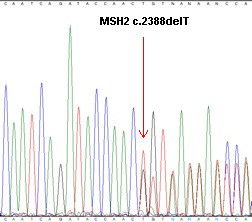
Sequence analysis of MSH2 gene (Genebank accession number U04045) with c.2388delT (p.Thr796ThrfsX15) in exon 14.
Neither sister (III:3) nor children (IV:5, IV:6) of the proband were found to be carriers of the familial deleterious MSH2 mutation.
Discussion
LS can be difficult to diagnose, due to a lack of specific phenotypic features, and the need for a high level of clinical suspicion when encountering cardinal LS features where extracolonic cancers are integral to the syndrome [6]. Our presented carrier case with a novel MSH2 gene mutation clearly demonstrates the broad extent of LS phenotypic expression and highlights several important clinical aspects.
Firstly, the patient over time presented with five primary cancers (in different organs) all of them, except that of breast, typical for the tumor spectrum of LS. Early-stage ovarian mucinous carcinoma was the first manifestation of LS spectrum in the proband. The lifetime risk of ovarian cancer in Lynch syndrome is approximately 10-12% [12], with higher risks (36%) reported in MSH2 carriers [13]. Recent estimates of age-specific cumulative cancer risks in Lynch syndrome families suggest lower than published elsewhere ovarian cancers risk (24% ovarian cancer risk in MSH2) [14]. MMR-deficient ovarian cancers are biologically and clinically different from BRCA deficient cancers, the former overrepresented by non-serous histology types, early-stage and early-onset disease [12,15].
For LS women, the lifetime risk for endometrial cancer (40-80%) is substantially higher than that for colorectal cancer (30-60%) [12]; the risk being greater in MSH2 and MSH6 carriers [16]. We assume that the increased occurrence of endometrial cancer in the proband III:4 was eliminated by radical hysterectomy performed after the diagnosis of ovarian cancer at the age of 38, although the other family member IV:1 developed endometrial cancer at an earlier age (35 years).
The majority of colonic cancers seen in LS patients have a proximal predilection for site of occurrence and affect the right colon [6], while rectal cancer (left-sided) is seen in ~20% of MLH1 and MSH2 carriers [2]. Our patient presented with firstly left sided then later right sided colonic cancer. Our patient's CRCs showed typical histopathological features of LS: mucinous histology and lymphocytic infiltration. It was observed that patients with an initial left-sided CRC developed a metachronous colon tumor in a statistically significant shorter time span (medium 12 years) than patients with a first right-sided CRC (medium 23 years) [2]. Furthermore, one third of patients having had surgery for a LS-associated CRC will have a second primary CRC within 10 years of surgical treatment if the surgery was less than a subtotal colectomy [6]. Consistently, our patient had developed right-sided metachronous CRC 14 years after her first presentation for rectal cancer.
Cancers of the upper urinary tract (renal pelvis/ureter) occur more frequently in LS, with a lifetime risk of 4% compared with < 1% in the general population [17], and are more common in MSH2 mutation carriers, reaching up to 9% for women and 20% for men who are such mutation carriers [18,19]. Upper urinary tract cancers are rarely diagnosed before the age of 40 years [2]. In our proband carcinoma of the ureter was the most aggressive cancer type, unfortunately metastasizing to the liver - a poor prognostic factor.
Breast cancer is not generally regarded as a tumor specific to LS, and lifetime risks are not increased in LS patients, although MMR may have effects on the progression of breast tumors [7,16]. Of note, is that the occurrence of both ovarian and breast cancer in our patient led to an erroneous assumption of the involvement of BRCA mutations by one physician. We have additionally screened (by high resolution melting analysis) BRCA1/2 genes for five common mutations in Lithuania (4 in BRCA1 and 1 in BRCA2). On the other hand, mucinous ovarian cancer is not found to be associated with BRCA1/2 genes [12,15]. About half of breast cancers arising in MMR gene mutation carriers have MMR proteins deficiencies and thus breast tissue samples may be used to improve identification of patients at risk [7,20]. Unfortunatelly, we were unable to perform additional MMR immunohistochemical staining because of the unavailability of necessary material.
We were unable to confirm neither the history nor find appearance of sebaceous tumors (Muirr-Torre variant), which can be seen in MSH2 or MLH1 carriers.
Additionally, the most striking feature of our case report is that our MSH2 mutation carrier, who experienced multiple cancers, is surviving 25 years after her first cancer diagnosis. Improved survival with CRC, compared to unselected sporadic CRC cases, has been consistently reported in LS patients [21,22], although this feature is also common for non-syndromic MSI instable CRCs. The data for other extracolonic LS cancers are scarce, and one study found no survival rate differences between ovarian cancer patients with LS and those with sporadic ovarian cancers [23]. Our survival data for CRC strongly supports increased survival in LS/MMR defective cases and sharply contrasts with previous observations for ovarian cancer. A very recent study found that the risk of dying from (diagnosed) ovarian cancer in germline MMR deficient patients is about 2% [24]. Our results are in line with the notion that MMR genes may predispose to a biologically different type of ovarian cancer than BRCA1/2 or in the general population, characterized by early stage disease at presentation and favorable prognosis [24]. After the MSH2 mutation confirmation in 1998 (at the age of 50), our case was regarded consistent with LS and therefore 12 years of survival data were collected prospectively. Given the absence of published survival data for extracolonic cancers in LS patients, our report of multiple primary tumors occurring in the 12-25 years interval might suggest that these patients do not succumb to other extracolonic cancers, provided they are on regularly follow-up.
Currently, only colonoscopic surveillance is proven to reduce the incidence of mortality from CRC and so is recommended every 1-2 years beginning at the age of 20-25 years [5]. Our case demonstrates that an optimal colonoscopy interval lies closer to 1 year, since a second asymptomatic stage II CRC was diagnosed only 1 year and 4 months after the last normal colonoscopy.
The other LS patient (IV:1) was recommended to be followed up by annual colonoscopy, abdominal ultrasound, urinalysis, gastroduodenoscopy and H. pylori detection and eradication. However, effective surveillance protocols for other extracolonic cancers need still to be identified [19].
In conclusion, prolonged survival in our case of a carrier of the novel MSH2 mutation affected by multiple cancers, raises current challenges in the management of individuals with LS. Obviously, from a single case we can not unequivocally draw conclusions about multiple tumor phenotype correlations with the particular mutation, without having to admit possible influences of modifier genes [25], which may also have effects on tumor spectra to be encountered and survival rates of probands. Additionally, host immune system responses might be enchanced to MMR-deficient tumors [26].
Consent
Written informed consent was obtained from the patient for publication of this case report. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RJ contributed to the genetic counselling of the familly, molecular genetic testing and data analysis, drafted and reviewed the manuscript; PE provided colonoscopic surveillance for the familly members, collected clinical data and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Ramunas Janavicius, Email: rjanavicius@gmail.com.
Pavel Elsakov, Email: elena.jelsakova@seb.lt.
Acknowledgements
We acknowledge Raimundas Meškauskas and Vaida Šimonytė (National Center of Pathology) for pathological reports and performing immunohistechemical analysis. Our thanks to Dr. Richard J Cervin for critical review of this paper, all treating physicians and case report family members.
References
- Peltomaki P, Vasen H. Mutations associated with HNPCC predisposition -- Update of ICG-HNPCC/INSiGHT mutation database. Disease markers. 2004;20(4-5):269–276. doi: 10.1155/2004/305058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecke T, Schulmann K, Engel C, Holinski-Feder E, Pagenstecher C, Schackert HK, Kloor M, Kunstmann E, Vogelsang H, Keller G, Dietmaier W, Mangold E, Friedrichs N, Propping P, Krüger S, Gebert J, Schmiegel W, Rueschoff J, Loeffler M, Moeslein G. German HNPCC Consortium. Genotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: a report by the German HNPCC Consortium. J Clin Oncol. 2006;24(26):4285–4292. doi: 10.1200/JCO.2005.03.7333. [DOI] [PubMed] [Google Scholar]
- Vasen HF, van der Meulen-de Jong AE, de Vos Tot Nederveen Cappel WH, Oliveira J. Familial colorectal cancer risk: ESMO clinical recommendations. Ann Oncol. 2009;20(Suppl 4):51–53. doi: 10.1093/annonc/mdp127. [DOI] [PubMed] [Google Scholar]
- Lindor NM, McMaster ML, Lindor CJ, Greene MH. Concise handbook of familial cancer susceptibility syndromes - second edition. Journal of the National Cancer Institute. 2008;38:1–93. doi: 10.1093/jncimonographs/lgn001. [DOI] [PubMed] [Google Scholar]
- Vasen HF, Moslein G, Alonso A, Bernstein I, Bertario L, Blanco I, Burn J, Capella G, Engel C, Frayling I, Friedl W, Hes FJ, Hodgson S, Mecklin JP, Møller P, Nagengast F, Parc Y, Renkonen-Sinisalo L, Sampson JR, Stormorken A, Wijnen J. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer) Journal of medical genetics. 2007;44(6):353–362. doi: 10.1136/jmg.2007.048991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clinical genetics. 2009;76(1):1–18. doi: 10.1111/j.1399-0004.2009.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanley S, Fung C, Milliken J, Leary J, Barnetson R, Schnitzler M, Kirk J. Breast cancer immunohistochemistry can be useful in triage of some HNPCC families. Familial cancer. 2009;8(3):251–255. doi: 10.1007/s10689-008-9226-4. [DOI] [PubMed] [Google Scholar]
- Vasen HF, Morreau H, Nortier JW. Is breast cancer part of the tumor spectrum of hereditary nonpolyposis colorectal cancer? American journal of human genetics. 2001;68(6):1533–1535. doi: 10.1086/320610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzawski G, Suchy J, Kladny J, Safranow K, Jakubowska A, Elsakov P, Kucinskas V, Gardovski J, Irmejs A, Sibul H, Huzarski T, Byrski T, Debniak T, Cybulski C, Gronwald J, Oszurek O, Clark J, Góźdź S, Niepsuj S, Słomski R, Pławski A, Łacka-Wojciechowska A, Rozmiarek A, Fiszer-Maliszewska Ł, Bebenek M, Sorokin D, Stawicka M, Godlewski D, Richter P, Brozek I, Wysocka B, Jawień A, Banaszkiewicz Z, Kowalczyk J, Czudowska D, Goretzki PE, Moeslein G, Lubiński J. Germline MSH2 and MLH1 mutational spectrum in HNPCC families from Poland and the Baltic States. Journal of medical genetics. 2002;39(10):E65. doi: 10.1136/jmg.39.10.e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findeisen P, Kloor M, Merx S, Sutter C, Woerner SM, Dostmann N, Benner A, Dondog B, Pawlita M, Dippold W, Wagner R, Gebert J, von Knebel Doeberitz M. T25 repeat in the 3' untranslated region of the CASP2 gene: a sensitive and specific marker for microsatellite instability in colorectal cancer. Cancer research. 2005;65(18):8072–8078. doi: 10.1158/0008-5472.CAN-04-4146. [DOI] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annual review of biochemistry. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- Manchanda R, Menon U, Michaelson-Cohen R, Beller U, Jacobs I. Hereditary non-polyposis colorectal cancer or Lynch syndrome: the gynaecological perspective. Current opinion in obstetrics & gynecology. 2009;21(1):31–38. doi: 10.1097/GCO.0b013e32831c844d. [DOI] [PubMed] [Google Scholar]
- Green J, O'Driscoll M, Barnes A, Maher ER, Bridge P, Shields K, Parfrey PS. Impact of gender and parent of origin on the phenotypic expression of hereditary nonpolyposis colorectal cancer in a large Newfoundland kindred with a common MSH2 mutation. Diseases of the colon and rectum. 2002;45(9):1223–1232. doi: 10.1007/s10350-004-6397-4. [DOI] [PubMed] [Google Scholar]
- Bonadona V, Bonaiti B, Olschwang S, Grandjouan S, Huiart L, Longy M, Guimbaud R, Buecher B, Bignon YJ, Caron O, Colas C, Noguès C, Lejeune-Dumoulin S, Olivier-Faivre L, Polycarpe-Osaer F, Nguyen TD, Desseigne F, Saurin JC, Berthet P, Leroux D, Duffour J, Manouvrier S, Frébourg T, Sobol H, Lasset C, Bonaïti-Pellié C. French Cancer Genetics Network. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. Jama. 2011;305(22):2304–2310. doi: 10.1001/jama.2011.743. [DOI] [PubMed] [Google Scholar]
- Pal T, Permuth-Wey J, Kumar A, Sellers TA. Systematic review and meta-analysis of ovarian cancers: estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin Cancer Res. 2008;14(21):6847–6854. doi: 10.1158/1078-0432.CCR-08-1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasen HF, Stormorken A, Menko FH, Nagengast FM, Kleibeuker JH, Griffioen G, Taal BG, Moller P, Wijnen JT. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol. 2001;19(20):4074–4080. doi: 10.1200/JCO.2001.19.20.4074. [DOI] [PubMed] [Google Scholar]
- Gylling AH, Nieminen TT, Abdel-Rahman WM, Nuorva K, Juhola M, Joensuu EI, Jarvinen HJ, Mecklin JP, Aarnio M, Peltomaki PT. Differential cancer predisposition in Lynch syndrome: insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis. 2008;29(7):1351–1359. doi: 10.1093/carcin/bgn133. [DOI] [PubMed] [Google Scholar]
- Watson P, Vasen HF, Mecklin JP, Bernstein I, Aarnio M, Jarvinen HJ, Myrhoj T, Sunde L, Wijnen JT, Lynch HT. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. International journal of cancer. 2008;123(2):444–449. doi: 10.1002/ijc.23508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koornstra JJ, Mourits MJ, Sijmons RH, Leliveld AM, Hollema H, Kleibeuker JH. Management of extracolonic tumours in patients with Lynch syndrome. The Lancet oncology. 2009;10(4):400–408. doi: 10.1016/S1470-2045(09)70041-5. [DOI] [PubMed] [Google Scholar]
- Walsh MD, Buchanan DD, Cummings MC, Pearson SA, Arnold ST, Clendenning M, Walters R, McKeone DM, Spurdle AB, Hopper JL, Jenkins MA, Phillips KD, Suthers GK, George J, Goldblatt J, Muir A, Tucker K, Pelzer E, Gattas MR, Woodall S, Parry S, Macrae FA, Haile RW, Baron JA, Potter JD, Le Marchand L, Bapat B, Thibodeau SN, Lindor NM, McGuckin MA, Young JP. Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010;16(7):2214–2224. doi: 10.1158/1078-0432.CCR-09-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23(3):609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- Elsakov P, Kurtinaitis J. Survival from colorectal carcinoma in HNPCC families as compared to the general population in Lithuania--initial results. Familial cancer. 2006;5(4):369–371. doi: 10.1007/s10689-006-0007-7. [DOI] [PubMed] [Google Scholar]
- Crijnen TE, Janssen-Heijnen ML, Gelderblom H, Morreau J, Nooij MA, Kenter GG, Vasen HF. Survival of patients with ovarian cancer due to a mismatch repair defect. Familial cancer. 2005;4(4):301–305. doi: 10.1007/s10689-005-6573-2. [DOI] [PubMed] [Google Scholar]
- Grindedal EM, Renkonen-Sinisalo L, Vasen H, Evans G, Sala P, Blanco I, Gronwald J, Apold J, Eccles DM, Sanchez AA, Sampson J, Järvinen HJ, Bertario L, Crawford GC, Stormorken AT, Maehle L, Moller P. Survival in women with MMR mutations and ovarian cancer: a multicentre study in Lynch syndrome kindreds. Journal of medical genetics. 2011;47(2):99–102. doi: 10.1136/jmg.2009.068130. [DOI] [PubMed] [Google Scholar]
- Scott RJ. Modifier genes and HNPCC: variable phenotypic expression in HNPCC and the search for modifier genes. Eur J Hum Genet. 2008;16(5):531–532. doi: 10.1038/ejhg.2008.46. [DOI] [PubMed] [Google Scholar]
- Kloor M, Michel S, von Knebel Doeberitz M. Immune evasion of microsatellite unstable colorectal cancers. International journal of cancer. 2011;127(5):1001–1010. doi: 10.1002/ijc.25283. [DOI] [PubMed] [Google Scholar]