

Abstract
Purpose
Based on the previous indications of founder ATP-binding cassette sub-family A member 4 gene (ABCA4) mutations in a South African subpopulation, the purpose was to devise a mechanism for identifying common disease-causing mutations in subjects with ABCA4-associated retinopathies (AARs). Facilitating patient access to this data and determining the frequencies of the mutations in the South African population would enhance the current molecular diagnostic service offered.
Methods
The majority of subjects in this study were of Caucasian ancestry and affected with Stargardt macular dystrophy. The initial cohort consisted of DNA samples from 181 patients, and was screened using the ABCR400 chip. An assay was then designed to screen a secondary cohort of 72 patients for seven of the most commonly occurring ABCA4 mutations in this population. A total of 269 control individuals were also screened for the seven ABCA4 mutations.
Results
Microarray screening results from a cohort of 181 patients affected with AARs revealed that seven ABCA4 mutations (p.Arg152*, c.768G>T, p.Arg602Trp, p.Gly863Ala, p.Cys1490Tyr, c.5461–10T>C, and p.Leu2027Phe) occurred at a relatively high frequency. The newly designed genetic assay identified two of the seven disease-associated mutations in 28/72 patients in a secondary patient cohort. In the control cohort, 12/269 individuals were found to be heterozygotes, resulting in an estimated background frequency of these mutations in this particular population of 4.46 per 100 individuals.
Conclusions
The relatively high detection rate of seven ABCA4 mutations in the primary patient cohort led to the design and subsequent utility of a multiplex assay. This assay can be used as a viable screening tool and to reduce costs and laboratory time. The estimated background frequency of the seven ABCA4 mutations, together with the improved diagnostic service, could be used by counselors to facilitate clinical and genetic management of South African families with AARs.
Introduction
Stargardt disease (STGD; OMIM 248200) is a juvenile-onset form of macular dystrophy (MD) characterized by loss of photoreceptor cells in the macula, resulting in a severe reduction of central vision [1]. The macula is situated in the central region of the retina and contains a high concentration of cone photoreceptor cells [2]. STGD, which can have either an autosomal recessive or an autosomal dominant mode of inheritance [3], is one of the most frequent causes of MD in childhood, accounting for approximately 7% of all retinal degenerative diseases (RDDs) [4], with an estimated incidence of 1 in 10,000 individuals [5].
Clinical features of the disease include progressive bilateral degeneration of the macula and the retinal pigment epithelium. The appearance of orange-yellow flecks located around the macula or the mid-retinal periphery is a distinctive feature of STGD [1]. Fluorescein angiography has shown the choroid to be dark in many patients, which indicates the accumulation of lipofuscin. Lipofuscin consists of a combination of different fluorescent by-products of the visual cycle and appears to have toxic effects on the retinal pigment epithelium [6].
To date, the ATP (ATP)-binding cassette, subfamily A (ABC1), member 4 gene, ABCA4, which codes for the retina-specific ATP-binding cassette transporter, is the only gene known to be involved with autosomal recessive STGD (arSTGD) [1].
The ABCA4 protein is expressed in the outer segment of the rod and cone photoreceptor cells in the retina [5,7]. Protein misfolding, decreased functioning, and mislocalization due to ABCA4 mutations have been described for ABCA4-associated retinopathies (AARs). Mutations in ABCA4 have been found to be associated not only with arSTGD but also with other clinical forms of RDDs, including fundus flavimaculatus, cone-rod dystrophy, and autosomal recessive retinitis pigmentosa [1]. More than 500 mutations have been identified in ABCA4, including single-base substitutions, duplications, and deletions [8].
Previous research performed in the Division of Human Genetics at the University of Cape Town, South Africa identified possible ABCA4 founder mutations, occurring at a relatively high frequency in the South African Caucasian population, specifically those of Afrikaner descent [9]. With the development of the ABCR400 microarray [10], an effective, rapid screening tool became available for the genetic confirmation of diagnosis of AARs. We previously evaluated the utility of the ABCR400 microarray and stressed the importance of validating the chip data by verifying mutations and familial investigations, before delivering results to patients [11]. Additional samples have subsequently been screened, and the validated chip data, including mutation spectrum and mutation frequencies, are reported here. Importantly, based on the frequencies of mutations identified in South African patients with STGD, it may be possible to target testing and provide a cost-effective diagnostic service.
Methods
RDD patients and their family members were referred to the Division of Human Genetics from throughout South Africa, by ophthalmologists and the lay support society, Retina South Africa. Individuals were invited to participate in the RDD research program, and informed consent was obtained from patients according to the tenets of the Declaration of Helsinki (2008). The consent forms and patient information sheets used in the RDD project had previously been approved by the University of Cape Town Research Ethics Committee (FHS HREC REF 226/2010). Biologic material, demographic information, relevant clinical details and diagnoses were archived for each patient, together with information as supplied, relating to additional health issues.
ABCR400 microarray screening
A total of 181 samples from affected probands with AARs were screened using the ABCR400 chip (Asper Ophthalmics, Tartu, Estonia). This includes the 132 samples previously described [11] and an additional 49 samples. The ethnic categorization of the patient cohort was as follows: 152 Caucasians, 17 indigenous Black Africans, seven Indians, and five individuals of mixed ancestry. Most (n=133) of the patients had a diagnosis of arSTGD, while the remainder were diagnosed with MD (n=19) or isolated cases of RP (n=24), or were probands from families diagnosed with autosomal recessive retinitis pigmentosa (n=5). All results obtained from the ABCR400 microarray screening were verified, and familial mutation cosegregation analysis was performed wherever possible, as previously described [11]. Briefly, PCR was performed to amplify the exons harbouring each mutation detected using the microarray. Probands were tested using restriction enzyme digests or direct sequencing (ABI Prism 3100 automated sequencer; Applied Biosystems, Foster City, CA) to verify the presence of each mutation. Cosegregation studies were performed to test for the presence of each mutation in samples from family members, using restriction enzyme digests, denaturing high-performance liquid chromatography (dHPLC) analysis (WAVE® Nucleic Acid Fragment Analysis System; Transgenomic Inc., Omaha, NE), allele-specific PCR (AS-PCR), or direct sequencing. Proband samples were included in the co-segregation analysis, having been amplified in a separate reaction to the initial verification experiment.
Detection of seven ABCA4 mutations
A locally-developed diagnostic assay was established to interrogate seven common ABCA4 mutations in a cohort of samples from 72 unrelated patients who had not previously been screened using the ABCR400 microarray. Most of the patients (n=70) were affected with STGD, while the remainder had a diagnosis of cone-rod dystrophy (n=2). The ethnic breakdown of the patient cohorts was as follows: 60 were Caucasian, eight individuals were of mixed ancestry, three were Indian, and one was of indigenous Black African origin.
In addition, the assay was used to screen a cohort of 269 Caucasian control individuals, 169 of whom were specifically of Afrikaner descent. Individuals were defined as being Afrikaner based on their family name and stated ancestry upon recruitment. Furthermore, for 119 of these 169 individuals, pedigrees indicated that this sub-cohort had been in South Africa for two or more generations. The pedigrees verified Afrikaner descent, showing the individuals to be of European origin, with distinct input from the Dutch, German, and French population groups. This control cohort comprised individuals who had not been previously assessed for the presence of an RDD.
Seven exons (exons 5, 6, 13, 17, 30, 39, and 44) of the ABCA4 gene were amplified using PCR (Table 1). The PCR products were screened for the seven common ABCA4 mutations with a multiplex assay intended to streamline our former approach to mutation validation [11]. Four of the mutations were detected with SNaPshot PCR (p.Cys1490Tyr, p.Arg602Trp, c.5461–10T>C, and p.Leu2027Phe mutations; Table 2, Figure 1, Figure 2, and Figure 3) using the SNaPshot® Multiplex Ready Reaction Mix (Applied Biosystems, Warrington, UK), resolved on the ABI 3100 Genetic Analyzer (Applied Biosystems) and subsequently analyzed using the GeneMapper 3.0 GeneScan software (Applied Biosystems). The p.Arg152* (Figure 4) and c.768G>T (Figure 5) mutations were screened for with dHPLC analysis using the WAVE® Nucleic Acid Fragment Analysis System (Transgenomic Inc.). AS-PCR was used to screen for the p.Gly863Ala mutation (Table 3), and the PCR products were separated using non-denaturing dHPLC conditions on the WAVE® System (Figure 6). Direct cycle sequencing was performed using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) on samples shown to have dHPLC profile alterations (Figure 5). These samples were resolved on the ABI 3100 Genetic Analyzer (Applied Biosystems). Family studies were performed wherever possible (depending on the availability of samples) for probands found to carry at least two mutations.
Table 1. Information pertaining to the primers used to amplify the exons containing the seven most common ABCA4 mutations.
| Exon (mutation) | Primer 5′-3′ | Annealing temperature | Mutation detection technique |
|---|---|---|---|
| Exon 5 (p.Arg152*) |
F: gacccatttccccttcaac |
60 °C |
dHPLC, Cycle sequencing using the reverse primer |
| |
R: aggctgggtgcttccctc |
|
|
| Exon 6 (c.768G>T) |
F: ggtgtctttcctaccacag |
57.9 °C |
dHPLC, Cycle sequencing using the forward primer |
| |
R: aggaatcaccttgcaattgg |
|
|
| Exon 13 (p.Arg602Trp) |
F: agctatccaagcccgttcc |
63 °C |
SNaPshot PCR |
| |
R: ccattagcgtgtcatggag |
|
|
| Exon 17 (p.Gly863Ala) |
F: ctgcggtaaggtaggataggg |
60 °C |
Allele-specific PCR |
| |
R: cacaccgtttacatagagggc |
|
|
| Exon 30 (p.Cys1490Tyr) |
F: gtcagcaactttgaggctg |
63 °C |
SNaPshot PCR |
| |
R: tccctctgtggcaggcag |
|
|
| Intron38/Exon39 (c.5461–10T>C) |
F: gccccacctgctgaagag |
63 °C |
SNaPshot PCR |
| |
R: tcccagctttggacccag |
|
|
| Exon 44 (p.Leu2027Phe) |
F: gaagcttctccagccctagc |
63 °C |
SNaPshot PCR |
| R: tgcactctcatgaaacaggc |
Table 2. Information pertaining to the primers designed for the snapshot multiplex reaction.
| Exon (mutation) | Primer length (bp) with tail | Primer sequence (with tail, 5′-3′)* |
|---|---|---|
| 13 (p.Arg602Trp) |
34 |
R: tgttccagtgccacgaacccgccccagatgtacc |
| 30 (p.Cys1490Tyr) |
32 |
R: cttcgtggttactgagcttctccctggtgctg |
| 39 (Intron 38) (c.5461–10T>C) |
37 |
F: ccgatgtagttgaccccgtttccaacagtcctacttc |
| 44 (p.Leu2027Phe) | 41 | R: tactctggatcttagtaggtaaagatgttctcgtcctgtga |
*The nucleotide sequence in bold is the sequence designed to bind complementary to the genomic DNA sequence. The nucleotide sequence not written in bold is the random nucleotide tail added to the primer sequence.
Figure 1.

An electropherogram of the multiplex SNaPshot reaction shows the results obtained for a sample that is heterozygous for the p.Arg602Trp and p.Cys1490Tyr mutations. The p.Arg602Trp alleles are indicated by blue and green peaks at 36 bp and 37 bp, respectively. The p.Cys1490Tyr alleles are indicated by black and red peaks at 38 bp and 39 bp, respectively. The c.5461–10 wild-type allele is represented by a red peak at 41.5 bp. The p.Leu2027 wild-type allele is represented by a blue peak at 45 bp. The LIZ Size Standard can be seen at 35 bp.
Figure 2.

An electropherogram of the multiplex SNaPshot reaction showing the results obtained for a sample that is heterozygous for the p.Leu2027Phe mutation. The p.Arg602 wild-type allele is indicated by a blue peak at 36 bp. The p.Cys1490 wild-type allele is indicated by a black peak at 38 bp. The c.5461–10 wild-type allele is represented by a red peak at 41.5 bp. The p.Leu2027Phe alleles are represented by blue and green peaks at 45 bp and 46 bp, respectively. The LIZ Size Standard can be seen at 35 bp.
Figure 3.

An electropherogram of the multiplex SNaPshot reaction showing the results obtained for a sample that is heterozygous for the c.5461–10T>C mutation. The p.Arg602 wild-type allele is indicated by a blue peak at 36 bp. The p.Cys1490 wild-type allele is indicated by a black peak at 38 bp. The c.5461–10T>C alleles are represented by a black and red peak at 40 bp and 41.5 bp, respectively. The p.Leu2027 wild-type allele is represented by a blue peak at 45 bp. The LIZ Size Standard can be seen at 35 bp.
Figure 4.
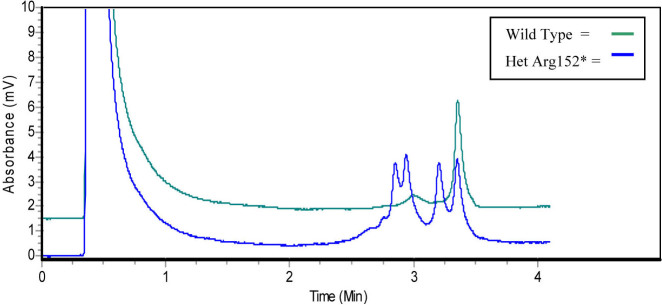
A chromatogram, generated using denaturing high-performance liquid chromatography, shows the wild type profile, and the profile obtained when a heterozygous p.Arg152* mutation is present.
Figure 5.
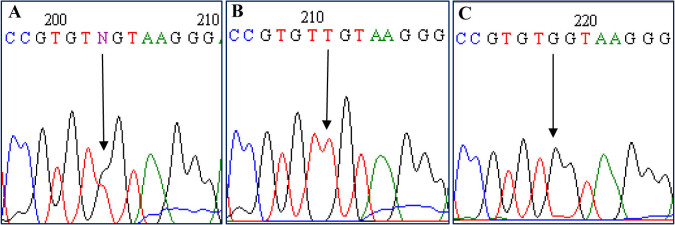
Sequencing electropherograms show (A) the heterozygous c.768G>T mutation, (B) homozygous c.768G>T mutation, and (C) the wild-type sequence following denaturing high-performance liquid chromatography screening.
Table 3. Information pertaining to the primers designed for AS-PCR for Gly863Ala mutation detection.
| Exon (mutation) | Primer name (bp) | Primer length | Primer sequence* (5′-3′) |
|---|---|---|---|
| 17 |
Gly863Ala-Rc |
26 |
tttttgaagtggggttccatagtcag |
| Gly863Ala-Rg | 36 | gcgtgcttggggtatgaagtggggttccatagtcac |
Gly863Ala-Rc: The AS-PCR primer designed specifically to bind to the nucleotide, cytosine (C), when exon 17 does not contain the p.Gly863Ala mutation, and is therefore known as the wild type primer. Gly863Ala-Rg: The AS-PCR primer designed specifically to bind to the nucleotide, guanine (G), when exon 17 contains the p.Gly863Ala mutation, and is therefore known as the mutation-specific primer. *The nucleotide sequence in bold is the sequence designed to bind complementary to the genomic DNA sequence. The nucleotide sequence not written in bold is the random nucleotide tail added to the primer sequence.
Figure 6.
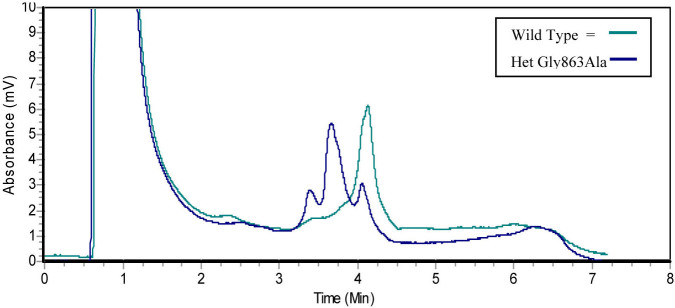
A chromatogram, generated using denaturing high-performance liquid chromatography following allele-specific PCR, shows the wild type profile and the profile obtained when a heterozygous p.Gly863Ala mutation is present.
Results
ABCR400 microarray screening
Of the 181 samples screened using the ABCR400 microarray, a total of 99 (54.69%) were characterized completely, i.e., to the point of identifying at least two disease-causing mutations (specifically, nine were homozygotes and 90 were presumed to be compound heterozygotes, although only 41 could be verified as having biallelic mutations through family studies). The remainder of the samples were either partially characterized, i.e., with either a single, heterozygous mutation (n=49; 27.07%) or no mutation (n=33; 18.23%). The identification of causative mutations in approximately 55% of the cohort in the current study is comparable to our initial findings, where 60% of the cohort was characterized [11].
A total of 51 distinct ABCA4 mutations were identified. However, seven mutations (p.Arg152*, c.768G>T, p.Arg602Trp, p.Gly863Ala, p.Cys1490Tyr, c.5461–10T>C, and p.Leu2027Phe) occurred at a significantly higher frequency, compared to the other variants in the cohort (Table 4).
Table 4. Spectrum of ABCA4 mutations detected using the ABCR400 microarray in 181 South African probands.
| Amino acid change | Nucleotide change | No. of alleles detected | Frequency |
|---|---|---|---|
| p.Cys54Tyr |
c. 161 G>A |
2 |
0.55% |
| p.Arg152* |
c. 454 C>T |
12 |
3.31% |
| p.Arg152Gln |
c. 455 G>A |
3 |
0.83% |
| p.Gly172Ser |
c. 514 G>A |
1 |
0.28% |
| p.Arg212Cys |
c. 634 C>T |
1 |
0.28% |
| p.Lys223Gln |
c. 667 A>C |
1 |
0.28% |
| p.V256V (Splice) |
c. 768 G>T |
18 |
4.97% |
| p.Pro291Leu |
c. 872 C>T |
1 |
0.28% |
| p.Trp439* |
c. 1317 G>A |
1 |
0.28% |
| p.Ala538Asp |
c. 1613 C>A |
1 |
0.28% |
| p.Leu541Pro |
c. 1622 T>C |
1 |
0.28% |
| p.Arg602Trp |
c. 1804C>T |
30 |
8.29% |
| p.Val643Met |
c. 1927 G>A |
1 |
0.28% |
| p.Arg653Cys |
c. 1957 C>T |
1 |
0.28% |
| p.Arg681* |
c. 2041 C>T |
3 |
0.83% |
| p.Val767Asp |
c. 2300 T>A |
1 |
0.28% |
| p.Trp855* |
c.2564_2571delGGTACCTT |
2 |
0.55% |
| p.Gly863Ala |
c. 2588 G>C |
11 |
3.04% |
| p.Val931Met |
c. 2791 G>A |
1 |
0.28% |
| p.Asn965Ser |
c. 2894 A>G |
4 |
1.10% |
| p.Val989Ala |
c. 2966 T>C |
1 |
0.28% |
| p.Gly991Arg |
c. 2971 G>C |
1 |
0.28% |
| p.Thr1019Met |
c. 3056 C>T |
1 |
0.28% |
| p.Ala1038Val |
c. 3113 C>T |
3 |
0.83% |
| p.Glu1087Lys |
c. 3259 G>A |
1 |
0.28% |
| p.Arg1108Cys |
c. 3322 C>T |
2 |
0.55% |
| p.Leu1201Arg |
c. 3602 T>G |
4 |
1.10% |
| p.Arg1300Gln |
c. 3899 G>A |
4 |
1.10% |
| p.Pro1380Leu |
c. 4139 C>T |
3 |
0.83% |
| p.Trp1408Arg |
c. 4222 T>C |
1 |
0.28% |
| - |
c. 4253+5G>A |
1 |
0.28% |
| p.Phe1440Ser |
c. 4319 T>C |
1 |
0.28% |
| p.Arg1443His |
c. 4328 G>A |
1 |
0.28% |
| p.Cys1490Tyr |
c.4469 G>A |
54 |
14.92% |
| p.Gln1513Pro fs*42 |
c. 4535 insC |
1 |
0.28% |
| p.Ala1598Asp |
c. 4793C>A |
1 |
0.28% |
| p.Arg1640Trp |
c. 4918 C>T |
2 |
0.55% |
| p.Ser1642Arg |
c. 4926 C>G |
1 |
0.28% |
| p.V1681_C1685del |
c. 5041 del15 |
1 |
0.28% |
| - |
c. 5461–10T>C |
24 |
6.63% |
| - |
c. 5714+5 G>A |
2 |
0.55% |
| p.Pro1948Leu |
c. 5843 C>T |
1 |
0.28% |
| p.Gly1961Glu |
c. 5882 G>A |
4 |
1.10% |
| p.Leu2027Phe |
c.6079 C>T |
30 |
8.29% |
| p.Arg2030* |
c. 6088 C>T |
1 |
0.28% |
| p.Arg2030Gln |
c. 6089 G>A |
3 |
0.83% |
| p.Arg2038Trp |
c. 6112 C>T |
1 |
0.28% |
| p.Arg2107His |
c. 6320 G>A |
2 |
0.55% |
| p.Arg2118Glu fs*27 |
c. 6352 delA |
1 |
0.28% |
| p.Cys2150Tyr |
c. 6449 G>A |
1 |
0.28% |
| p.Gln2220* | c. 6658 C>T | 1 | 0.28% |
Of the 99 completely characterized subjects (i.e., both disease-causing mutations identified), 64 showed two mutated alleles from this prevalent set of seven mutations (n=128 alleles), while 23 had a single allele from this high-incidence subset (n=23).
Of the 49 partially characterized samples (i.e., where only one disease-causing mutation was identified), 28 had one of the prevalent mutations (n=28). In total, 179 of the 362 alleles (49.45%) represented the seven common mutations.
Based on the mutation frequencies observed, one would expect approximately 35.2% (0.65×0.55) of samples from South African Caucasian STGD patients to be potentially fully characterized by screening for only the seven mutations.
Detection of seven ABCA4 mutations
A total of 78/144 (54.17%) disease-causing mutations were detected in the secondary cohort of 72 patients screened (Table 5). Twenty-eight patients (38.89%) were found to carry two mutations. The majority of the patients found to carry two mutations were presumed to be compound heterozygotes (n=23 individuals, 82.14%), with the remaining five homozygotes (17.86%). In these 28 individuals, ABCA4 is thus presumed to be the disease-causing gene, although family studies to prove the mutations were biallelic could be performed for only seven compound heterozygous individuals. Twenty-two patients (30.55%) were found to carry one ABCA4 mutation, meaning that another rarer ABCA4 mutation may exist or that a different gene causes the condition. Finally, 22 patients (30.55%) were shown not to carry any of the seven mutations, indicating that either two rarer ABCA4 mutations exist or that a different gene could cause the condition.
Table 5. The results from screening both the patient and control cohort for seven ABCA4 mutations, showing the number and frequency of mutant alleles detected per mutation.
| |
Mutant alleles |
||||||
|---|---|---|---|---|---|---|---|
| Cohort | p.Cys1490Tyr | p.Arg602Trp | p.Leu2027Phe | c.5461–10T>C | c.768G>T | p.Gly863Ala | p.Arg152* |
| Patient (n=72; 144 alleles) |
16 (11.11%) |
10 (6.94%) |
12 (8.33%) |
13 (9.03%) |
13 (9.03%) |
7 (4.86%) |
7 (4.86%) |
| Control (total; n=269; 538 alleles) |
2 (0. 37%) |
2 (0. 37%) |
2 (0. 37%) |
0 |
2 (0. 37%) |
3 (0.56%) |
1 (0.19%) |
| a) Caucasian (n=100; 200 alleles) |
0 |
0 |
1 (0.5%) |
0 |
1 (0.5%) |
2 (1%) |
0 |
| b) Afrikaner (n=169; 338 alleles) | 2 (0.59%) | 2 (0.59%) | 1 (0.296%) | 0 | 1 (0.296%) | 1 (0.296%) | 1 (0.296%) |
Of the 269 control samples screened, 12 (4.46%) were shown to carry a heterozygous mutation (Table 5). Four heterozygotes (4%) were detected in the Caucasian sub-cohort, with eight heterozygotes (4.73%) detected in the Afrikaner sub-cohort.
Discussion
Through ABCR400 microarray analysis of 181 AAR samples, seven specific mutations accounted for approximately 50% of the disease-causing alleles.
The seven common mutations were all regarded as pathogenic contributors to disease. Functional analysis of the p.Arg602Trp and p.Cys1490Tyr mutations showed that these missense alleles result in misfolding and mislocalization of the ABCA4 protein, as well as a marked reduction in ATPase activity [12]. The c.768G>T mutation, although isocoding (Val256Val), has been shown to affect splicing and results in a lack of the mRNA transcript [13]. The same study showed that the p.Gly863Ala mutation, which occurs in the first nucleotide and hence the splice acceptor site of exon 17, results in a mixture of mutant ABCA4 proteins. The presence of this mutation has been reported to reduce the splice potential at the normal splice site, and increase the splice score at a cryptic site three base pairs downstream [13]. The encoded protein therefore either lacks the p.Gly863 amino acid (due to cryptic splicing) or carries the missense mutation [13,14]. The p.Arg152* mutation leads to a premature stop codon in exon 5, thereby causing inactivation of the ABCA4 allele [3]. Functional analysis of the Nucleotide Binding Domain 2 of ABCA4, which contains the p.Leu2027Phe mutation, has indicated that mutations in this domain cause inhibition of ATP hydrolysis [14]. Although the c.5461–10T>C mutation has been shown not to cause aberrant splicing of exon 39 [15], the mutation may affect splicing of another exon or may affect the protein in another way. Several studies have reported this rare sequence variant as being likely to cause retinal disease or existing in linkage disequilibrium with a pathogenic mutation [13,15,16]. Furthermore, two recent studies investigating the pathogenic contributions of ABCA4 mutations reported the c.5461–10T>C mutation to be a relatively severe allele [17,18].
A study has shown that there were several possible ABCA4 founder mutations, occurring at a relatively high frequency in the South African Caucasian population [9]. More than 90% of the South African cohort interrogated by September et al. [9] was of Afrikaner descent, and 84% of the microarray cohort in the present study was of Caucasian ancestry. The Caucasian population in South Africa originated from the immigration of settlers from Europe, with the Afrikaner population originating from individuals from Holland, Germany, and France [19]. Afrikaners form a distinct, biologically homogeneous group, and several inherited diseases have been described due to founder mutations [20]. A locally developed assay that allows targeted screening of the seven common mutations in Caucasians (particularly those of Afrikaner descent) with STGD was developed to aid in diagnostic and predictive testing offered to families with AARs. To further improve the clinical utility of this diagnostic assay, a control cohort was screened to determine the carrier frequencies of the mutations in this population, thereby providing valuable risk information for genetic counseling.
Patient cohort screening
In a screening of 72 unrelated patients affected with AARs, at least one of the seven ABCA4 mutations was detected in 50 (69.44%) patients; this included 28 (38.89%) individuals in whom two mutations were identified (i.e., these individuals are presumed to be completely characterized, although it was not possible to prove the mutations were biallelic in all cases).
The p.Cys1490Tyr mutation was found to be the most frequently occurring mutation, detected in 19.44% of patient samples. This mutation was also found to be most commonly occurring in the September et al. study [9], as well as in the ABCR400 microarray analysis (Table 4), where the majority of the patients were affected with STGD. This mutation is therefore the most common in the South African STGD cohort.
Of the seven ABCA4 mutations, p.Arg152* and p.Gly863Ala occurred least frequently; each was detected in seven (9.72%) patient samples. These mutations were also found to be the least commonly detected in the September et al. study [9], as well as in the results obtained from microarray analysis using the ABCR400 chip screening. These two mutations are therefore the least commonly occurring mutations in the South African STGD cohort.
As discussed by September et al. [9], the majority of the common mutations have been found to occur at a higher frequency in the South African population compared to populations in Europe [15,21]. With the exception of the p.Gly863Ala mutation, which appears to have a founder effect in the Netherlands [13,15], the results obtained from the current study are in agreement with September et al.’s conclusions [9].
Control cohort screening
Previous research has suggested the presence of founder mutations in the South African population of Caucasian Afrikaner descent. Therefore, the Afrikaner-specific sub-cohort was of particular interest in the current study. The results obtained from the control cohort screening indicate that the carrier frequency of the p.Cys1490Tyr, p.Arg602Trp, and p.Gly863Ala mutations is slightly higher compared to the other mutations (p.Leu2027Phe, c.768G>T, and p.Arg152*), with the c.5461–10T>C mutation not detected at all. The identification of 12/269 heterozygotes in the control cohort reflected a background frequency of carriers in this population of 4.46 per 100 individuals (i.e., approximately 5% of South African Caucasian individuals may be carriers of one of the seven ABCA4 mutations screened in the present study). This is higher than other reports, estimating the carrier frequency to be between 5% and 10% for all ABCA4 mutations (n>500) tested for on the ABCR400 microarray [10]. When the control cohort is subdivided, the observed carrier frequencies were 4% and 4.8% in the Caucasian and Afrikaner sub-cohorts, respectively. However, as much as an emphasis was made to recruit “Afrikaner” controls (n=169), the Caucasian control cohort (n=100) was not selected based on excluding any “Afrikaner” ancestry, and the findings may represent an unavoidable level of “admixture.” It is tempting to speculate that if the Caucasian sub-cohort had consisted only of individuals of British or English heritage, that an even higher difference in frequency between the two subgroups might have been observed. Despite this, the results obtained could aid in genetic counseling when risk prediction is discussed with patients and their family members.
Conclusion
The assays designed for this project used the existence of seven common ABCA4 mutations in the South African population, specifically in those individuals of Afrikaner descent, and may be employed to target or direct testing for research and diagnostic purposes. These assays will potentially reduce costs to the research laboratory and patient. The incorporation of the seven mutations into a single test could be appealing in a diagnostic setting, where the test could be recommended to patients by counselors, medical geneticists, and ophthalmologists.
The carrier frequency of common ABCA4 mutations in this particular population of Caucasian/Afrikaner descent was indicated to be 4.46 per 100 individuals. This information can be used to assist counselors in relating risk to patients and their families during a genetic counseling session. Future work regarding ABCA4 mutations could involve investigating the frequencies of these seven mutations in other population groups, such as mixed ancestry and indigenous Black African, for comparison with the frequencies observed in the Caucasian/Afrikaner population.
The molecular diagnosis of an AAR can be made only if homozygous or compound heterozygous ABCA4 mutations are identified in an individual. The ABCR400 microarray and the other analysis methods presented here do not screen for previously uncharacterized mutations. Although the seven mutations were found in a relatively large proportion of South African patients screened, novel variants might also be found to be diseasing-causing in a significant proportion of individuals affected with STGD. Therefore, carefully worded, meaningful result reports must be delivered to patients by counselors, as part of the ophthalmic genetic service offered [22]. When mutations have not been proven to be biallelic (in trans) due to a lack of samples from additional family members, the reports state, “Since no parental DNA was available for verification, there is a possibility that two or all of the mutations are on the same chromosome.” Furthermore, the reports for patients screened for ABCA4 mutations state that “animal studies suggest that light exposure may accelerate vision loss; wearing sunglasses in bright light or sunshine may help protect vision; the investigators also agree that people with ABCA4-associated RDDs should avoid vitamin A supplementation” [23]. The molecular definition of AARs is a valuable part of clinical care for the affected families, as precautions that may limit disease progression should be taken until treatment options become available.
Acknowledgments
We are most grateful to the patients with AARs and their families who consented to being part of the RDD research program. The authors thank Retina South Africa, The Medical Research Council, and UCT for their contribution to funding for this research. The control samples were kindly provided by the Sports Science Institute of South Africa, the Division of Medical Biochemistry, as well as by the Division of Lipidology at UCT.
References
- 1.Allikmets R. Stargardt disease: from gene discovery to therapy. In: Tombran-Tink J, Barnstable CJ, editors. Ophthalmology research: Retinal degenerations: biology, diagnostics, and therapeutics. New Jersey: Humana Press; 2007. p.105–118. [Google Scholar]
- 2.Smit AL, van Dijk DE. Introduction to modern biology. 1st edn. Cape Town: Maskew Miller LTD.; 1970. p.506–514. [Google Scholar]
- 3.Zhang K, Kniazeva M, Hutchinson A, Han M, Dean M, Allikmets R. The ABCR gene in recessive and dominant Stargardt diseases: a genetic pathway in macular degeneration. Genomics. 1999;60:234–7. doi: 10.1006/geno.1999.5896. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan J, Gerber S, Larget-Piet D, Rozet JM, Dollfus H, Dufier JL, Odent S, Postel-Vinay A, Janin N, Briard ML, Frézal J, Munnich A. A gene for Stargardt’s disease (fundus flavimaculatus) maps to the short arm of chromosome 1. Nat Genet. 1993;5:308–11. doi: 10.1038/ng1193-308. [DOI] [PubMed] [Google Scholar]
- 5.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–46. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 6.Boon CJF, Klevering BJ, Keunen JEE, Hoyng CB, Theelen T. Fundus autofluorescence imaging of retinal dystrophies. Vision Res. 2008;48:2569–77. doi: 10.1016/j.visres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Molday LL, Rabin AR, Molday RS. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat Genet. 2000;25:257–8. doi: 10.1038/77004. [DOI] [PubMed] [Google Scholar]
- 8.Aguirre-Lamban J, Riveiro-Alvarez R, Maia-Lopes S, Cantalapiedra D, Vallespin E, Avila-Fernandez A, Villaverde-Montero C, Trujillo-Tiebas MJ, Ramos C, Ayuso C. Molecular analysis of the ABCA4 gene for reliable detection of allelic variations in Spanish patients: identification of 21 novel variants. Br J Ophthalmol. 2009;93:614–21. doi: 10.1136/bjo.2008.145193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.September AV, Vorster AA, Ramesar RS, Greenberg LJ. Mutation spectrum and founder chromosomes for the ABCA4 gene in South African patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2004;45:1705–11. doi: 10.1167/iovs.03-1167. [DOI] [PubMed] [Google Scholar]
- 10.Jaakson K, Zernant J, Külm M, Hutchinson A, Tonisson N, Glavac D, Ravnik-Glavac M, Hawlina M, Meltzer MR, Caruso RC, Testa F, Maugeri A, Hoyng CB, Gouras P, Simonelli F, Lewis RA, Lupski JR, Cremers FP, Allikmets R. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003;22:395–403. doi: 10.1002/humu.10263. [DOI] [PubMed] [Google Scholar]
- 11.Roberts LJ, Ramesar RS, Greenberg J. Clinical utility of the ABCR400 microarray: basing a genetic service on a commercial gene chip. Arch Ophthalmol. 2009;127:549–54. doi: 10.1001/archophthalmol.2009.47. [DOI] [PubMed] [Google Scholar]
- 12.Wiszniewski W, Zaremba CM, Yatsenko AN, Jamrich M, Wensel TG, Lewis RA, Lupski JR. ABCA4 mutations causing mislocalization are found frequently in patients with severe retinal dystrophies. Hum Mol Genet. 2005;14:2769–78. doi: 10.1093/hmg/ddi310. [DOI] [PubMed] [Google Scholar]
- 13.Maugeri A, van Driel MA, van de Pol DJ, Klevering BJ, van Haren FJ, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Dahl N, Brunner HG, Deutman AF, Hoyng CB, Cremers FP. The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am J Hum Genet. 1999;64:1024–35. doi: 10.1086/302323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun H, Smallwood PM, Nathans J. Biochemical defects in ABCR protein variants associated with human retinopathies. Nat Genet. 2000;26:242–6. doi: 10.1038/79994. [DOI] [PubMed] [Google Scholar]
- 15.Rivera A, White K, Stöhr H, Steiner K, Hemmrich N, Grimm T, Jurklies B, Lorenz B, Scholl HP, Apfelstedt-Sylla E, Weber BH. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am J Hum Genet. 2000;67:800–13. doi: 10.1086/303090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klevering BJ, Deutman AF, Maugeri A, Cremers FP, Hoyng CB. The spectrum of retinal phenotypes caused by mutations in the ABCA4 gene. Graefes Arch Clin Exp Ophthalmol. 2005;243:90–100. doi: 10.1007/s00417-004-1079-4. [DOI] [PubMed] [Google Scholar]
- 17.Schindler EI, Nylen EL, Ko AC, Affatigato LM, Heggen AC, Wang K, Sheffield VC, Stone EM. Deducing the pathogenic contribution of recessive ABCA4 alleles in an outbred population. Hum Mol Genet. 2010;19:3693–701. doi: 10.1093/hmg/ddq284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cideciyan AV, Swider M, Aleman TS, Tsybovsky Y, Schwartz SB, Windsor EA, Roman AJ, Sumaroka A, Steinberg JD, Jacobson SG, Stone EM, Palczewski K. ABCA4 disease progression and a proposed strategy for gene therapy. Hum Mol Genet. 2009;18:931–41. doi: 10.1093/hmg/ddn421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramesar R, September A, Rebello G, Greenberg J, Goliath R. Migratory history of populations and its use in determining research directions for retinal degenerative disorders. In: Anderson RE, LaVail MM, Hollyfield JG, editors. New insights into retinal degenerative diseases. Kluwer Academic/Plenum Publishers; 2001. p. 335–38. [Google Scholar]
- 20.Botha MC, Beighton P. Inherited disorders in the Afrikaner population of southern Africa. Part I. Historical and demographic background, cardiovascular, neurological, metabolic and intestinal conditions. S Afr Med J. 1983;64:609–12. [PubMed] [Google Scholar]
- 21.Passerini I, Sodi A, Giambene B, Mariottini A, Menchini U, Torricelli F. Novel mutations in of the ABCR gene in Italian patients with Stargardt disease. Eye (Lond) 2010;24:158–64. doi: 10.1038/eye.2009.35. [DOI] [PubMed] [Google Scholar]
- 22.Greenberg J, Roberts L, Bruwer Z, Schoeman M, Loggenberg K, Loubser F. Delivery of an ophthalmic genetic service in South Africa. South African Ophthalmology Journal. 2010;5:14–19. [Google Scholar]
- 23.Radu RA, Yuan Q, Hu J, Peng JH, Lloyd M, Nusinowitz S, Bok D, Travis GH. Accelerated accumulation of lipofuscin pigments in the RPE of a mouse model for ABCA4-mediated retinal dystrophies following Vitamin A supplementation. Invest Ophthalmol Vis Sci. 2008;49:3821–9. doi: 10.1167/iovs.07-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]