

Abstract
A convergent synthesis of highly substituted and stereodefined dihydroindanes is described from alkoxide-directed Ti-mediated cross-coupling of internal alkynes with substituted 4-hydroxy-1,6-enynes (substrates that derive from 2-directional functionalization of readily available epoxy alcohol derivatives). In addition to describing a new and highly stereoselective approach to bimolecular [2+2+2] annulation that delivers products not available with other methods in this area of chemical reactivity, evidence is provided to support annulation by way of regioselective alkyne–alkyne coupling, followed by metal-centered [4+2] rather than stepwise alkene insertion and reductive elimination. Overall, the reaction proceeds with exquisite stereochemical control and defines a convenient, convergent, and enantiospecific entry to fused carbocycles of great potential value in target-oriented synthesis and medicinal chemistry.
INTRODUCTION
Highly substituted and stereodefined hydroindanes define a structural motif that is encountered in natural and synthetic small molecules of broad pharmacological and biological relevance. Examples include complex natural products like cortistatin,1 batrachotoxin,2 ouabain3 and cephalostatin4 (Figure 1A), as well as a large collection of less functionalized steroids that are prescribed daily for pharmaceutical intervention across a diverse landscape of therapeutic areas (Figure 1B).5 Robust chemical methods based on cycloaddition,6 cation-olefin cyclization,7 and Robinson annulation8 are typically embraced as strategies of choice for the assembly of such systems, yet these venerable methods can be challenged when confronted with the goal of accessing enantiodefined, highly substituted and/or oxygenated systems.9
Figure 1.

Dihydroindanes in natural products and pharmaceuticals.
Pioneered by Vollhardt, metal-mediated [2+2+2] annulation has surfaced as a useful strategy for the synthesis of dihydroindanes.10 First demonstrated in a wholly intramolecular fashion, recent studies have begun to describe the utility of such processes in intermolecular settings,11 where the strategic benefit of convergency can dramatically enhance both the efficiency and flexibility of synthetic pathways to complex carbocyclic targets. This area of chemical reactivity, however, remains in its infancy, with intermolecular reactions being plagued by challenges associated with overcoming barriers associated with chemical reactivity (i.e. sluggish reactivity of substrates bearing substituted alkenes and/or sterically hindered internal alkynes), controlling regioselectivity (i.e. site of C–C bond formation on unsymmetrical systems) and stereoselection.12 As such, the utility of [2+2+2] annulation in target-oriented synthesis has been broadly constrained to the world of intramolecular cycloisomerization chemistry.10
In the context of a program aimed at the control of intermolecular metallacycle-mediated C–C bond formation, we have been actively engaged in the study of alkoxide-directed approaches to achieve fine control of metallacycle reactivity. These studies have led to the description of new intermolecular or convergent coupling reactions that have overcome the substantial limitations associated with prior art [including the difficulty in achieving sufficient reactivity and selectivity to accomplish bimolecular union of differentially substituted internal alkynes (1 + 2 → 3; Figure 2A)13 and the cross-coupling of internal alkynes with substituted alkenes (4 + 2 → 5; Figure 2B)14]. Here, we describe a general solution to the asymmetric synthesis of densely functionalized hydroindanes that builds on these earlier successes. In short, we report a highly stereoselective alkoxide-directed Ti-mediated intermolecular annulation between readily available 4-hydroxy-1,6-enynes and internal alkynes as a strategy to access densely functionalized hydroindanes (Figure 2C). This reaction, while related to previous metallacycle-mediated transformations (i.e. [2+2+2] annulation chemistry), is to our knowledge the first that is capable of delivering stereodefined angularly substituted hydroindanes in a convergent and highly selective manner using unsymmetric and non-tethered reaction partners.
Figure 2.

Metallacycle-mediated cross-coupling of alkenes and alkynes.
As depicted in Figure 3, Ti-alkoxide-based methods have been described for intermolecular [2+2+2], yet these methods yield substituted aromatics15 (Figure 3A) or nearly symmetric cyclohexadienes16 (Figure 3B). In this latter case, intramolecular diyne cyclization delivers a symmetrical metallacyclopentadiene that is converted to a carbocyclic product by way of a process that avoids any challenges with regard to regioselection. Other metallacycle-mediated chemistry has been reported to access angularly substituted hydroindane products as depicted in Figures 3C and 3D.10c, 17 These methods represent impressive demonstrations of intramolecular chemistry but, like many powerful modes of reactivity suitable for intramolecular C–C bond formation, these metallacycle-mediated bond constructions have not been demonstrated to be similarly useful in intermolecular settings. Given the significance of convergency in realizing step-economical pathways to complex molecules, this restriction to intramolecularity marks a significant limitation in the synthetic utility of these latter methods.
Figure 3.

Established metallacycle-mediated annulation chemistry of relevance to the present advance.
In short, the chemistry described herein marks a substantial advance in metallacycle-mediated annulation chemistry, offering a reaction process that enables bimolecular [2+2+2] annulation as an entry to complex angularly substituted hydroindanes – structural motifs that are encountered in a diverse array of natural products and synthetic molecules of pharmacological relevance.
RESULTS
Initial investigation of intermolecular metallacycle-mediated coupling chemistry was focused on the union of internal alkynes with notoriously sluggish reaction partners – i.e. other internal alkynes and substituted alkenes. While a number of metallacycle-mediated reactions of this ilk are known, all available transformations are substantially limited in substrate scope. While reductive cross-coupling of alkynes is typically accomplished with one terminal alkyne,18 related union of internal alkynes with substituted alkenes has remained useful in only the simplest of cases – i.e. with unhindered terminal mono-substituted alkenes.19 In fact, from a broader perspective, few reactions in organic chemistry are suitable for the regioselective carbometalation of an electronically unactivated alkene or alkyne; especially if such systems are 1,2-disubstituted.
In an attempt to overcome the well established barrier to bimolecular reactivity of preformed metal–alkyne complexes with other electronically unactivated π-systems, we speculated that an alkoxide-directed approach as depicted in Figures 4A and 4B may offer a convenient solution. In this way, rapid and reversible ligand exchange would serve as a mechanism to render the carbometalation reaction intramolecular, and provide a means for stepwise encapsulation of the metal center by an orchestrated sequence of metal–ligand, metal–carbon, and carbon–carbon bond-forming processes. Importantly, in addition to providing a pathway to overcome the barriers to reactivity associated with scores of potential metallacycle-mediated cross-coupling processes, this strategy would define a mechanism to control regioselection that is distinct from established processes based on steric or electronic differentiation of the reacting π-systems.
Figure 4.

Alkoxide-directed alkyne–alkyne and alkene–alkyne reductive cross-coupling.
To accomplish such a transformation, we accepted that catalytic versions of metallacycle-mediated coupling would not be reasonable as a general solution to the problem. This conclusion derived from the expected difficulty of favoring cross-coupling over homodimerization in a system where both reactive π-systems are available (in large excess) to the catalytic metal center. Also, we aimed to achieve ligand-directed C–C bond formation with a ubiquitous free hydroxy group rather than with phosphines, thioethers, or terminal alkenes.20 The combination of these factors led to the selection of established and inexpensive Ti(IV) alkoxides as the metal of choice to accomplish alkoxide-directed metallacycle-mediated cross-coupling as generalized in Figures 4B and 4C. In addition to Ti(Oi-Pr)4 being easy to handle and purify in the absence of a glovebox, this reagent is inexpensive, non-toxic, and on aqueous workup delivers byproducts that are easily removed from product mixtures (TiO2, i-PrOH, and MgX2 salts–from the RMgX-mediated reduction of the Ti(IV) alkoxide). For these reasons, we moved forward with the development of stoichiometric Ti(IV)-mediated alkoxide-directed metallacycle-mediated cross-coupling chemistry.
As summarized in Figure 5, this strategy for the control of metallacycle-mediated C–C bond formation proved to be quite effective and general.13, 14 In the case of alkyne–alkyne coupling (Figure 5A), successful reactions were achieved with homopropargylic and bis-homopropargylic alcohols, delivering 1,3-diene products as single regio- and stereoisomers. In the case of products 9 and 10, the regioselectivity reported (≥ 42:1 and ≥ 65:1) is based on the signal-to-noise ratio of each 1H NMR spectrum of the crude product mixture, as no evidence could be found for presence of the other regioisomeric product. Symmetrical non-conjugated alkynes are also viable coupling partners in this diene synthesis (→ 11; → 12). Further, while demonstrating a powerful means to overcome the barriers associated with reactivity for the cross-coupling process, this directing group strategy proved quite effective for overriding steric effects in the site-selective C–C bond forming process (i.e. for 13–15, C–C bond formation occurs at the site distal to the free hydroxyl, independent of the nature of the proximal substituent – Et, i-Pr, t-Bu).
Figure 5.

Examples of alkoxide-directed reductive cross-coupling.
This general strategy also proved to be effective in coupling substrates that contain a hindered secondary alcohol directing group (i.e. → 16). And, most interestingly, coupling of two unsymmetrical alkyne coupling partners proved to be highly selective, delivering tetrasubstituted 1,3-dienes 17 and 18 as single isomers. Here, exquisite regioselectivity is observed in the carbometalation of both π-systems. This observation was a welcomed surprise, as related coupling reactions of internal alkynes with carbonyl electrophiles do not typically proceed with such high levels of regioselection.21 While the mechanistic origins of this phenomenon remain to be determined, we suspect that the transition states associated with the present coupling reactions are significantly more product-like, as one would suspect that these processes are less exothermic than the analogous reactions of metal–alkyne complexes with aldehydes. As such, the closer distance between the reactive loci in the transition state should result in an enhancement of regioselection due to steric differences between the alkyne termini.
We note that these reductive cross-coupling reactions uniformly proceed with exceptional levels of selectivity, albeit in modest yield (51–68%). This compromise is associated with the frequent inability to push these reactions to completion and/or the observation of homodimer derived from the alkynyl alcohol coupling partner. This second observation likely arises from the use of a slight excess of Ti(Oi-Pr)4 to efficiently convert the first alkyne coupling partner (2.5 eq) to the metal–alkyne complex.
Moving on to the more demanding coupling reactions between alkynes and alkenes, we were delighted to find that the general approach taken with alkyne–alkyne coupling was also effective here. As illustrated in Figure 5B, a range of products can be generated from the union of internal alkynes with: (1) terminal-(→ 19), (2) (E)-disubstituted-(→ 20), (3) (Z)-disubstituted-(→ 20), and (4) 1,1-disubstituted alkenes (→ 21). In all cases, the site of C–C bond formation is completely controlled by the position of the free hydroxy group in the homoallylic alcohol coupling partners. Further, as seen in the alkyne–alkyne coupling process, the union of unsymmetrical alkynes with unsymmetrical homoallylic alcohols also proceeds with exquisite levels of regioselection with respect to both reacting π-systems (→ 25 and 26). In these final cases, we observed a general lack of influence of double asymmetric relationships on the stereochemical course of bond formation.
With a firm foundation of preliminary data that confirms the ability to employ a hydroxy-group directing strategy to enable intermolecular metallacycle-mediated union of preformed metal–alkyne complexes with internal alkynes or substituted alkenes, we moved on to investigate the inherent reactivity of substrates that could react by either mode of reactivity in isolation, or by a process that engaged all three π-systems in an annulation process.
As illustrated in Figure 6A, preformation of a Ti-alkyne complex of 28 (Ti(Oi-Pr)4, c-C5H9MgCl, Et2O, −78 to −30 °C), followed by addition of the lithium alkoxide of an enyne coupling partner (27) may result in either: 1) alkene–alkyne coupling (to deliver A),14 2) alkyne–alkyne coupling (to furnish B),13 or 3) [2+2+2] annulation (to produce the functionalized carbocycle C).15, 16 As illustrated in Scheme 6B, alkene substitution plays a dominant role in affecting product distribution.22 If the alkene is unsubstituted, reaction proceeds by alkene–alkyne coupling to deliver 30 (eq 1). Alternatively, substrates that contain terminal substitution on the alkene react by alkyne–alkyne coupling (eqs 2 and 3). Finally, a substrate bearing a simple 1,1-disubstituted alkene (35) undergoes reaction by yet a different course – here, convergent union engages all three π-systems and proceeds by way of a highly stereoselective [2+2+2] annulation, delivering the functionalized dihydroindane product 36 in 83% yield as a single stereoisomer.
Figure 6.

Path selectivity in alkoxide-directed metallacycle-mediated cross-coupling. Reaction conditions: (a) (PMBOCH2CH2C)2, Ti(Oi-Pr)4, c-C5H9MgCl, (−78 to −30 °C), then add enyne as the corresponding Li-alkoxide (−30 °C), b. Identical procedure as described for “a”, except that this reaction was warmed to 0 °C – If this reaction (35 → 36) is quenched at −30 °C, little change in overall yield was observed (77%). For entries 2 and 3, if these reactions were warmed to 0 °C prior to quenching, no hydroindane could be identified from the product mixture.
At this point, we speculated that the unique sequence of bond-forming events inherent to these alkoxide-directed coupling reactions that was previously described as a “stepwise encapsulation of the metal center by metal–ligand, metal–carbon, and carbon–carbon bond-forming reactions” may define a powerful new direction for the control of metal-centered [2+2+2] chemistry. Based on this perspective, we pursued the study of regio- and stereoselection in this unique annulation process in hopes of achieving a robust strategy for the synthesis of densely functionalized and stereodefined hydroindane systems that are not accessible with current [2+2+2] annulation methods.
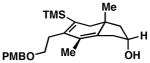
As illustrated in Table 1, a range of 4-hydroxy-1,6-enynes and substituted alkynes are effective coupling partners in this stereoselective annulation process. Further, this reaction proceeds in an enantiospecific fashion.23a As depicted in entry 1, union of the optically active enyne 37 (95% ee) with Me2PhSi-alkyne 38 occurs in 74% yield to deliver a mixture of regioiomeric hydroindane products, the major isomer of which is the stereodefined hydroindane 39 (95% ee; dr ≥ 20:1). While regiochemical control in the functionalization of alkyne 38 was not particularly high in this case (rs = 2:1), the stereochemically-defined product 39 possesses substitution of significance in the context of de novo steroid synthesis [see C8, C9 and C11 (steroid numbering)]. In efforts to explore the scope of this annulation process, we observed enhancements in regioselection as a function of the coupling partners employed. First, regioselection was seen to increase slightly when using the TMS-substituted alkyne 41 that contains an aliphatic substituent (CH2CH2OPMB) rather than Ph (as in 38). Coupling of 40 with alkyne 41 proceeds in 75% yield to deliver a 3:1 mixture of regioisomeric carbocycles, the major isomer of which is the hydroindane product 42 (dr ≥ 20:1; entry 2). Regioselection also improved in the coupling reaction of TMS-substituted enyne 44 with 4-hydroxy-1,6-enyne 43 (entry 3). Here, the annulation event proceeds with 4:1 regioselection, ≥ 20:1 diastereoselection, and the major isomer 45 can be isolated in 53% yield as a single isomer. Finally, when the alkene of the 4-hydroxy-1,6-enyne possesses a more sterically demanding substituent (as compared to Me), regioselection also increases (entry 4). Here, union of the benzyl-substituted substrate 46 with TMS-alkyne 47 proceeds with 5:1 regioselection, ≥ 20:1 diastereoselection, and the major product 48 can be isolated in 55% yield as a single isomer.23b While in entries 3 and 4 of Table 1, the major isomer could be separated from the minor regioisomer, it is important to note that in all cases, purification of the major isomer is facile after chemoselective protodesilylation of the major products’ vinylsilane (See SI for details).
Table 1.
Initial studies regarding regioselection and enantio-specificity.
 | |||||
|---|---|---|---|---|---|
| entry | enyne | alkyne | yield (%) | dr rs |
producta,b |
| 1 |
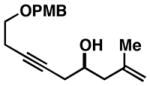 37 (95% ee) |
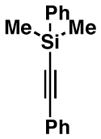 38 |
74c | ≥20:1 2:1 |
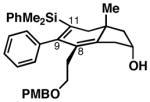 39 (95% ee) |
| 2 |
40 |
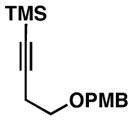 41 |
75c | ≥20:1 3:1 |
 42 |
| 3 |
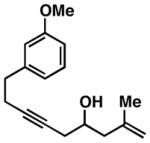 43 |
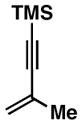 44 |
53d | ≥20:1 4:1 |
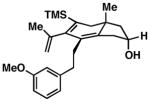 45 |
| 4 |
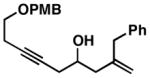 46 |
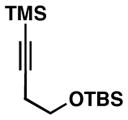 47 |
55d | ≥20:1 5:1 |
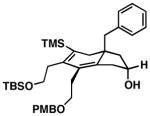 48 |
Reaction conditions:
alkyne, Ti(Oi-Pr)4, c-C5H9MgCl, toluene (−78 to −30 °C).
cool to −78 °C and add lithium alkoxide of enyne, warm to a final temperature of between −15 and 23 °C.
yield reported is for the mixture of regioisomers.
yield reported is for the major regioisomer. See Supporting Information for additional information.
Notably, this annulation process can be employed to generate hydroindane products possessing C17 β-substitution (Table 2). As depicted in entry 1, coupling of enyne 49 with alkyne 50 results in the formation of hydroindane products possessing a C17 β-Me substituent in 63% yield and ≥20:1 diastereoselection, albeit as a 3:1 mixture of regioisomers. Interestingly, when exploring the utility of this annulation process for the synthesis of hydroindanes possessing C17 β-aryl substitution (a prominent architectural feature of the cortistatins), we discovered that this process can proceed with exquisite regiochemical control. As illustrated in entry 2, coupling of hydroxy-enyne 52 with alkyne 41 delivers hydroindane product 53 in 64% yield, with ≥ 20:1 ds and 13:1 rs. While the mechanistic origin of this long-range effect on regioselection remains unclear, all enyne substrates bearing an allylic Ph-substituent underwent Ti-mediated annulation reactions with high levels of regioselectivity (typically ≥ 20:1), and consistently exquisite levels of diastereoselection (uniformly ≥ 20:1) (entries 2–7).
Table 2.
C17 β-aryl substitution and regioselection.
 | |||||
|---|---|---|---|---|---|
| entry | enyne | alkyne | yield (%) | dr rs |
producta,b |
| 1 |
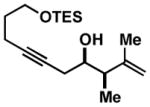 49 |
50 |
63c | ≥20:1 3:1 |
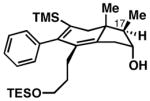 51 |
| 2 |
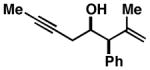 52 |
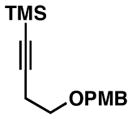 41 |
64 | ≥20:1 13:1 |
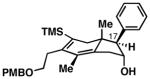 53 |
| 3 |
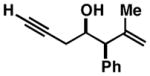 54 |
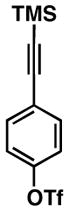 55 |
52 | ≥20:1 ≥20:1 |
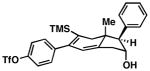 56 |
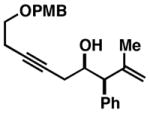
|
|
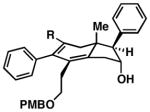
|
|||
| 4 | 57 |
38 R = SiMe2Ph |
57 | ≥20:1 ≥20:1 |
58 R = SiMe2Ph |
| 5 | 57 |
50 R = SiMe3 |
57 | 17:1 ≥20:1 |
59 R = SiMe3 |
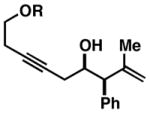
|
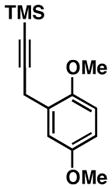
|
|
|||
| 6 |
57 R = PMB |
60 | 54 | ≥20:1 ≥20:1 |
61 R = PMB |
| 7 |
62 R = TBS |
60 | 54 | ≥20:1 ≥20:1 |
63 R = TBS |
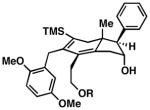
| 8 |
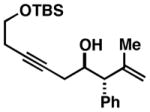 64 |
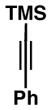 50 |
22 | –d –d |
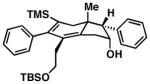 65 |
Reaction conditions:
alkyne, Ti(Oi-Pr)4, c-C5H9MgCl, toluene (−78 to −30 °C).
cool to −78 °C and add lithium alkoxide of enyne, warm to a final temperature of between −15 and 23 °C.
yield reported is for the mixture of regioisomers.
Precise selectivities could not be determined from the 1H NMR spectrum of the crude product – hydroindane product 64 was isolated as a single regio- and stereoisomer. See Supporting Information for additional information
While this annulation process is quite effective at generating 17-β-aryl substituted hydroindanes as essentially single regio and stereoisomers (i.e. 53, 56, 58, 59, 61 and 63), the process is sensitive to the relative stereochemistry of the enyne starting material. As illustrated in entry 8 of Table 2, annulation of enyne 64 with TMS-phenylacetylene (50) produced a complex mixture of products from which only 22% yield of the C17 α-substituted hydroindane product (65) could be obtained. While this stereochemical pattern certainly represents a preparative limitation to the current chemistry, product 65 could easily be isolated as a single regio- and stereoisomer.
Mechanistic Hypothesis
Metal-centered [2+2+2] annulation chemistry has long been proposed to occur by initial formation of a metallacyclopentadiene, followed by either:
Figure 7.

Potential pathways for metal-centered annulation.
In our efforts to explore the scope of this Ti-mediated annulation, we have come upon an experiment that distinguishes between these two pathways, and further provides evidence in support of a sequential [4+2]/cycloreversion in preference to alkene insertion/reductive elimination.
As illustrated in Figure 8A, coupling of enyne 66 with alkyne 55 delivers a carbocyclic product (67) that lacks the PMB ether of the starting material and contains an exocyclic alkene. This observation is consistent with the empirical model depicted in Figure 8B, where initial alkyne–alkyne coupling delivers metallacyclopentadiene 68 in a regioselective fashion.13 Subsequent stereoselective annulation is then proposed to occur through a sequence of steps that includes alkoxide-promoted cleavage of the oxametallacyclopentane (68 → 69), followed by stereoselective intramolecular [4+2] (69 → 70) — a highly stereoselective process that is thought to be controlled by intramolecular coordination of the pendant alkoxide to a ligand on Ti. Unlike previous examples depicted in Table 1, intermediate 70 is suitably functionalized to undergo vinylogous elimination of the PMB ether to generate an unstable tertiary allyltitanium species 71. Finally, 1,3-metallotropic shift generates allyltitanium species 72 which, on protonolysis with allylic transposition, is poised to deliver the observed product 67. Alternatively, if annulation proceeded by a sequence defined by alkene insertion and reductive elimination (Scheme 7B and Figure 8C), a suitable intermediate is not readily apparent to support the facile elimination of the PMB ether [i.e. 73 (Figure 8B) vs. 70 (Figure 8C)].24
Figure 8.

Development of an empirical model for the Ti-mediated intermolecular hydroindane synthesis.
Overall, alkoxide-directed intermolecular [2+2+2] annulation between an internal alkyne and a 1,6-enyne defines a unique and powerful approach to controlling regio- and stereoselection in this class of chemical reactivity. The virtues of stepwise encapsulation of the metal center (through a process of sequential ligand exchange and intramolecular carbometalation) set the stage for a highly controlled series of bond forming events en route to a functionalized carbocyclic system of great potential utility in natural product synthesis. Further, the mechanistic course of this process (which is more consistent with [4+2]/cycloreversion than alkene insertion/reductive elimination) may afford the opportunity to divert chemical reactivity away from the expected [2+2+2] products (i.e. Table 1) to stereodefined products that take advantage of the unique reactivity of bridged polycyclic allylic metal intermediate (as demonstrated in Figure 8A).
Utility of the carbocyclic products
The present annulation process delivers complex carbocycles that can be differentially functionalized to address a range of common substitution patterns about the hydroindane core of natural products and small molecules of pharmacological relevance. As depicted in Scheme 6, deoxygenation of the D-ring hydroxy group proceeds readily by the Barton procedure (61 → 75).25 Alternatively, C11 functionality (steroid numbering) can easily be introduced by protodesilylation26 and hydroboration (61→ 76). Finally, a sequence of oxidation, isomerization and protodesilylation is suitable to access a CD-ring system bearing a dienone (59 → 77). While certainly not comprehensive, the transformations depicted in Scheme 6 serve to introduce the great potential utility of the functionalized dihydroindane products derived from the Ti-mediated annulation reaction in the stereoselective synthesis of complex carbocycles.
CONCLUSION
We have described a convenient and highly stereoselective pathway to a range of functionalized hydroindanes by the union of simple 4-hydroxy-1,6-enynes with silyl-substituted alkynes. Since the enyne-containing starting materials are easily derived from two-directional functionalization of chiral epoxy alcohol derivatives (including epichlorohydrin), the synthetic pathway secured is also amenable to the production of optically active hydroindanes that bear a variety of functionality of likely utility in target-oriented synthesis. While mechanistic ambiguity has traditionally surrounded related [2+2+2] annulation chemistry, our studies have led to the conclusion that sequential [4+2] cycloaddition/cycloreversion is likely operative in preference to alkene insertion/reductive elimination. From a preparative perspective, the convergent annulation described is efficient (isolated yields ranging from 48–83%), regioselective (up to ≥ 20:1), and highly stereoselective (dr is uniformly ≥ 20:1). We are unaware of another intermolecular metallacycle-mediated annulation reaction capable of delivering stereodefined products of related structure to that observed here — structural motifs that are core skeletal components of a vast array of pharmacologically active natural and synthetic substances. Based on the considerations discussed, the current science defines a significant advance in metallacycle-mediated annulation chemistry, and marks the establishment of an exceptionally concise means for the convergent assembly of densely functionalized hydroindanes. As such, we anticipate that the current findings will be of great potential utility in chemical synthesis and medicinal chemistry.
Supplementary Material
Figure 9.

Some functionalization processes suitable for elaboration of the dihydroindane products.
Acknowledgments
We gratefully acknowledge financial support of this work by the National Institutes of Health –NIGMS (GM80266 and GM80266-04S1). The authors also acknowledge Hayley Browdy for assistance in synthesizing the panel of enynes depicted in Figure 6B.
Footnotes
SUPPORTING INFORMATION PARAGRAPH:
Experimental procedures and tabulated spectroscopic data for new compounds (PDF) are available free of charge via the Internet at http://pubs.acs.org/paragonplus/submission/jacsat/.
References
- 1.Narayan ARH, Simmons EM, Sarpong R. Eur J Org Chem. 2010:3553–3567.Flyer AN, Si C, Myers AG. Nature Chem. 2010;2:886–892. doi: 10.1038/nchem.794.Aoki S, Watanabe Y, Sanagawa M, Setiawan A, Kotoku N, Kobayashi M. J Am Chem Soc. 2006;128:3148–3149. doi: 10.1021/ja057404h.Shenvi RA, Guerrero CA, Shi J, Li CC, Baran PS. J Am Chem Soc. 2008;130:7241–7243. doi: 10.1021/ja8023466.Lee HM, Nieto-Oberhuber C, Shair MD. J Am Chem Soc. 2008;130:16864–16866. doi: 10.1021/ja8071918.Nicolaou KC, Peng XS, Sun YP, Polet D, Lim CS, Chen DYK. J Am Chem Soc. 2009;131:10587–10597. doi: 10.1021/ja902939t.For an approach to the core skeleton of the cortistatins by coupling of a functionalized hydroindane with an aromatic A-ring precursor, see: Dai M, Danishefsky SJ. Tetrahedron Lett. 2008;49:6610–6612. doi: 10.1016/j.tetlet.2008.09.018.
- 2.(a) Daly JW, Witkop B, Bommer P, Biemann K. J Am Chem Soc. 1965;87:124–126. doi: 10.1021/ja01079a026. [DOI] [PubMed] [Google Scholar]; (b) Dumbachaer JP, Beehler BM, Spande TF, Garraffo HM, Daly JW. Science. 1992;258:799–801. doi: 10.1126/science.1439786. [DOI] [PubMed] [Google Scholar]; (c) Tokuyama T, Daly J, Witkop B. J Am Chem Soc. 1969;91:3931–3938. doi: 10.1021/ja01042a042. [DOI] [PubMed] [Google Scholar]; (d) Albuquerque EX, Daly JW, Witkop B. Science. 1971;172:995–1002. doi: 10.1126/science.172.3987.995. [DOI] [PubMed] [Google Scholar]; (e) Kurosu M, Marcin LR, Grinsteiner TJ, Kishi Y. J Am Chem Soc. 1998;120:6627–6628. [Google Scholar]
- 3.(a) Prassas I, Diamandis EP. Nat Rev Drug Discov. 2008;7:926–935. doi: 10.1038/nrd2682. [DOI] [PubMed] [Google Scholar]; (b) Newman RA, Yang P, Pawlus AD, Block KI. Mol Interv. 2008;8:36–49. doi: 10.1124/mi.8.1.8. [DOI] [PubMed] [Google Scholar]; (c) Riganti C, Campia I, Kopecka J, Gazzano E, Doublier S, Aldieri E, Bosia A, Ghigo D. Curr Med Chem. 2011;18:872–885. doi: 10.2174/092986711794927685. [DOI] [PubMed] [Google Scholar]; (d) Overman LE, Rucker PV. Heterocycles. 2000;52:1297–1314. [Google Scholar]; (e) Reddy MS, Zhang H, Phoenix S, Deslongchamps P. Chem Asian J. 2009;4:725–741. doi: 10.1002/asia.200800429. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lee S, LaCour TG, Fuchs PL. Chem Rev. 2009;109:2275–2314. doi: 10.1021/cr800365m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) LaCour TG, Guo C, Bhandaru S, Boyd MR, Fuchs PL. J Am Chem Soc. 1998;120:692–707. [Google Scholar]; (c) Moser BR. J Nat Prod. 2008;71:487–491. doi: 10.1021/np070536z. [DOI] [PubMed] [Google Scholar]; (d) Rudy A, Lopez-Anton N, Dirsch VM, Vollmar AM. J Nat Prod. 2008;71:482–486. doi: 10.1021/np070534e. [DOI] [PubMed] [Google Scholar]; (e) Fortner KC, Kato D, Tanaka Y, Shair MD. J Am Chem Soc. 2010;132:275–280. doi: 10.1021/ja906996c. [DOI] [PubMed] [Google Scholar]
- 5.Zeleen FJ. Principles of Medical Biology, Volume 8B, Molecular and Cellular Pharmacology. Stamford: JAI Press, Inc; 1997. Medicinal Chemistry of Steroids; pp. 427–463.For reviews on the synthesis of steroids, see: Chapelon AS, Moraleda D, Rodriguez R, Ollivier C, Santelli M. Tetrahedron. 2007;63:11511–11616.Jankowski P, Marczak S, Wicha J. Tetrahedron. 1998;54:12071–12150.
- 6.(a) Parker KA, Iqbal T. J Am Chem Soc. 1982;47:337–342. [Google Scholar]; (b) Wilson SR, Jacob L. J Org Chem. 1992;57:4380–4385. [Google Scholar]; (c) Wender PA, Smith TE. Tetrahedron. 1998;54:1255–1275. [Google Scholar]; (d) Taber TF, Song Y. J Org Chem. 1996;61:7508–7512. doi: 10.1021/jo9610974. [DOI] [PubMed] [Google Scholar]
- 7.(a) Snider BB, Kirk TC. J Am Chem Soc. 1983;105:2364–2368. [Google Scholar]; (b) Johnson WS, Elliot JD, Hanson GJ. J Am Chem Soc. 1984;106:1138–1139. [Google Scholar]; (c) Hatakeyama S, Numata H, Osanai K, Takano S. J Chem Soc, Chem Commun. 1989:1893–1895. [Google Scholar]; (d) Browder CC, West FG. Synlett. 1999;9:1363–1366. [Google Scholar]
- 8.(a) Hajos ZG, Parrish DR. J Org Chem. 1974;39:1612–1615. [Google Scholar]; (b) Stork G, Shiner CS, Winkler JD. J Am Chem Soc. 1982;104:310–312. [Google Scholar]; (c) Stork G, Winkler JD, Shiner CS. J Am Chem Soc. 1982;104:3767–3768. [Google Scholar]; (d) Yamamoto K, Iijima M, Ogimura Y, Tsuji J. Tetrahedron Lett. 1984;25:2813–2816. [Google Scholar]; (e) Kotoku N, Sumii Y, Hayashi T, Kobayashi M. Tetrahedron Lett. 2008;49:7078–7081. [Google Scholar]
- 9.For example, see: References 1d, 5b, 5c, 6b, 6d, 7b, 7c, 8d, 8e and Corey EJ, Huang AX. J Am Chem Soc. 1999;121:710–714.Rychnovsky SD, Mickus DE. J Org Chem. 1992;57:2732–2736.Clasby MC, Craig D, Jaxa-Chamiec AA, Lai JYQ, Marsh A, Slawin AMZ, White AJP, Williams DJ. Tetrahedron. 1996;52:4769–4802.Nemoto H, Matsuhashi N, Imaizumi M, Nagai M, Fukumoto K. J Org Chem. 1990;55:5625–5631.
- 10.Sternberg E, Vollhardt KPC. J Am Chem Soc. 1980;102:4839–4841.Sternberg E, Vollhardt KPC. J Org Chem. 1982;47:3447–3450.Clinet JC, Duñach E, Vollhardt KPC. J Am Chem Soc. 1983;105:6710–6712.Butenschön H, Winkler M, Vollhardt KPC. J Chem Soc, Chem Commun. 1986:388–390.Grotjahn DB, Vollhardt KPC. J Am Chem Soc. 1986;108:2091–2093.Germanas J, Aubert C, Vollhardt KPC. J Am Chem Soc. 1991;113:4006–4008.Johnson EP, Vollhardt KPC. J Am Chem Soc. 1991;113:381–382.For reviews, see: Vollhardt KPC. Acc Chem Res. 1977;10:1–8.Aubert C, Buisine O, Petit M, Slowinski F, Malacria M. Pure Appl Chem. 1999;71:1463–1470.Rubin M, Sromek AW, Gevorgyan V. Synlett. 2003:2265–2291.Gandon V, Aubert C, Malacria M. Chem Commun. 2006:2209–2217. doi: 10.1039/b517696b.. For examples of catalytic asymmetric intramolecular [2+2+2] annulation, see: Shibata T, Kurokawa H, Kanda K. J Org Chem. 2007;72:6521–6525. doi: 10.1021/jo070762d.Shibata T, Tahara Y. J Am Chem Soc. 2006;128:11766–11767. doi: 10.1021/ja0639160.For recent reviews of [2+2+2] annulations, see: Shibata T, Tsuchikama K. Org Biomol Chem. 2008;6:1317–1323. doi: 10.1039/b720031e.Tanaka K. Chem Asian J. 2009;4:508–518. doi: 10.1002/asia.200800378.Leboeuf D, Gandon V, Malacria M. Transition Metal-Mediated [2+2+2] Cycloaddtions. In: Ma S, editor. Handbook of Cyclization Reactions. Vol. 1. Wiley–VCH; Weinheim: 2009. p. 367.
- 11.(a) Evans PA, Lai KW, Sawyer JR. J Am Chem Soc. 2005;127:12466–12467. doi: 10.1021/ja053123y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shibata T, Arai Y, Tahara Y. Org Lett. 2005;7:4955–4957. doi: 10.1021/ol051876j. [DOI] [PubMed] [Google Scholar]
- 12.For recent reviews that describe these challenges, see: Reichard HA, McLaughlin M, Chen MZ, Micalizio GC. Eur J Org Chem. 2010:391–409. doi: 10.1002/ejoc.200901094.Reichard HA, Micalizio GC. Chem Sci. 2011;2:573–589. doi: 10.1039/C0SC00394H.
- 13.Ryan J, Micalizio GC. J Am Chem Soc. 2006;128:2764–2765. doi: 10.1021/ja057352w. [DOI] [PubMed] [Google Scholar]
- 14.(a) Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440–1443. doi: 10.1002/anie.200603515. [DOI] [PubMed] [Google Scholar]; (b) Canterbury DP, Micalizio GC. J Am Chem Soc. 2010;132:7602–7604. doi: 10.1021/ja102888f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki D, Urabe H, Sato F. J Am Chem Soc. 2001;123:7925–7926. doi: 10.1021/ja0161913.Suzuki D, Tanaka R, Urabe H, Sato F. J Am Chem Soc. 2002;124:3518–3519. doi: 10.1021/ja017766z.Tanaka R, Yuza A, Watai Y, Suzuki D, Takayama Y, Sato F, Urabe H. J Am Chem Soc. 2005;127:7774–7780. doi: 10.1021/ja050261e.For earlier examples demonstrating the ability of Ti-aryloxides to promote [2+2+2] annulation en route to substituted aromatics, see: Hill JE, Balaich G, Fanwick PE, Rothwell IP. Organometallics. 1993;12:2911–2924.
- 16.Sung MJ, Pang JH, Park SB, Cha JK. Org Lett. 2003;5:2137–2140. doi: 10.1021/ol034592c.For early examples demonstrating the ability of Ti-aryloxides to generate cyclohexadienes from the coupling of two alkynes and one alkene, see: Balaich GJ, Rothwell IP. J Am Chem Soc. 1993;115:1581–1583.Johnson ES, Balaich GJ, Rothwell IP. J Am Chem Soc. 1997;119:7685–7693.
- 17.Wender PA, Smith TE. Tetrahedron. 1998;54:1255–1275. [Google Scholar]
- 18.Hamada T, Suzuki D, Urabe H, Sato F. J Am Chem Soc. 1999;121:7342–7344.(b) see Ref. 15cBuchwald SL, Watson BT, Buchwald SL, Nielsen RBJ. Am Chem Soc. 1989;111:2870–2874.Shimp HL, Micalizio GC. Org Lett. 2005;7:5111–5114. doi: 10.1021/ol052241n.Perez LJ, Shimp HL, Micalizio GC. J Org Chem. 2009;74:7211–7219. doi: 10.1021/jo901451c.For Zr-mediated cross-coupling of two different internal, yet symmetrical, alkynes, see: Xi Z, Hara R, Takahashi R. J Org Chem. 1995;60:4444–4448.
- 19.For addition reactions of preformed benzyne–ZrCp2 complexes to simple alkenes, see: Aoki K, Peat AJ, Buchwald SL. J Am Chem Soc. 1998;120:3068–3073.For addition reactions of preformed Ti–vinylsilane complexes to alkynes, see: Mizojiri R, Urabe H, Sato F. J Org Chem. 2000;65:5217–6222. doi: 10.1021/jo000925x.For a review of Co-catalyzed coupling chemistry of alkynes with alkenes, see: Jeganmohan M, Cheng C-H. Chem Eur J. 2008;14:10876–10886. doi: 10.1002/chem.200800904.
- 20.For a review of directed reactions in organic chemistry, see: Hoveyda AH, Evans DA, Fu GC. Chem Rev. 1993;93:1307–1370.For a recent example of an alkene-directed metallacycle-mediated coupling, see: Moslin RM, Miller KM, Jamison TF. Tetrahedron. 2006;62:7598–7610.For examples of heteroatom-directed intermolecular Pauson–Khand reactions, see: Gibson SE, Mainolfi N. Angew Chem Int Ed. 2005;44:3022–3037. doi: 10.1002/anie.200462235.
- 21.(a) Harada K, Urabe H, Sato F. Tetrahedron Lett. 1995;36:3203–3206. [Google Scholar]; (b) Bahadoor AB, Flyer A, Micalizio GC. J Am Chem Soc. 2005;127:3694–3695. doi: 10.1021/ja050039+. [DOI] [PubMed] [Google Scholar]; (c) Bahadoor AB, Micalizio GC. Org Lett. 2006;8:1180–1184. doi: 10.1021/ol0600786. [DOI] [PubMed] [Google Scholar]
- 22.For an example of how alkene substitution plays a significant role in dictating site selectivity in the reductive cross-coupling of 1,5-dienes with alkynes, see: Diez PS, Micalizio GC. J Am Chem Soc. 2010;132:9576–9578. doi: 10.1021/ja103836h.
- 23.(a) See the Supporting Information for the opposite antipode – a demonstration that supports the enantiospecific nature of the process. (b) Overall, isolated yields of the annulation products typically range from 52–83%. These less than quantitative yields reflect: (i) incomplete consumption of the enyne, (ii) alkyne–alkyne coupling without annulation (the product of this is a functionalized triene – i.e. Figure 6B, eqs 2 and 3), and occasionally (iii) partial deprotection of the PMB/silyl ether of the annulation product.
- 24.Metallacycle-mediated [2+2+2] annulation is generally thought to proceed through initial formation of a metallacyclopentadiene followed by either alkene insertion/reductive elimination or [4+2]/cycloreversion. We have opted to invoke a model based on [4+2]/cycloreversion, where stereochemistry is controlled by a coordination event between the homoallylic alkoxide and an alkoxide bound to Ti. This proposition does not preclude the involvement of an alkene insertion mechanism, and only intends to offer an empirical model to predict the stereochemical course of annulation and that is consistent with the observations summarized in Figure 8A. Future studies are aimed at deconvoluting these mechanistically distinct pathways.
- 25.Crich D, Quintero L. Chem Rev. 1989;89:1413–1432. [Google Scholar]
- 26.Fleming I, Dunogues J, Smithers R. Org React. 1989;37:57–193. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.