

Abstract
Background
Transplant arteriosclerosis (TA) is the pathognomonic feature of chronic rejection, the primary cause of allograft failure. We have shown that the NF-κB inhibitory protein A20 exerts vasculoprotective effects in endothelial (EC) and smooth muscle cells (SMC), and hence is a candidate to prevent TA. We sought direct proof for this hypothesis.
Methods
Fully mismatched, C57BL/6 (H2b) into BALB/c (H2d), aorta to carotid allografts were pre-perfused with saline, recombinant A20 adenovirus (rAd.A20) or rAd.βgalactosidase, implanted, harvested 4 weeks after transplantation, and analyzed by histology, immunohistochemistry, and immunofluorescence staining. We measured indoleamine 2,3-dioxygenase (IDO), IL-6 and TGFβ mRNA and protein levels in non-transduced, and rAd.A20 or rAd.βgal-transduced human SMC cultures following cytokine treatment.
Results
Vascular overexpression of A20 significantly reduced TA lesions. This correlated with decreased graft inflammation and increased apoptosis of neointimal SMC. Paradoxically, T cell infiltrates increased in A20-expressing allografts, including the immunoprivileged media, which related to A20 preventing IDO upregulation in SMC. However, infiltrating T cells were predominantly T regulatory cells (CD25+/FoxP3+). This agrees with A20 inhibiting IL-6 and promoting TGFβ production by medial SMC and in SMC cultures exposed to cytokines, thereby favoring differentiation of regulatory over pathogenic T cells.
Conclusions
In summary, A20 prevents immune-mediated remodeling of vascular allografts, therefore reduces TA lesions by affecting apoptotic and inflammatory signals and modifying the local cytokine milieu to promote an immunoregulatory response within the vessel wall. This highlights a novel function for A20 in local immunosurveillance, which added to its vasculoprotective effects, supports its therapeutic promise in TA.
Keywords: Graft arteriosclerosis, A20/tnfaip3, Smooth muscle cells, Tregs, IL-6
Despite progress in controlling acute allograft rejection, 5% of kidney and heart allografts fail each year post-transplantation. This is mostly due to chronic rejection, with transplant arteriosclerosis (TA) being one of its pathognomonic features (1). TA is particularly dramatic in cardiac transplant recipients, affecting >50% of patients at 10 years, and is the principal cause of late death and allograft dysfunction (2). TA is a form of accelerated atherosclerosis resulting from chronic inflammation initiated by both immune and non-immune insults (3, 4). Much progress was made in defining molecular signals promoting TA, including IFNγ, IL-17, IL-6, chemokines, antibodies, and complement (5-9). However, current therapies focused on increasing immunosuppression largely failed to circumvent the natural course of TA, while further immunocompromising the host (10).
We propose an alternative approach to preventing TA through modifying the vessel wall so it resists transplant-related injury. We previously demonstrated that gene therapy with the ubiquitin-editing (11) and NF-κB inhibitory protein A20 (12) prevents and cures neointimal hyperplasia in a rat carotid balloon injury model (13, 14) and protects from accelerated atherosclerosis in diabetic ApoE-null mice (14). A20 does so by maintaining vascular homeostasis, thanks to its anti-inflammatory and anti-apoptotic functions in EC (15-17) and to inhibiting SMC activation and proliferation while promoting neointimal SMC apoptosis (13, 14, 18). We propose that A20-based therapies to the vessel wall could protect from TA. In this study, we provide direct in vivo proof for the protective effect of A20 against TA in a mouse aorta to carotid artery allograft model. Vascular allografts are adequate alternatives to cardiac allografts for the study of TA (19), as they offer the advantages of easier quantification of vascular changes and better accessibility for testing vascular-targeted therapies.
RESULTS
Overexpression of A20 in Vascular Aortic Allografts Prevents Development of TA
Saline and rAd.βgal-treated aorta to carotid allografts across full MHC mismatch, showed significant neointimal hyperplasia by 4 weeks, with intima to media (I/M) ratios reaching 1.92±0.50 (n=5 mice/group) and 1.74±0.45 (n=6), respectively (Fig. 1A&B). In contrast, overexpression of A20 significantly inhibited neointima formation with an I/M ratio of 0.40±0.08 (n=5; P<0.05; Fig. 1A&B). Syngeneic (C57BL/6 to C57BL/6) allografts had minimal neointima (I/M=0.27±0.1), reflecting the vessel response to procurement and surgery injury. We kept the concentration of rAd. administered per allograft below the toxic range (5×107 pfu), as recommended (20). This limited inflammatory responses to viral antigens, thereby containing viral-triggered neutrophil infiltration while still permitting transgene expression for up to 4 weeks following transduction (Fig. 1C). Since both saline and rAd.βgal-treated aortic allografts showed comparable TA lesions and cell infiltrate profile (given minimal viral toxicity), these groups were combined for statistical analysis of immunohistochemistry (IHC) and immunofluorescence (IF) grading, and referred to as controls.
Figure 1.
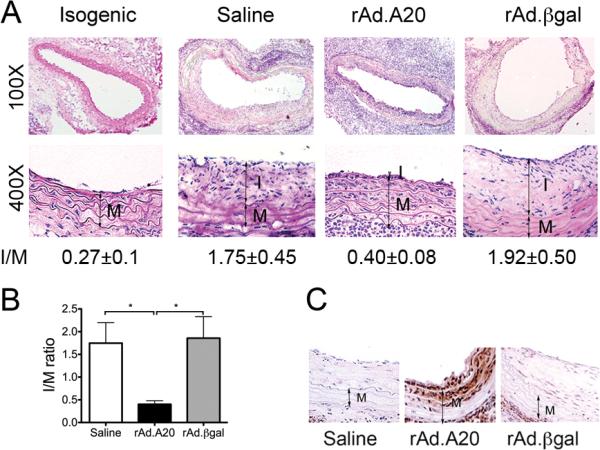
Overexpression of A20 decreases neointima formation in aorta to carotid allografts. A. Representative photomicrographs of hematoxylin-eosin stained whole and partial vessel cross-sections of allografted vessels 4 weeks after transplantation show lesions of TA. B. Morphometric analysis of intima/media (I/M) ratios demonstrates a significant decrease in neointima formation in rAd.A20-treated allografts, as compared to saline and rAd.βgal controls. 6-10 sections per vessel were measured using Image J. Results represent mean±SE of 5 to 6 mice per group. C. Overexpression of A20 in the grafts was confirmed by immunostaining (IS) that showed A20 expression in EC and the two first layers of medial SMC of the allograft five days after transduction. I-intima, M-media, original magnification X400. *p<0.05.
Overexpression of A20 in Aortic Allografts Decreases Inflammation and Increases Apoptosis of Neointimal SMC
The immunostaining intensity of the NF-κB dependent pro-inflammatory adhesion molecule VCAM-1 was significantly decreased in neointimal and medial layers of A20-overexpressing aortic allografts when compared to controls (Fig. 2A; p=0.015). This was associated with a decrease in graft infiltrating monocytes/macrophages (trend) and neutrophils (p=0.03) (Fig. 2). Furthermore, intensity of immunostaining of inducible nitric oxide synthase (iNOS) was two-fold higher in medial SMC of A20-overexpressing allografts than in controls (Fig. 2B, p=0.0005). This correlated with heightened apoptosis of neointimal SMC, contrasting with scarce apoptosis in control allografts, as evaluated by Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL, Fig. 2C; p=0.02). These results corroborate what we showed in a rat carotid balloon angioplasty model, i.e. that overexpression of A20 in SMC increases iNOS expression, which specifically causes apoptosis of neointimal SMC (13). We confirmed that apoptosis occurred in neointimal SMC by showing overlapping IF staining of the executioner caspase 3 with the SMC-specific marker, SMC alpha-actin (SMA) (Fig. 2B). We also noted few TUNEL+ EC in control but not A20-overexpressing aortic allografts, which agrees with the anti-apoptotic function of A20 in EC and reinforces its vasculoprotective role, in part through its opposite anti versus pro-apoptotic effects in EC and neointimal SMC (17).
Figure. 2.

Overexpression of A20 decreases inflammation in aortic allografts while increasing apoptosis in neointimal SMC. Representative IHC photomicrographs show significantly A. Decreased VCAM-1 immunostaining in the neointimal and medial layers of rAd.A20 transduced vascular allografts at 4 weeks after transplant, as opposed to saline and rAd.βgal-treated vascular allografts; This correlated with decreased infiltration of the vascular allografts by monocytes/macrophages and neutrophils, as evaluated by immunostaining with F4/80 and Gr-1 antibodies, respectively. B. Increased iNOS immunostaining in the media of rAd.A20-treated aortic allografts correlating with C increased number of TUNEL positive (blue) neointimal SMC per mm2, as adjusted by Adobe scaling, when compared to saline and rAd.βgal treated allografts. Overlay of IF staining for smooth muscle cell α-actin SMA (red), Caspase 3 (green) and 4’,6-diamidino-2-phenylindole (DAPI, nuclear staining, blue) confirm that most TUNEL+ cells are neointimal SMC (yellow overlay). In A, B, and C, each bar represents mean±SE of immunostaining score, or number of F4/80, Gr-1, and TUNEL+ cells/HPF of 3-4 mice in rAd.A20-treated group and 4-7 mice in the control group. The control group represents combined saline and rAd.βgal treated vascular allografts (2-3 saline and 3-4 rAd.βgal-treated). I-intima, and M-media, original magnification X400 for VCAM-1, iNOS, TUNEL, and Caspase 3/SMA, and X200 for F4/80 and Gr-1. *p<0.05, ***p<0.001.
A20 Protects from TA Despite Increased Number of Graft Infiltrating T Cells and Breach of Media Immunoprivilege
Immunostaining for total (CD3), helper (CD4), and cytotoxic (CD8) T cells revealed substantial T cell infiltrates in all allografts, regardless of treatment (Fig. 3 A). We even noted a trend towards higher number of CD3+, and CD4+ T cells in A20-overexpressing versus control aortic allografts. This trend reached significance for CD8+ T cells (p=0.04). T cells in A20 overexpressing allografts were present across all vascular layers including the usually immunoprivileged media, indicating a breach of this immunoprivilege in these grafts as compared to controls (Fig. 3 A). Similarly, we noted a significant increase in B cell infiltrates mostly in the adventitia of A20 overexpressing allografts (p=0.009; Fig. 3A).
Figure 3.

Overexpression of A20 increases T and B-cell infiltration in vascular aortic allografts and breaks media immunoprivilege by inhibiting inflammation-induced IDO upregulation. A. Representative photomicrographs show increased numbers of CD3+, CD4+ and CD8+ T cells, as well as B220+ B cells in rAd.A20 as compared to saline and rAd.βgal treated aortic allografts, albeit this increase was only significant for CD8+, and B cells. Graphs depict numbers of CD3+, CD4+, CD8+, B220+ cells/ high power field (HPF). Each bar represents the mean±SE of CD3+, CD4+, CD8+, or B220+ cells/ HPF of 3-4 mice in rAd.A20-treated group and 4 mice in the control group combining 2 saline and 2 rAd.βgal-treated allografts. Breach of media immunoprivilege relates to overexpression of A20 in SMC preventing upregulation of IDO in response to inflammatory insults, as shown in B. HCASMC cultures, and C. in media of aortic allografts in vivo. B. Representative Western blots of cell lysates from non-transduced (NT), rAd.A20, and rAd.βgal transduced SMC stimulated for 24 hours with IL-17 (20 ng/mL), a triple cytokine (TC) cocktail (400 U/ml TNFα, 100 U/ml IL-1β, 400 U/ml IFNγ), or a combination of TC and IL-17, immunoblotted with anti-IDO and anti-GAPDH (loading control) antibodies. Bar graphs represent densitometric quantification of migrating bands from 3 independent experiments, expressed as mean±SE fold induction of control (C=non transduced, untreated cells). C. Representative immunofluorescence staining of IDO demonstrates increased expression in media of saline and rAd.βgal treated, but not rAd.A20-treated aortic allografts. Color codes are as follows: Red-IDO, blue-4’,6-diamidino-2-phenylindole (DAPI) nuclear staining, green-elastic lamina auto fluorescence. I-intima, M-media, and A-adventitia, original magnification X400. *p<0.05, **p<0.01, ***p<0.001.
Media immunoprivilege results from cytokine-induced expression of indoleamine 2-3 dioxygenae (IDO), the rate-limiting enzyme in tryptophan metabolism (21, 22). Accordingly, we evaluated whether overexpression of A20 in human coronary artery SMC (HCASMC) cultures affects cytokine-induced upregulation of IDO. Overexpression of A20 in HCASMC inhibited IDO upregulation, as measured by Western blot (WB) analysis, following stimulation with a triple cytokine (TC) cocktail (TNFα, IL-1β, and IFNγ), IL-17, or TC + IL-17, as compared to nontransduced (NT) or rAd.βgal-transduced cells (Fig. 3B). We confirmed by IF staining that A20-overexpressing allografts showed minimal IDO expression in their media, while saline and rAd.βgal-treated allografts did (Fig.3C).
Overexpression of A20 Shifts the Immune Response to Aortic Allografts from a Pathogenic Th1/Th17 Towards an Immunoregulatory Phenotype
Despite a significant T cell infiltrate, A20 overexpressing allografts were essentially devoid of IFNγ and IL-17 IF staining, as opposed to controls (p=0.0002 for IFNγ and p=0.03 for IL-17; Fig. 4A), indicating a blunted Th1 and Th17 response. Accordingly, we checked whether T cells infiltrating A20-overexpressing allografts were of the immunoregulatory phenotype (Tregs). Immunostatining for the Treg-specific transcription factor, Forkhead box P3 (FoxP3), and for the IL-2 receptor, CD25, demonstrated significant enrichment of CD25/FoxP3+ T cells in rAd.A20-treated allografts as compared to controls (p=0.0008 for FoxP3 and p=0.02 for CD25; Fig. 4B).
Figure 4.

Overexpression of A20 favors an immunoregulatory T Cell response. A. Representative immunofluorescence staining shows decreased adventitial expression of IFNγ and IL-17 in rAd.A20-treated aortic allografts, as compared to saline and rAd.βgal-treated vascular allografts. Color codes are as follows: red-IFNγ or IL-17, blue-DAPI nuclear staining, green-elastic laminae auto-fluorescence. Graphs depict grading scores for the IF staining within vascular allograft. Each bar represents the mean±SE IF score from 3 mice in the rAd.A20-treated group and 4 mice in the control group (2 saline and 2 rAd.βgal-treated) for IFNγ statining, and from 4 mice in the rAd.A20-treated group and 6 mice in the control group (3 saline and 3rAd.βgal-treated) for IL-17 staining. B. Representative immunohistochemistry photomicrographs show increased number of CD25+ and FoxP3+ T cells/HPF in rAd.A20-treated aortic allografts, as compared to controls (saline and rAd.βgal treated) vascular allografts. Each bar represents the mean±SE number of CD25+ and FoxP3+ cells/HPF from 4 mice in the rAd.A20-treated group and 5 mice in the control group (2 saline and 3 rAd.βgal-treated.) I-intima, M-media and A-adventitia, original magnification X400. *p<0.05, **p<0.01, ***p<0.001.
A20 Skews the Immune Response in Aortic Allografts by Modulating SMC Cytokine Signature
We evaluated the expression of the Treg/Th17-driving cytokines, TGFβ and IL-6, in aortic allografts. We identified IL-6 IF staining in medial and neointimal SMC of control allografts, suggesting that these cells were the major source of vascular IL-6 (Fig. 5A). In contrast, IL-6 IF staining in medial SMC of rAd.A20-treated allografts was significantly decreased (p=0.03). Decreased in vivo IL-6 expression in A20-overexpressing allografts could stem from reduced local inflammatory cues (less IFNγ) driving IL-6 upregulation, and/or from decreased SMC responses to such inflammatory signals. Having already shown the first (Fig. 4A), we confirmed the latter by demonstrating that overexpression of A20 in HCASMC cultures significantly inhibited cytokine-mediated upregulation of IL-6 mRNA (p<0.001, Fig. 5B) and protein secretion (p<0.01 and <0.05; Fig. 5 B) when compared to NT and rAd.βgal transduced HCASMC.
Figure 5.

A20 overexpression decreases IL-6 and increases TGFβ expression in medial SMC of aortic allografts, and in cytokine treated HCASMC. A. Representative IF staining shows a significant decrease in IL-6 staining, and a trend towards increased staining of TGFβ in rAd.A20-treated medial SMC, as compared to controls (saline and rAd.βgal-treated vascular allografts). Graphs show grading scores for the IF staining (IS) within the media. Each bar represents the mean±SE grading score from 5 (rAd.A20) and 6 (3 saline and 3 rAd.βgal) different mice per group with the control group combining saline and rAd.βgal-treated vascular allografts. Color codes are as follows: Red-IL-6 or TGFβ, blue-DAPI nuclear staining, green-elastic lamina auto-fluorescence. I-intima, M-media, and A-adventitia. Original magnification in A and B X400; *p<0.05. B. IL-6 mRNA (6h) expression, as measured by real-time PCR (qPCR), and protein secretion (24h), as measured by ELISA, are significantly decreased in rAd. A20-transduced SMC cultures as compared with non-transduced (NT) or rAd.βgal-transduced SMC, 6 and 24 hours, respectively, following treatment with a triple cytokine cocktail (TC) including TNFα, (400 U/ml), IL1β (100U/ml) and IFNγ (400 U/ml). C. TGFβ mRNA expression, as measured by real-time PCR (qPCR), and protein secretion (24h), as measured by ELISA, are significantly increased in SMC transduced with rAd.A20, as compared with NT or rAd. βgal-transduced SMC, 6 and 24 hours, respectively, following treatment with TC. In both B and C, qPCR amplification was normalized against cyclophilin A (Cyp). In ELISA assays IL-6 (pg/mg of protein) and ng̃mgọfproteiṇ levels are expressed as fold change of non-transduced, untreated control (C), which allows normalizing differences between experiments and cell donors. Each bar represents the mean± SEM of 3 independent experiments performed using primary cells from 3 different donors. *p<0.05, **p<0.01, ***p<0.001.
There was also greater TGFβ IF staining in medial SMC of rAd.A20-tranduced allografts as compared to controls, although the difference failed to reach significance (Fig. 5B). However, overexpression of A20 in HCASMC cultures significantly increased TGFβ mRNA levels (p<0.001, and p<0.01) and protein secretion (p<0.001) following TC treatment, while these levels were moderately reduced in NT and rAd.βgal-transduced SMC (Fig. 5C).
Discussion
The work presented in this manuscript provides the first direct cause-effect relationship between A20 and vascular remodeling in TA. We demonstrate that A20 prevents TA, at least in part, through its direct vasculoprotective effects in EC and SMC. Overexpression of A20 in aortic allografts achieved a potent anti-inflammatory effect in EC and SMC, shutting down NF-κB activation, as evidenced by a significant decrease in the expression of NF-κB-dependent, VCAM-1. By promoting mononuclear leukocyte trafficking through the allograft vasculature, upregulation of VCAM-1 in EC and SMC is key to TA pathogenesis (23). Accordingly, A20-treated allografts had less monocyte/macrophage and neutrophil infiltrates.
In addition, overexpression of A20 in mouse vascular allografts drastically increased iNOS expression in medial SMC, which correlated with heightened apoptosis of neointimal SMC. This result is consistent with our previous data from a rat carotid balloon angioplasty model and from HCASMC cultures, demonstrating that overexpression of A20 increases iNOS mRNA and protein expression, leading to increased NO, an established pro-apoptotic stimulus to SMC (13). In fact, mere iNOS overexpression prevents TA in rat aortic allografts (24). Contrasting with neointimal SMC, A20 overexpression decreased EC apoptosis in aortic allografts. This is consistent with the potent anti-apoptotic effect of A20 in EC against a broad spectrum of immune and non-immune stimuli (17).
We had initially surmised that prevention of TA in A20-overexpressing vascular allografts could relate to the immune system being “blinded” towards inflammation-resistant, hence low “danger” allografts (25). Our data invalidate this hypothesis by demonstrating more T cell infiltrates in TA-free A20-overexpressing aortic allografts (including in the immunoprivileged media) than in controls, implying that immune recognition occurred and breached medial immunoprivilege. This paradox of A20 protecting from TA despite increasing T cell infiltration and breaching media immunoprivilege is in part reconciled by A20 decreasing IFNγ levels and signaling. The Th1 cytokine IFNγ has been posited as a critical mediator of TA. Clinically, Th1 cells dominate T cell infiltrates in human cardiac allografts undergoing chronic rejection (26). Furthermore, exogenous IFNγ alone is sufficient to induce TA in human vascular allografts transplanted into immuno-incompetent mice (27) through its direct pathogenic effects on EC and SMC (9). Thus, strategies to reduce IFNγ levels and/or signaling are highly attractive for preventing TA. Based on our data, overexpression of A20 in SMC of vascular allografts achieves both tasks: reducing IFNγ+ Th1 cells, hence local IFNγ production and interrupting IFNγ signaling.
Break of medial immunoprivilege in A20-overexpressing aortic allografts could also reflect decreased IFNγ levels/signaling. Media immunoprivilege results from IFNγ-mediated increase of IDO expression in SMC, as IDO depletes tryptophan, required for T cell proliferation and survival, and promotes the accumulation of its catabolites (Kyrunenines) that induce T cell apoptosis (21, 22, 28). Our in vivo and in vitro data demonstrate that overexpression of A20 in aortic allografts reduces both IFNγ levels (by decreasing the number of Th1 cells), and IFNγ signaling in SMC, thereby inhibiting IDO upregulation. We are currently investigating the molecular basis for A20-dependent interruption of IFNγ signaling in SMC.
In addition, A20-overexpressing aortic allografts had significantly fewer Th17 infiltrating cells. Th17 cells are key mediators of autoimmune disease, allograft rejection, and TA (5, 29). Mouse Th17 cells and Tregs share a common origin, and are both generated in response to TGFβ, with Th17 cells developing when IL-6 is simultaneously present to repress FoxP3. Consistent with a decreased number of Th17 cells in rAd.A20-treated allografts, IL-6 immunostaining was nearly absent in medial SMC. We confirmed that overexpression of A20 in HCASMC cultures inhibits cytokine-mediated upregulation of IL-6. This is consistent with IL-6 transcription relying in part on NF-κB activation, hence inhibited by the NF-κB-inhibitory protein A20 (12, 13, 30, 31). From a therapeutic standpoint, we propose that decreased IL-6 levels is fundamental to the protective effect of A20 against TA as it halts the generation of pathogenic Th17 cells, reduces inflammatory injury to EC/SMC, and contributes to promoting acceptance by favoring a Treg response.
In contrast to IL-6, we detected more TGFβ IF staining in the media of rAd.A20-treated allografts, while its expression was almost absent in controls. Here again, overexpresion of A20 increased TGFβ expression in cytokine-treated HCASMC, establishing in vitro causality in a human system. In the absence of IL-6, TGFβ drives Treg differentiation. Consequently, we noted significantly more FoxP3+ Tregs in A20-overexpressing aortic allografts than in controls. This is likely essential to protection from TA, since human Tregs were shown to protect vascular allografts from TA in a chimeric humanized mouse system (32). Since vascular A20 locally modulates cytokine production, we anticipate that differentiation of FoxP3+ T cells occurs within the graft, setting the basis for tissue-based immune surveillance (33). We wish to explore the molecular basis for A20-induced upregulation or maintenance of adequate TGFβ levels in SMC facing inflammation. Beyond protecting from TA, A20-induced modulation of the immune response could be relevant to graft acceptance, given the role of Tregs in inducing tolerance to allografts (34).
A20-overexpressing allografts showed also a significant increase in B cell infiltrates. The involvement of increased B cells (possibly of the protective phenotype) in A20-induced protection from TA needs further exploration. This intriguing result is in keeping with data observed in spontaneously accepted C57BL/6 to B10.BR mouse kidney allografts that showed a pattern of early and abundant cell infiltrates associating B and FOxP3+T cells, similar to what we observe (35).
Altogether, these results highlight a novel immunomodulatory function of A20 in the vessel wall through its multiple effects on cytokine expression and signaling, mainly in SMC. This, in addition to its direct vasculoprotective effects in EC and SMC, supports its therapeutic promise in preventing TA. The causal nature of these experiments lends mechanistic credibility to our correlational data, associating vascular A20 expression in human (Kreis et al, Necker Hospital, unpublished data) and rat kidney allografts, and hamster to rat heart xenografts, with the absence of TA (36, 37). Furthermore, our results emphasize the emerging paradigm of a central role for the allograft in determining its own fate, owing to its ability to mount an adequate “protective” response to immune and inflammatory injury. These findings are interesting in light of recently reported A20 gene polymorphisms, some of which leading to decreased A20 protein expression/function (38). Such A20 polymorphisms in donor grafts could determine susceptibility to TA.
Materials and Methods
Mouse Cervical Aortic Allograft
Aorta to carotid artery interposition vascular allografts were performed across total MHC mismatched C57BL/6 (H-2b) to BALB/c (H-2d) mice (Charles River Laboratories; Wilmington, MA), as described (39). Donor aortae were treated in situ, intraluminally, with saline or 5×107 plaque forming units (pfu) of rAd.A20 or rAd.βgal in 50μL of saline, for 15min, prior to harvesting. Grafts were recovered 4 weeks later for analysis. All animal procedures were conducted in accordance with the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals, and approved by the Beth Israel Deaconess Medical Center Institutional Committee for Use and Care of Laboratory Animal.
Histology, Immunohistochemistry and Immunofluorescence
For morphometric analysis, I/M ratio were measured using NIH ImageJ software on hematoxylin/eosin stained sections, as described in the supplementary methods (13). For IHC analysis, frozen sections were incubated with antibodies to human A20, and mouse iNOS (Santa Cruz Biotechnology Inc, Santa Cruz, CA), VCAM-1, CD4, CD8, B-220 and GR-1 (BD Biosciences, San Jose, CA), CD3 (AbD Serotec, Raleigh, NC), F4/80 (AbD Serotec, Raleigh, NC), CD25 and FoxP3 (Biolegend, San Diego, CA), followed by biotinylated secondary antibodies (Vector, Burlingame, CA) (18). For IF studies, antibodies to mouse IL-17 (Abcam, Cambridge, MA), IFNγ, anḍIL̃ 6 (BD Biosciences), TGFβ (Santa Cruz), IDO (Millipore, Billerica, MA), and Caspase-3 (Cell Signaling Technologies, Danvers, MA) were used, followed by Alexa Fluor 594 conjugated secondary antibodies (Invitrogen, Carlsbad, CA, USA). Cy-3 labeled anti-SMA antibody was from Sigma. We detected apoptosis using TUNEL (VasoTACS, Trevigen, Gaithersburg, MD). IHC and IF staining were analyzed in a blinded fashion by PS, SE, EK, and CGS. VCAM-1, iNOS, IFNγ, IL-6, and TGFβ staining were graded from 1 to 4 (1=lack, 2=minimal, 3= intermediate, and 4=high level of expression). CD3, CD4, CD8, CD25, and FoxP3 positive cells were scored by automated cell counting using Image J and expressed as cell number per high power field (HPF). TUNEL positive cells were scored by automated cell counting and expressed by mm2/media as scaled on Adobe Photoshop.
Cell Culture and Reagents
HCASMC (Lonza, Walkersville, MD; Genlantis, San Diego, CA) were cultured in SmGM-2 and used between passage 5 and 8. Human embryonic kidney (293) cells were obtained from American Type Culture Collection (Manassas, VI) and cultured as described (15). Human recombinant IL1-β, TNFα, IFNγ and IL-17, used at 100 U/ml, 400 U/ml, 400 U/ml, and 20ng/ml, respectively were purchased from R&D Systems (Minneapolis, MN, USA).
Recombinant Adenoviruses
We generated rAd.A20 using a plasmid provided by Dr. V. Dixit (Genentench, San Francisco, CA) (15). The rAd.βgal was a gift of Dr. Robert Gerard (University of Texas SW, Dallas, Texas). Recombinant adenoviruses were generated, produced and tittered on HEK293 cells, purified by cesium chloride density gradient centrifugation for in vivo administration (13), or by the AdenoPure LS Kit (Puresyn, Malvern, PA) for in vitro experiments. SMC at 90% confluency were transduced with rAd. at a MOI of 500, which leads to transgene expression in >95% of cells.
Western Blot Analysis
The following antibodies were used for WB: anti-human IDO (Millipore), A20/TNFAIP3 (Abcam), GAPDH (Calbiochem/ EMD Biosciences, Gibbstown, NJ), and appropriate secondary antibodies (ThermoScientific, Rockford, IL). Densitometry measurements of identified bands were performed using the ImageJ software and corrected to GAPDH density values.
IL-6 and TGFβ Enzyme-linked Immunosorbent Assay
IL-6 (R&D Systems) and TGFβ (eBioscience, San Diego, CA) were measured in supernatants of SMC using a quantitative sandwich immunoenzyme linked assay. Results were normalized by total protein amount and expressed as relative fold over control (non-transduced, untreated cells).
Quantitative Real-time Reverse-transcriptase Polymerase Chain Reaction (qPCR)
We extracted RNA using Qiagen RNeasy Mini Kit (Qiagen, Valencia, CA) and synthesized cDNA using iScript cDNA Synthesis Kit (BioRad USA, Hercules, CA, USA). qPCR reactions were prepared in duplicate using iTaq Fast SYBR Green Supermix with ROX (BioRad) and gene specific primers (Invitrogen), and performed on a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA). Gene expression was quantified using the relative quantification method of Pfaffl (40). We used Cyclophilin A as the house keeping gene after verifying that its expression did not vary in our experimental system (Fig. S1A) (41). A list of primers and extended methods are provided in supplementary material.
Statistics
Statistics were performed using Prism 5 software (GraphPad, La Jolla CA). Two-way ANOVA with post-hoc Bonferroni test was used to analyze TGFβ and IL-6 qPCR and ELISA. One-Way ANOVA with post-hoc Tukey test was used for I/M ratios. Unpaired t-test was used for IHC and IF values. P<0.05 was considered statistically significant.
Acknowledgements
We thank Dr. Vishva Dixit and Robert Gerard for providing the A20 plasmid and the recombinant β-galactosidase adenovirus. This research was supported by NIH grant RO1 HL080130 and in part by NIH grants RO1 DK063275 and RO1HL57791, as well by the Juvenile Diabetes Research Foundation grant 1-2007-567 to CF. JJS, MDF, SMD, CP and AL were supported by NIH T32 HL07734. PS was supported by a grant from the National Swiss Foundation and EM was supported by fellowship award from the National Council for Scientific and Technological Development (CNPq), Brasil.
Abbreviations
- βgal
βgalactosidase
- Cyp
cyclophilin A
- EC
endothelial cells
- FoxP3
Forkhead Box P3
- HEK
Human embryonic kidney cells
- IF
Immunofluorescence
- IHC
Immunohistochemistry
- iNOS
Induced Nitric Oxide Synthase
- IS
Immunostaining
- MOI
Multiplicity of infection
- NT
non-transduced
- qPCR
quantitative reverse-transcriptase polymerase chain reaction
- rAd.
Recombinant adenovirus
- SMC
smooth muscle cells
Footnotes
The authors declare no conflict of interest.
JJS participated in research design, performance of the surgeries, data collection and analysis, and manuscript writing; MDF participated in research design, performance of the surgeries, data collection and analysis; CGS participated in research design, data collection and analysis, in vitro experiments, figure preparation, statistical analysis and manuscript editing; CRP participated in research design, contributed to performance of the surgeries, data collection and analysis, in vitro experiments and manuscript editing; EC performed all pathology and immunohistochemistry (IHC) studies; HM performed in vitro experiments, analyzed the data and helped in figure preparation; SMD performed in vitro experiments, analyzed the data and helped in manuscript editing; LEC and ERM participated in data analysis, namely blinded reading of intima/media ratios; PS, SE, EK, and AL participated in data analysis, namely blinded reading of IHC slides; SD participated in research design, contributed to performance of in vitro experiments and data collection and analysis; CF participated in research design, data analysis, manuscript writing and sponsored the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cecka JM. The UNOS scientific renal transplant registry. Clin. Transpl. 1999;13:1. [PubMed] [Google Scholar]
- 2.Taylor DO, Edwards LB, Boucek MM, et al. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult heart transplant report--2007. J Heart Lung Transplant. 2007;26(8):769. doi: 10.1016/j.healun.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Potena L, Grigioni F, Ortolani P, et al. Relevance of cytomegalovirus infection and coronary-artery remodeling in the first year after heart transplantation: a prospective three-dimensional intravascular ultrasound study. Transplantation. 2003;75(6):839. doi: 10.1097/01.TP.0000054231.42217.A5. [DOI] [PubMed] [Google Scholar]
- 4.Hillebrands JL, Rozing J. Chronic transplant dysfunction and transplant arteriosclerosis: new insights into underlying mechanisms. Expert Rev Mol Med. 2003;5(2):1. doi: 10.1017/S146239940300557X. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell P, Afzali B, Lombardi G, Lechler RI. The T helper 17-regulatory T cell axis in transplant rejection and tolerance. Curr Opin Organ Transplant. 2009;14(4):326. doi: 10.1097/MOT.0b013e32832ce88e. [DOI] [PubMed] [Google Scholar]
- 6.Mitchell RN, Libby P. Vascular remodeling in transplant vasculopathy. Circ Res. 2007;100(7):967. doi: 10.1161/01.RES.0000261982.76892.09. [DOI] [PubMed] [Google Scholar]
- 7.Wehner J, Morrell CN, Reynolds T, Rodriguez ER, Baldwin WM., 3rd Antibody and complement in transplant vasculopathy. Circ Res. 2007;100(2):191. doi: 10.1161/01.RES.0000255032.33661.88. [DOI] [PubMed] [Google Scholar]
- 8.Belperio JA, Ardehali A. Chemokines and transplant vasculopathy. Circ Res. 2008;103(5):454. doi: 10.1161/CIRCRESAHA.108.182865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tellides G, Pober JS. Interferon-gamma axis in graft arteriosclerosis. Circ Res. 2007;100(5):622. doi: 10.1161/01.RES.0000258861.72279.29. [DOI] [PubMed] [Google Scholar]
- 10.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14(4):387. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 11.Wertz IE, O'Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 12.Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-κB-dependent mechanism. J. Biol. Chem. 1996;271:18068. doi: 10.1074/jbc.271.30.18068. [DOI] [PubMed] [Google Scholar]
- 13.Patel VI, Daniel S, Longo CR, et al. A20, a modulator of smooth muscle cell proliferation and apoptosis, prevents and induces regression of neointimal hyperplasia. FASEB J. 2006;20(9):1418. doi: 10.1096/fj.05-4981com. [DOI] [PubMed] [Google Scholar]
- 14.Shrikhande GV, Scali ST, da Silva CG, et al. O-glycosylation regulates ubiquitination and degradation of the anti-inflammatory protein A20 to accelerate atherosclerosis in diabetic ApoE-null mice. PLoS ONE. 2010;5(12):e14240. doi: 10.1371/journal.pone.0014240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferran C, Stroka DM, Badrichani AZ, et al. A20 inhibits NF-kappaB activation in endothelial cells without sensitizing to tumor necrosis factor-mediated apoptosis. Blood. 1998;91(7):2249. [PubMed] [Google Scholar]
- 16.Longo CR, Arvelo MB, Patel VI, et al. A20 protects from CD40-CD40 ligand-mediated endothelial cell activation and apoptosis. Circulation. 2003;108(9):1113. doi: 10.1161/01.CIR.0000083718.76889.D0. [DOI] [PubMed] [Google Scholar]
- 17.Daniel S, Arvelo MB, Patel VI, et al. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood. 2004;104(8):2376. doi: 10.1182/blood-2003-02-0635. [DOI] [PubMed] [Google Scholar]
- 18.Damrauer SM, Fisher MD, Wada H, et al. A20 inhibits post-angioplasty restenosis by blocking macrophage trafficking and decreasing adventitial neovascularization. Atherosclerosis. 2010;211(2):404. doi: 10.1016/j.atherosclerosis.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ensminger SM, Billing JS, Morris PJ, Wood KJ. Development of a combined cardiac and aortic transplant model to investigate the development of transplant arteriosclerosis in the mouse. J Heart Lung Transplant. 2000;19(11):1039. doi: 10.1016/s1053-2498(00)00195-9. [DOI] [PubMed] [Google Scholar]
- 20.Schulick AH, Newman KD, Virmani R, Dichek DA. In vivo gene transfer into injured carotid arteries. Optimization and evaluation of acute toxicity. Circulation. 1995;91(9):2407. doi: 10.1161/01.cir.91.9.2407. [DOI] [PubMed] [Google Scholar]
- 21.Mellor AL, Munn DH. Tryptophan catabolism and T-cell tolerance: immunosuppression by starvation? Immunol Today. 1999;20(10):469. doi: 10.1016/s0167-5699(99)01520-0. [DOI] [PubMed] [Google Scholar]
- 22.Cuffy MC, Silverio AM, Qin L, et al. Induction of indoleamine 2,3-dioxygenase in vascular smooth muscle cells by interferon-gamma contributes to medial immunoprivilege. J Immunol. 2007;179(8):5246. doi: 10.4049/jimmunol.179.8.5246. [DOI] [PubMed] [Google Scholar]
- 23.Sai S, Iwata A, Thomas R, Allen MD. Vascular cell adhesion molecule-1 up-regulation and phenotypic modulation of vascular smooth muscle cells predate mononuclear infiltration in transplant arteriopathy. J Thorac Cardiovasc Surg. 2001;122(3):508. doi: 10.1067/mtc.2001.113601. [DOI] [PubMed] [Google Scholar]
- 24.Shears LLn, Kawaharada N, Tzeng E, et al. Inducible nitric oxide synthase suppresses the development of allograft arteriosclerosis. J. Clin. Invest. 1997;100:2035. doi: 10.1172/JCI119736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296(5566):301. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 26.van Loosdregt J, van Oosterhout MF, Bruggink AH, et al. The chemokine and chemokine receptor profile of infiltrating cells in the wall of arteries with cardiac allograft vasculopathy is indicative of a memory T-helper 1 response. Circulation. 2006;114(15):1599. doi: 10.1161/CIRCULATIONAHA.105.597526. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Burns WR, Tang PC, et al. Interferon-gamma plays a nonredundant role in mediating T cell-dependent outward vascular remodeling of allogeneic human coronary arteries. FASEB J. 2004;18(3):606. doi: 10.1096/fj.03-0840fje. [DOI] [PubMed] [Google Scholar]
- 28.Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J Exp Med. 2002;196(4):459. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Afzali B, Lombardi G, Lechler RI, Lord GM. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148(1):32. doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsusaka T, Fujikawa K, Nishio Y, et al. Transcription factors NF-IL6 and NF-kappa B synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(21):10193. doi: 10.1073/pnas.90.21.10193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stein B, Yang MX. Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-kappa B and C/EBP beta. Molecular & Cellular Biology. 1995;15(9):4971. doi: 10.1128/mcb.15.9.4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nadig SN, Wieckiewicz J, Wu DC, et al. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat Med. 2010;16(7):809. doi: 10.1038/nm.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wahl SM, Wen J, Moutsopoulos N. TGF-beta: a mobile purveyor of immune privilege. Immunol Rev. 2006;213:213. doi: 10.1111/j.1600-065X.2006.00437.x. [DOI] [PubMed] [Google Scholar]
- 34.Rudensky AY, Campbell DJ. In vivo sites and cellular mechanisms of T reg cell-mediated suppression. J Exp Med. 2006;203(3):489. doi: 10.1084/jem.20060214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C, Cordoba S, Hu M, et al. Spontaneous acceptance of mouse kidney allografts is associated with increased Foxp3 expression and differences in the B and T cell compartments. Transpl Immunol. 2011;24(3):149. doi: 10.1016/j.trim.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Kunter U, Floege J, von Jurgensonn AS, et al. Expression of A20 in the vessel wall of rat-kidney allografts correlates with protection from transplant arteriosclerosis. Transplantation. 2003;75(1):3. doi: 10.1097/00007890-200301150-00002. [DOI] [PubMed] [Google Scholar]
- 37.Bach FH, Ferran C, Hechenleitner P, et al. Accomodation of vascularized xenografts: expression of “protective genes” by donor endothelial cells in a host Th2 cytokine environment. Nature Med. 1997;3:196. doi: 10.1038/nm0297-196. [DOI] [PubMed] [Google Scholar]
- 38.Boonyasrisawat W, Eberle D, Bacci S, et al. Tag Polymorphisms at the A20 (TNFAIP3) Locus Are Associated With Lower Gene Expression and Increased Risk of Coronary Artery Disease in Type 2 Diabetes. Diabetes. 2007;56(2):499. doi: 10.2337/db06-0946. [DOI] [PubMed] [Google Scholar]
- 39.Tomita Y, Zhang QW, Uchida T, et al. A technique of cervical aortic graft transplantation in mice. J Heart Lung Transplant. 2001;20(6):699. doi: 10.1016/s1053-2498(00)00219-9. [DOI] [PubMed] [Google Scholar]
- 40.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bustin SA. Why the need for qPCR publication guidelines?--The case for MIQE. Methods. 2010;50(4):217. doi: 10.1016/j.ymeth.2009.12.006. [DOI] [PubMed] [Google Scholar]